
A New Visualization and Analysis Method for a Convolved Representation of Mass Computational Experiments with Biological Models



Abstract
:1. Introduction
2. Materials and Methods
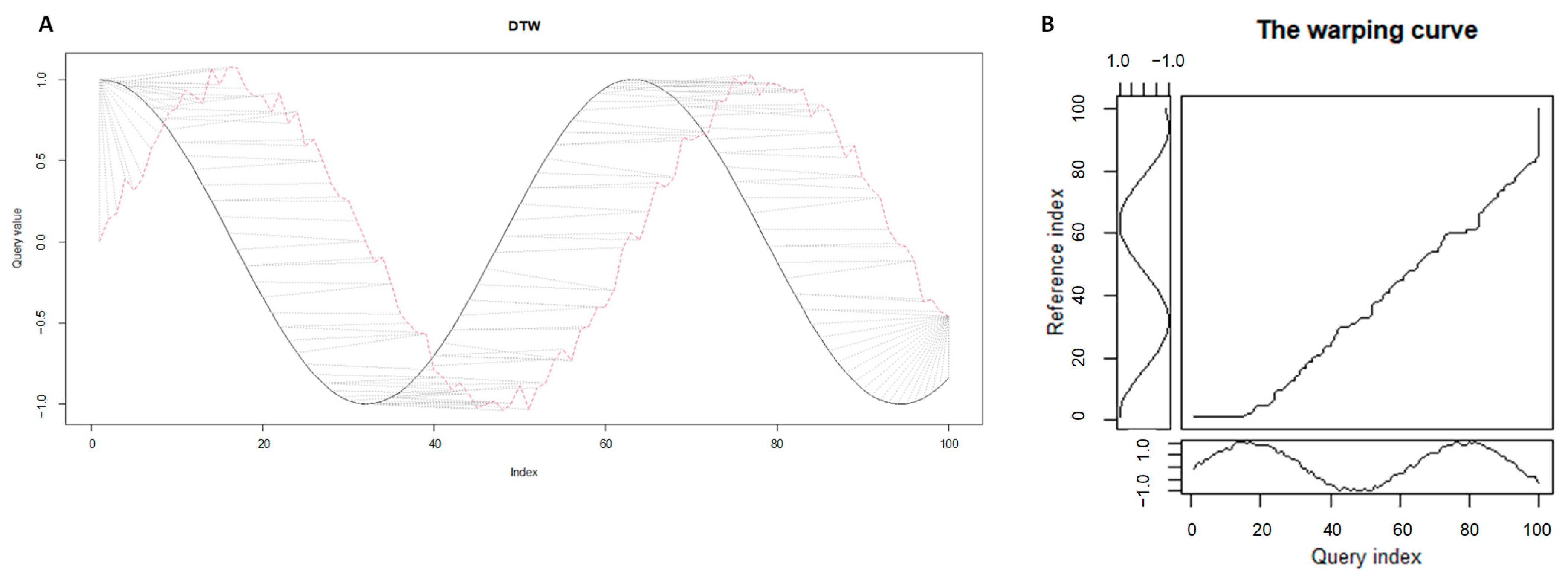
2.1. Time series Analysis with Dynamic Time Warping Algorithm
2.1.1. Formulation of Time Series Alignment Problem
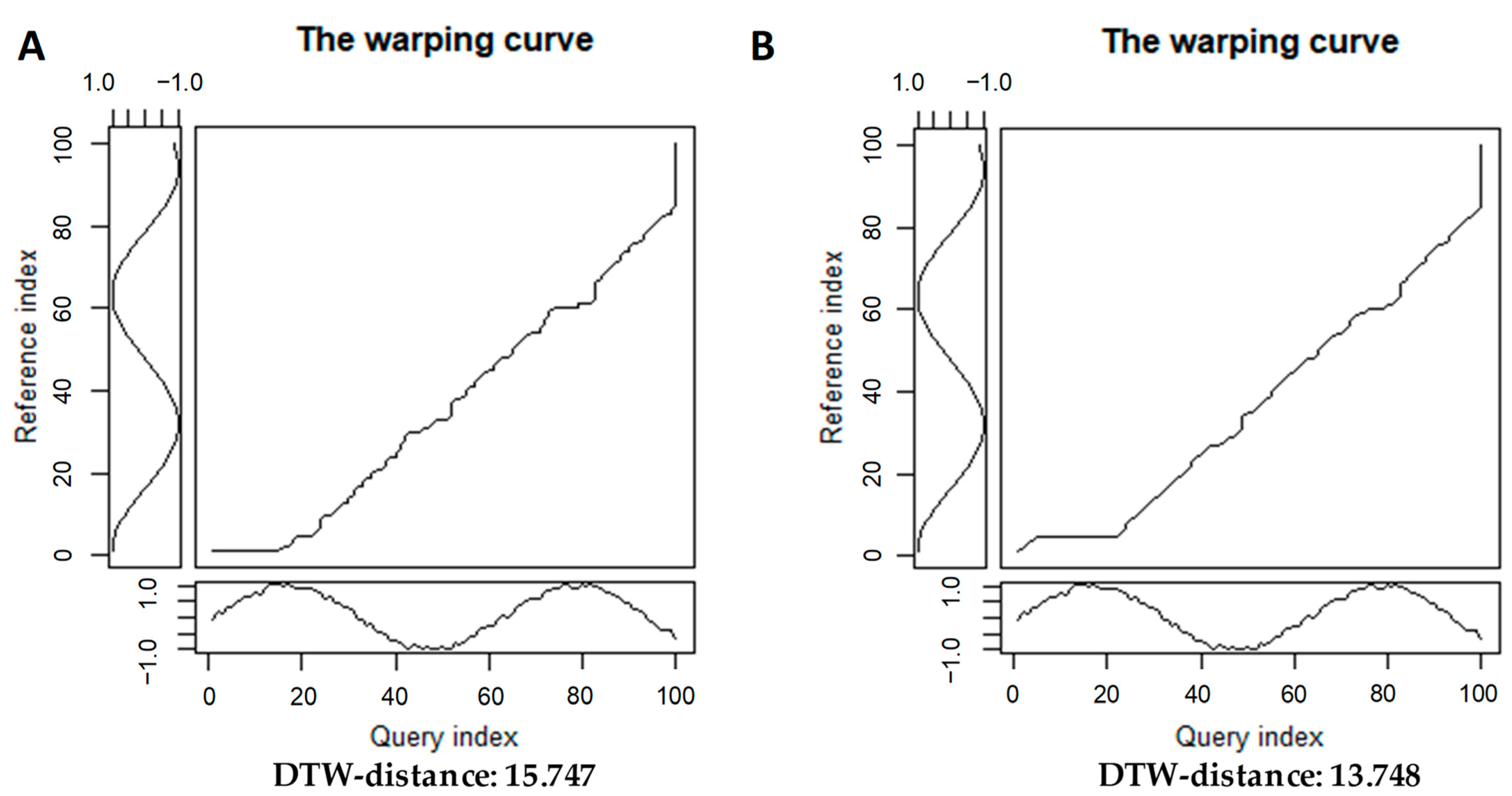
2.1.2. Time Series Alignments: Samples of Step Patterns and Local Slope Constraints for the Warping Curves
2.2. Dimensionality Reduction during Metric Multidimensional Scaling Using Principal Coordinate Analysis
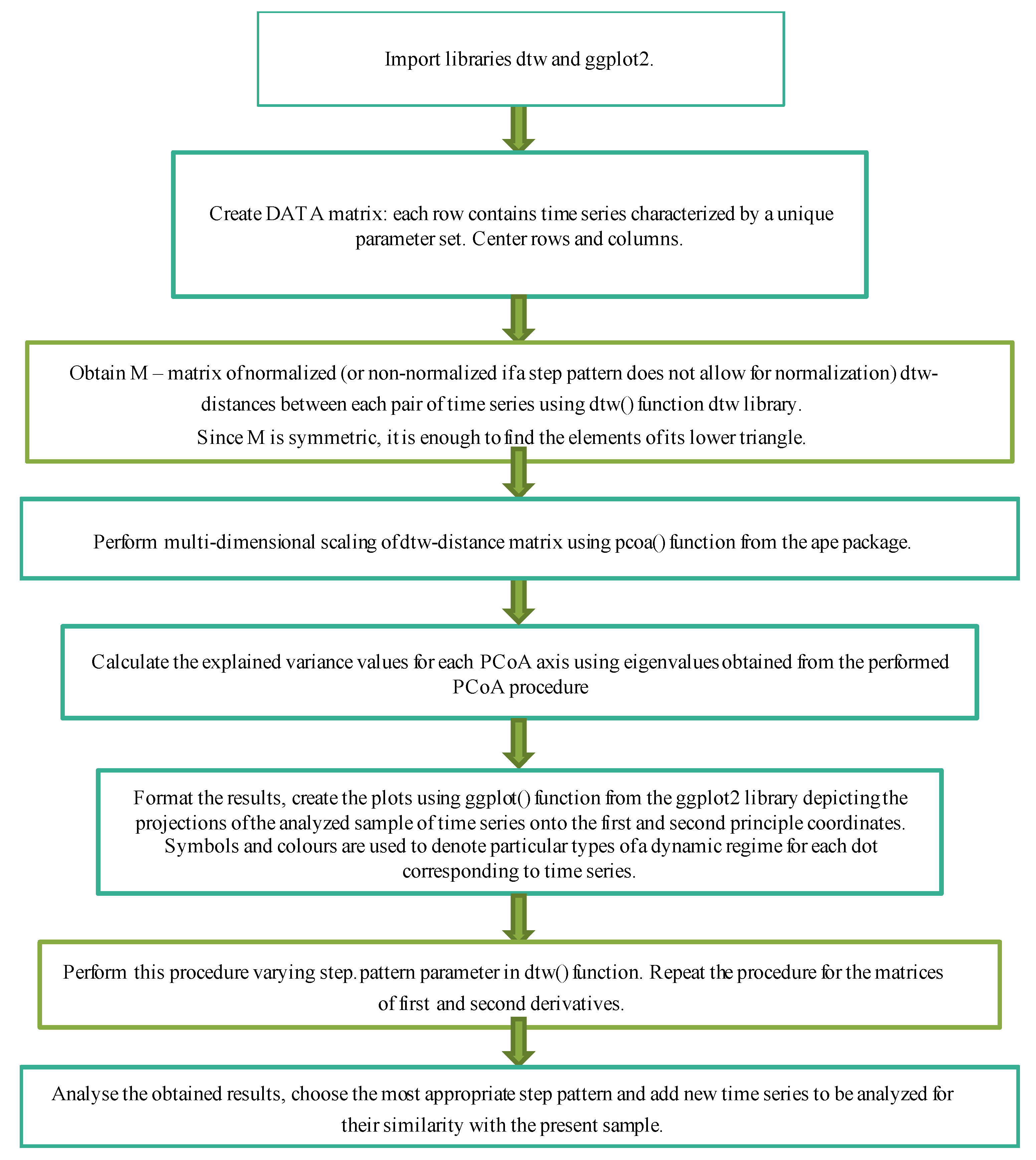
2.3. DTW+PCoA-Based Method for a Convolved Representation of Mass Computational Experiments
- to perform computational experiments with different sets of parameters under consideration;
- to obtain a matrix of DTW distances between all samples;
- to apply principal coordinates analysis to it;
- to qualitatively analyze the obtained results for each of the parameters or for the whole set, determining how the types of dynamic regimes of the model change depending on changes in its parameters.
3. Results
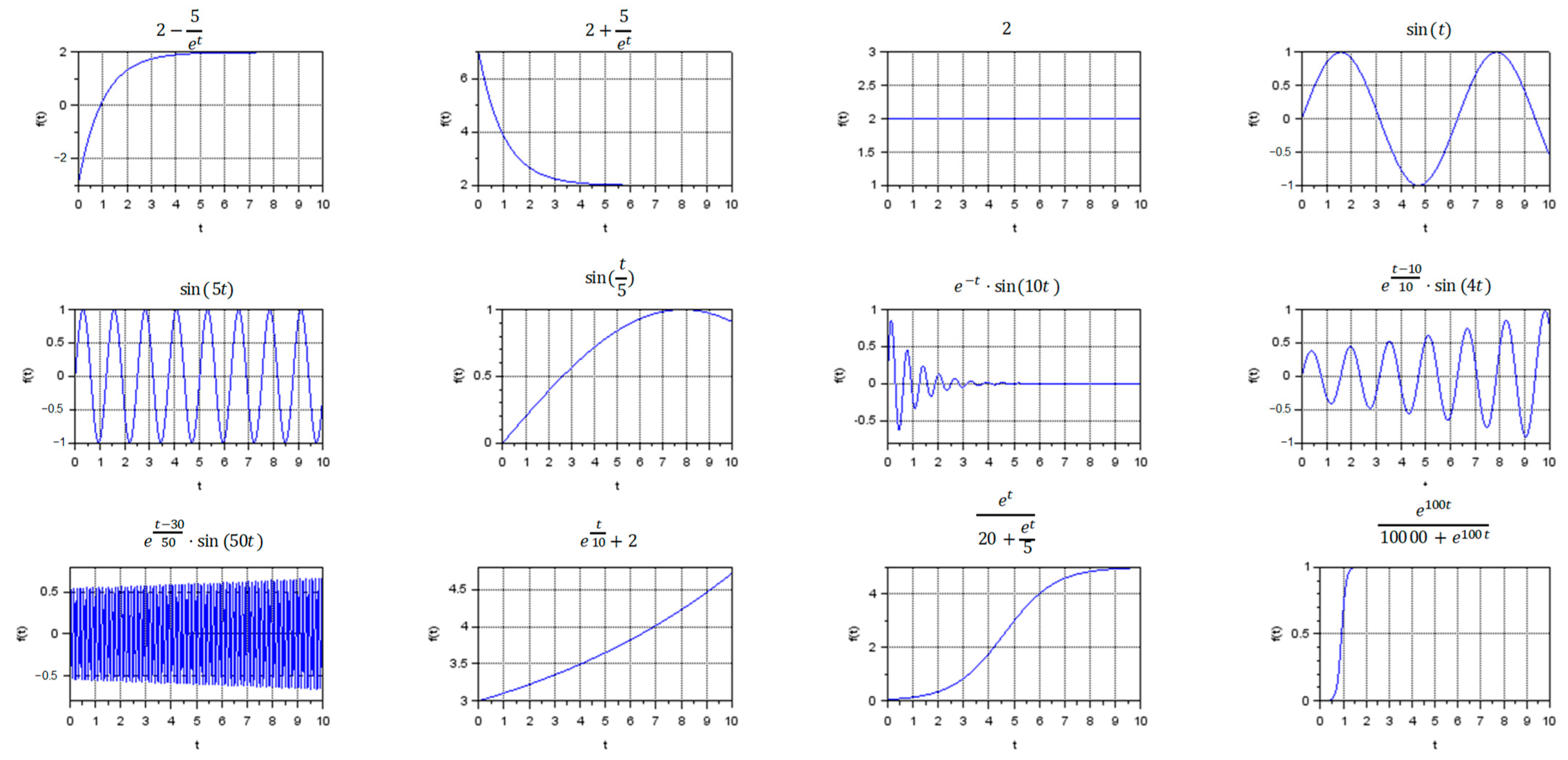
3.1. Visualization of Different Types of Dynamic Regimes
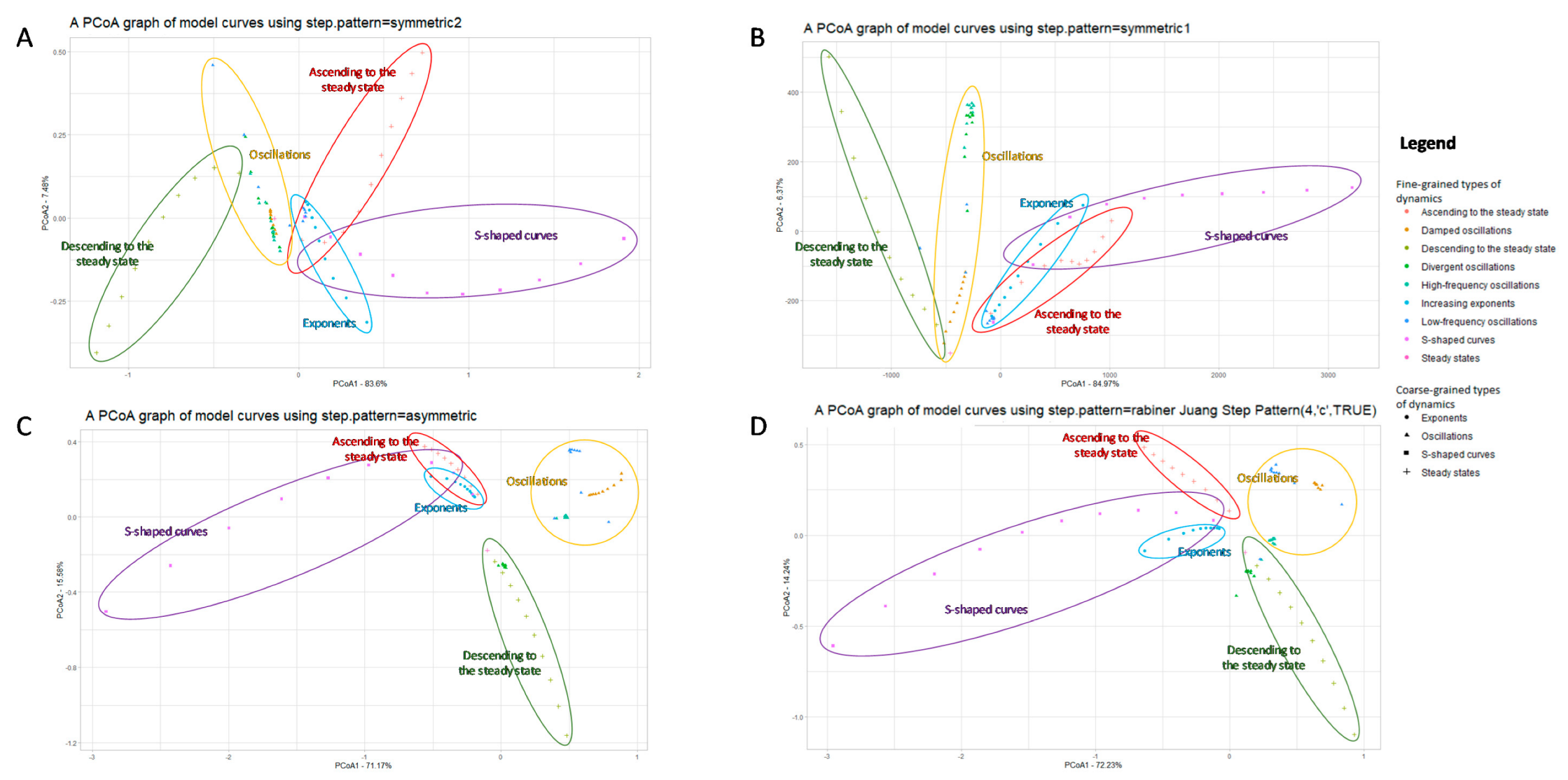
3.1.1. A Basic Application of DTW+PCoA-Based Method with Various Step Patterns
- Stationary regimes, including transient modes;
- Oscillatory regimes, including frequent and rare oscillations with the same magnitude, as well as damped or divergent oscillations with different frequency;
- Exponential growth and exponential decline;
- S-curves.
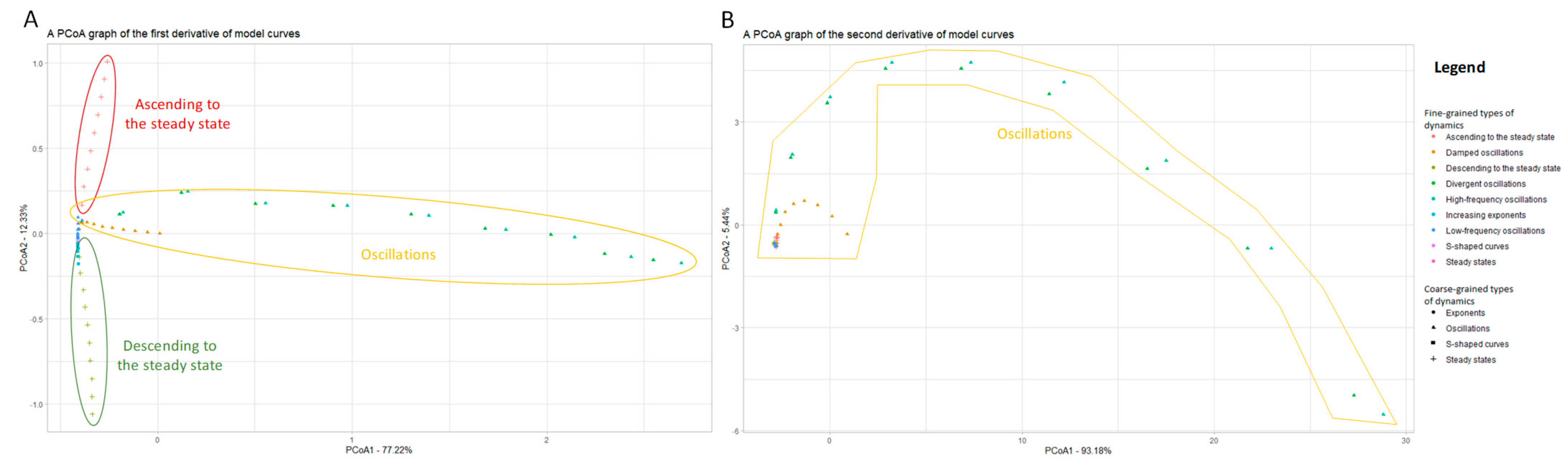
3.1.2. Using Approximations of the Derivatives to Include Additional Information on Curves
3.1.3. A Comparison with Standard Euclidean Principal Coordinate Analysis
3.2. Parametric Sensitivity Analysis of Dynamical Systems Models: Case Study of the Lotka–Volterra Model
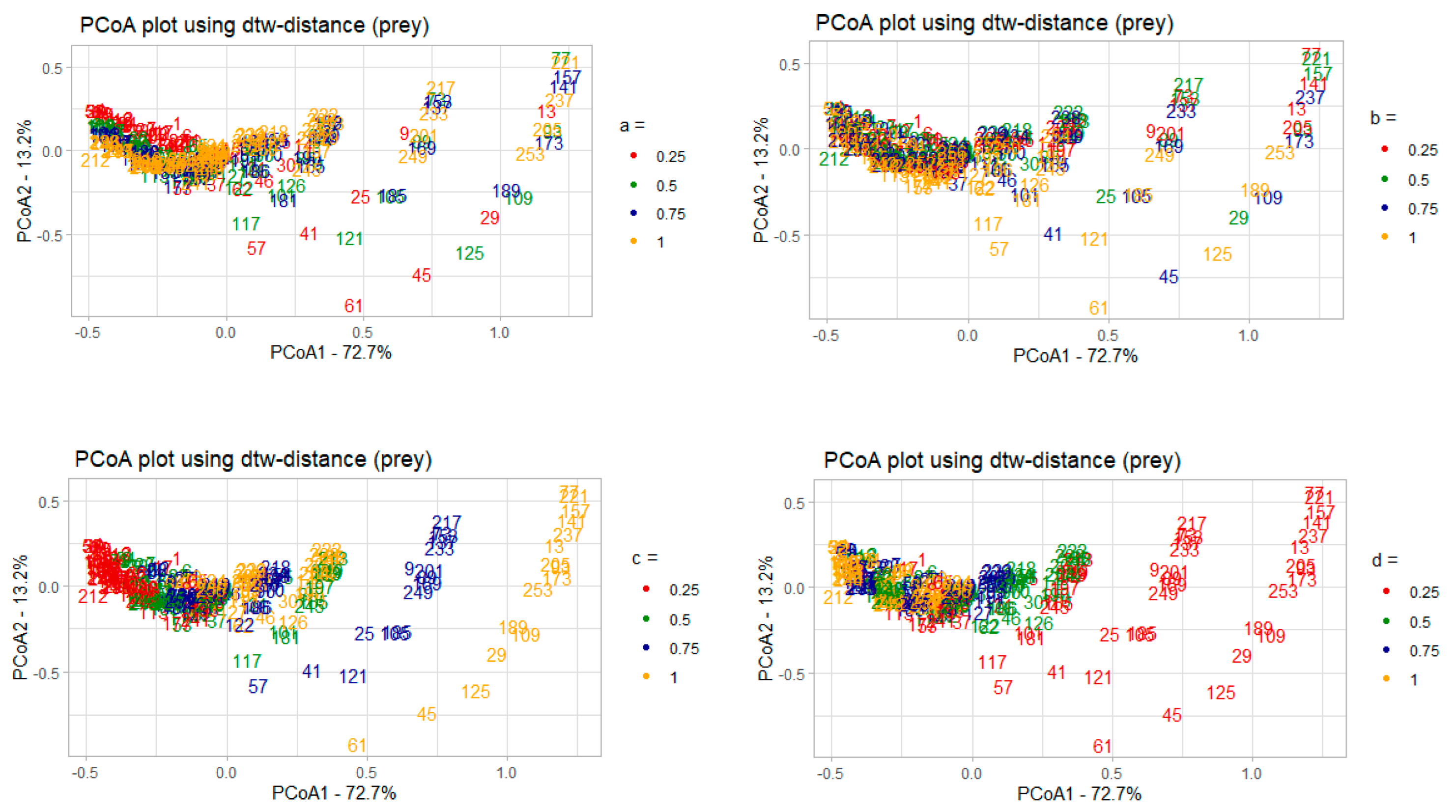
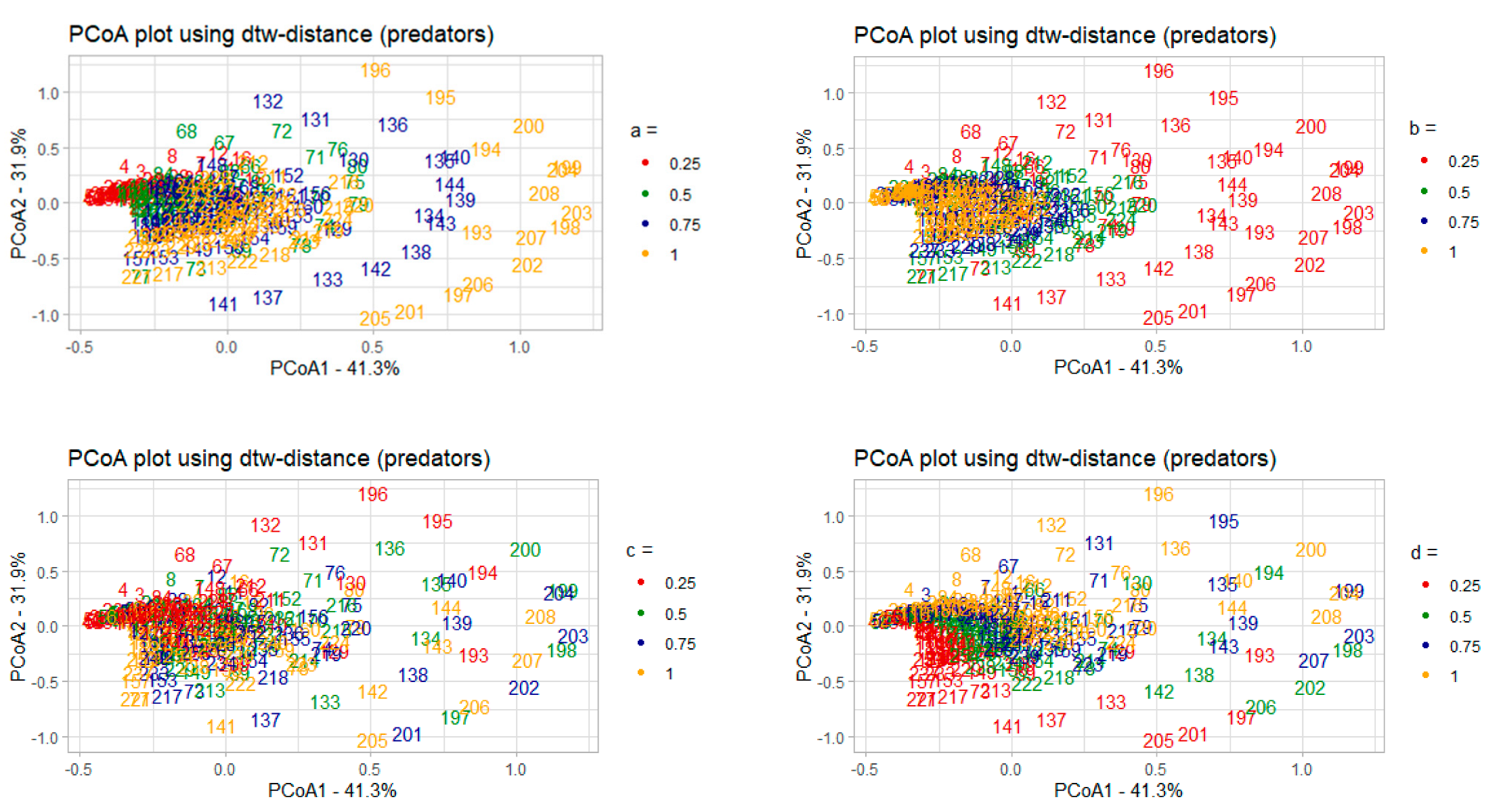
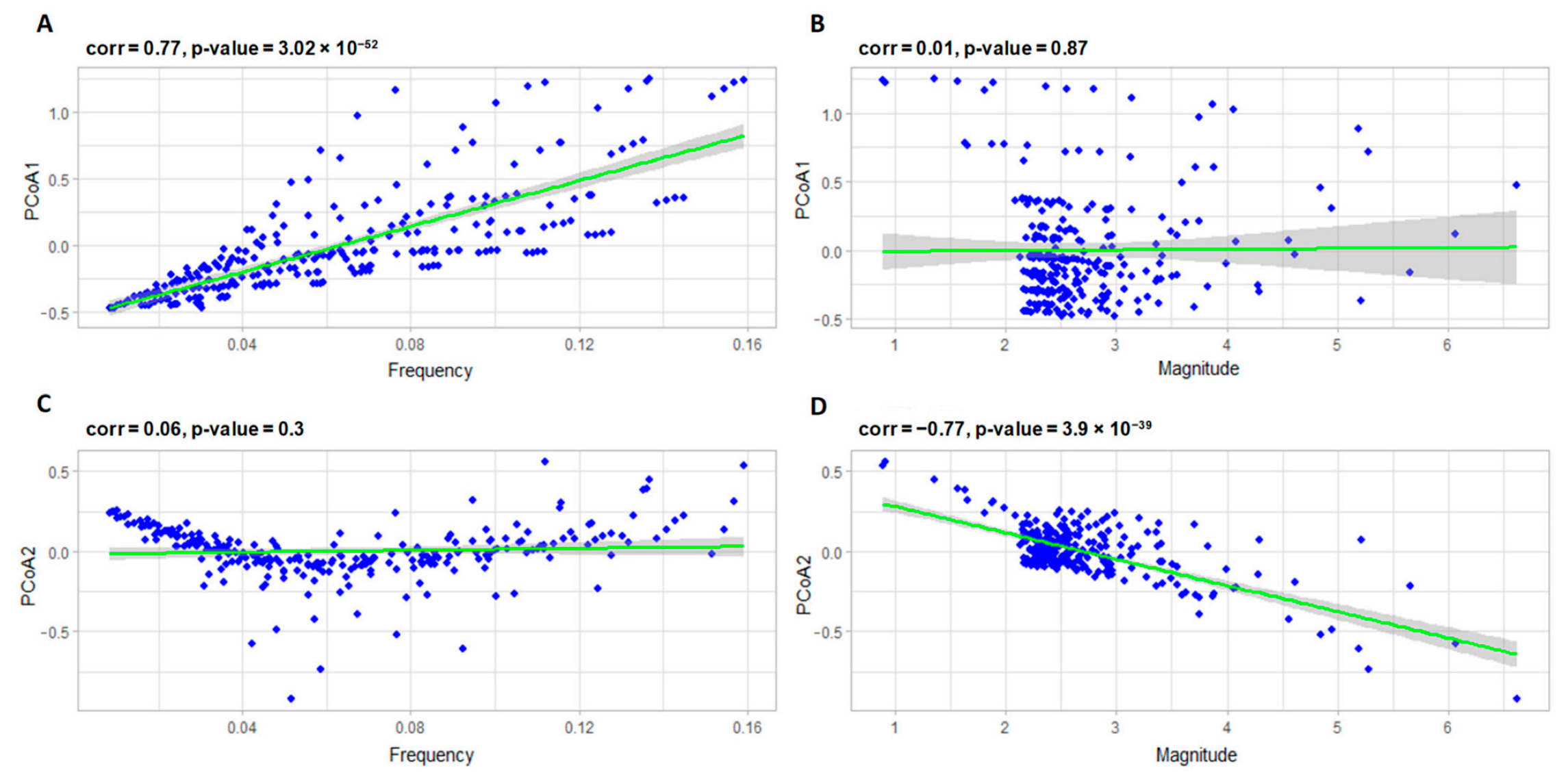
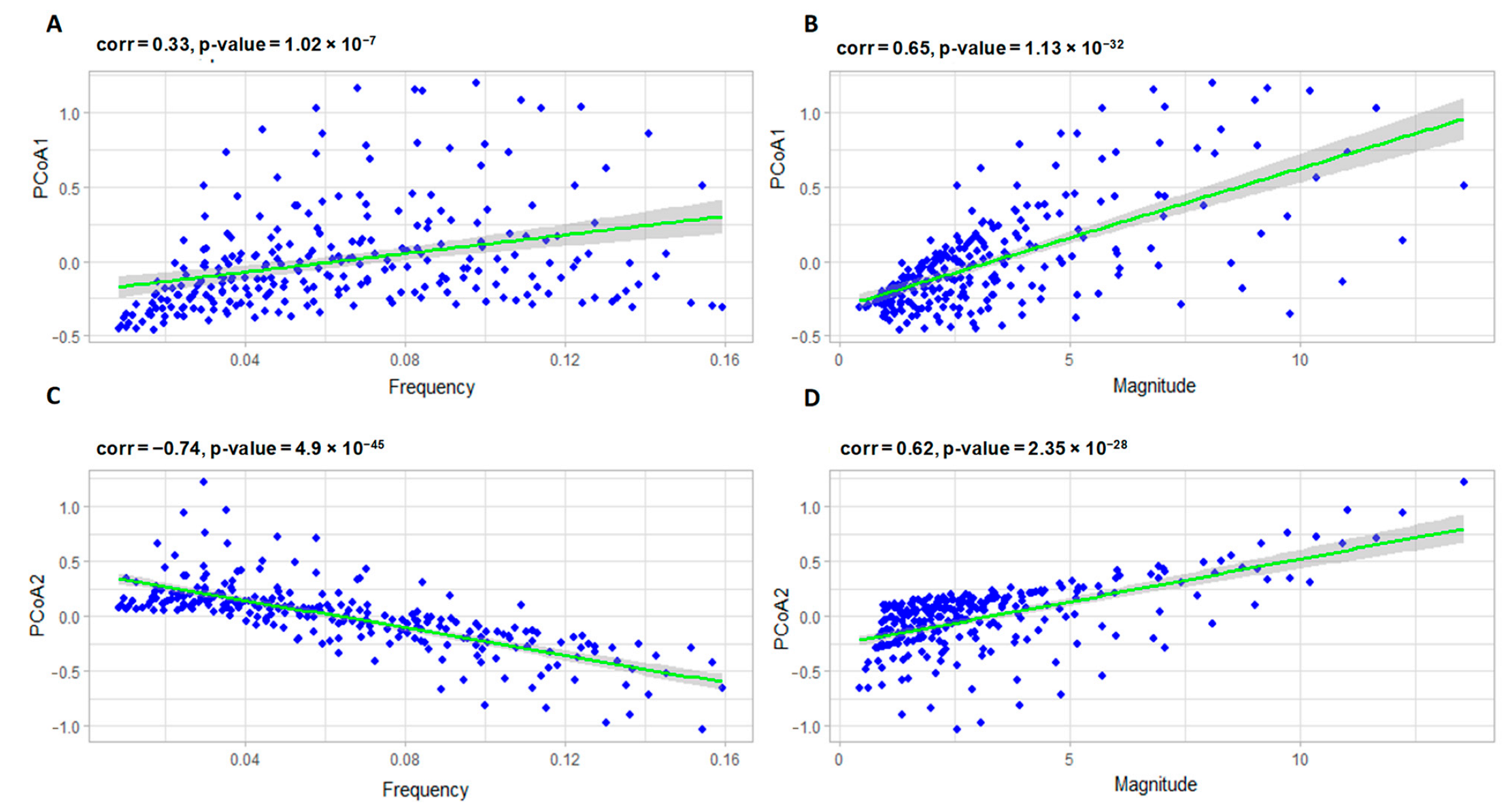
3.2.1. Correlation Analysis of the Model Parameters and PCoA Axes with Respect to the Predator and Prey Populations
- 𝑎–coefficient of prey growth;
- 𝑏—coefficient of loss of prey caused by interaction with predators;
- 𝑐—coefficient of loss of predators;
- 𝑑—coefficient of predator growth due to interaction with prey species.
3.2.2. Interpreting PCoA Axes in Terms of Characteristics of Solutions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Boza, G.; Barabás, G.; Scheuring, I.; Zachar, I. Eco-Evolutionary Modelling of Microbial Syntrophy Indicates the Robustness of Cross-Feeding over Cross-Facilitation. Sci. Rep. 2023, 13, 907. [Google Scholar] [CrossRef]
- Kuntal, B.K.; Gadgil, C.; Mande, S.S. Web-GLV: A Web Based Platform for Lotka-Volterra Based Modeling and Simulation of Microbial Populations. Front. Microbiol. 2019, 10, 288. [Google Scholar] [CrossRef]
- Krysiak-Baltyn, K.; Martin, G.J.O.; Stickland, A.D.; Scales, P.J.; Gras, S.L. Computational Models of Populations of Bacteria and Lytic Phage. Crit. Rev. Microbiol. 2016, 42, 942–968. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, X.; Liu, J.; Nememan, I.; Singh, A.H.; Weiss, H.; Levin, B.R. The Population Dynamics of Bacteria in Physically Structured Habitats and the Adaptive Virtue of Random Motility. Proc. Natl. Acad. Sci. USA 2011, 108, 4047–4052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeAngelis, D.L.; Mooij, W.M. Individual-Based Modeling of Ecological and Evolutionary Processes. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 147–168. [Google Scholar] [CrossRef]
- Kreft, J.; Picioreanu, C.; Wimpenny, J.W.T.; Loosdrecht, M.C.M. Van. Individual-Based Modelling of Biofilms. Microbiology 2001, 147, 2897–2912. [Google Scholar] [CrossRef] [Green Version]
- Lardon, L.A.; Merkey, B.V.; Martins, S.; Dötsch, A.; Picioreanu, C.; Kreft, J.U.; Smets, B.F. IDynoMiCS: Next-Generation Individual-Based Modelling of Biofilms. Environ. Microbiol. 2011, 13, 2416–2434. [Google Scholar] [CrossRef]
- Ayllón, D.; Railsback, S.F.; Vincenzi, S.; Groeneveld, J.; Almodóvar, A.; Grimm, V. InSTREAM-Gen: Modelling Eco-Evolutionary Dynamics of Trout Populations under Anthropogenic Environmental Change. Ecol. Modell. 2016, 326, 36–53. [Google Scholar] [CrossRef]
- Klimenko, A.; Matushkin, Y.; Kolchanov, N.; Lashin, S. Leave or Stay: Simulating Motility and Fitness of Microorganisms in Dynamic Aquatic Ecosystems. Biology 2021, 10, 1019. [Google Scholar] [CrossRef]
- Hellweger, F.L.; Clegg, R.J.; Clark, J.R.; Plugge, C.M.; Kreft, J.U. Advancing Microbial Sciences by Individual-Based Modelling. Nat. Rev. Microbiol. 2016, 14, 461–471. [Google Scholar] [CrossRef]
- Misirli, G.; Nguyen, T.; McLaughlin, J.A.; Vaidyanathan, P.; Jones, T.S.; Densmore, D.; Myers, C.; Wipat, A. A Computational Workflow for the Automated Generation of Models of Genetic Designs. ACS Synth. Biol. 2019, 8, 1548–1559. [Google Scholar] [CrossRef]
- Shmulevich, I.; Dougherty, E.R.; Kim, S.; Zhang, W. Probabilistic Boolean Networks: A Rule-Based Uncertainty Model for Gene Regulatory Networks. Bioinformatics 2002, 18, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Wimpenny, J.W.T.; Colasanti, R. A Unifying Hypothesis for the Structure of Microbial Biofilms Based on Cellular Automaton Models. FEMS Microbiol. Ecol. 1997, 22, 1–16. [Google Scholar] [CrossRef]
- Ashby, B.; Gupta, S.; Buckling, A. Spatial Structure Mitigates Fitness Costs in Host-Parasite Coevolution. Am. Nat. 2014, 183, E64–E74. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.C.; Kurth, W.; Grimm, V. Facilitating Parameter Estimation and Sensitivity Analysis of Agent-Based Models: A Cookbook Using NetLogo and R.; University of Surrey: Surrey, UK, 2014; Volume 17, p. 11. [Google Scholar] [CrossRef]
- Sedlmair, M.; Heinzl, C.; Bruckner, S.; Piringer, H.; Moller, T. Visual Parameter Space Analysis: A Conceptual Framework. IEEE Trans. Vis. Comput. Graph. 2014, 20, 2161–2170. [Google Scholar] [CrossRef] [PubMed]
- Malik-Sheriff, R.S.; Glont, M.; Nguyen, T.V.N.; Tiwari, K.; Roberts, M.G.; Xavier, A.; Vu, M.T.; Men, J.; Maire, M.; Kananathan, S.; et al. BioModels—15 Years of Sharing Computational Models in Life Science. Nucleic Acids Res. 2019, 48, D407–D415. [Google Scholar] [CrossRef] [Green Version]
- Olivier, B.G.; Snoep, J.L. Web-Based Kinetic Modelling Using JWS Online. Bioinformatics 2004, 20, 2143–2144. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, C.M.; Lawson, J.R.; Hunter, P.J.; Nielsen, P.F. The CellML Model Repository. Bioinformatics 2008, 24, 2122–2123. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A Platform for Integrating, Standardizing and Sharing Genome-Scale Models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef] [PubMed]
- Colléter, M.; Valls, A.; Guitton, J.; Gascuel, D.; Pauly, D.; Christensen, V. Global Overview of the Applications of the Ecopath with Ecosim Modeling Approach Using the EcoBase Models Repository. Ecol. Model. 2015, 302, 42–53. [Google Scholar] [CrossRef]
- Hamby, D.M. A Review of Techniques for Parameter Sensitivity Analysis of Environmental Models. Environ. Monit. Assess. 1994, 32, 135–154. [Google Scholar] [CrossRef]
- Ingalls, B. Sensitivity Analysis: From Model Parameters to System Behaviour. Essays Biochem. 2008, 45, 177–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zi, Z. Sensitivity Analysis Approaches Applied to Systems Biology Models. IET Syst. Biol. 2011, 5, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Trame, M.N.; Lesko, L.J.; Schmidt, S. Sobol Sensitivity Analysis: A Tool to Guide the Development and Evaluation of Systems Pharmacology Models. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Saltelli, A.; Homma, T. Importance Measures in Global Sensitivity Analysis of Model Output. Reliab. Eng. Sys. Saf. 1996, 52, 1–17. [Google Scholar]
- Keogh, E.J.; Pazzani, M.J. Derivative Dynamic Time Warping. In Proceedings of the SIAM International Conference on Data Mining, SDM 2001, Chicago, IL, USA, 5–7 April 2001. [Google Scholar]
- Giorgino, T. Computing and Visualizing Dynamic Time Warping Alignments in R: The Dtw Package. J. Stat. Softw. 2009, 31, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.F.; Waterman, M.S. Comparison of Biosequences. Adv. Appl. Math. 1981, 2, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Beijering, K.; Gooskens, C.; Heeringa, W. Predicting Intelligibility and Perceived Linguistic Distance by Means of the Levenshtein Algorithm. Linguist. Neth. 2008, 25, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Needleman, S.B.; Wunsch, C.D. A General Method Applicable to the Search for Similarities in the Amino Acid Sequence of Two Proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Legendre, P.; Gallagher, E.D. Ecologically Meaningful Transformations for Ordination of Species Data. Oecologia 2001, 129, 271–280. [Google Scholar] [CrossRef]
- Yang, D.; Dong, Z.; Lim, L.H.I.; Liu, L. Analyzing Big Time Series Data in Solar Engineering Using Features and PCA. Sol. Energy 2017, 153, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Терехина, А.Ю. Метoды Мнoгoмернoгo Шкалирoвания и Визуализации Данных (Обзoр). Автoмат. и телемех. 1973, 34, 80–94. [Google Scholar]
- Anderson, M.J.; Willis, T.J. Canonical Analysis of Principal Coordinates: A Useful Method of Constrained Ordination for Ecology. Ecology 2003, 84, 511–525. [Google Scholar] [CrossRef]
- Groth, D.; Hartmann, S.; Klie, S.; Selbig, J. Principal Components Analysis. Methods Mol. Biol. 2013, 930, 527–547. [Google Scholar] [CrossRef]
- Gower, J.C. Some Distance Properties of Latent Root and Vector Methods Used in Multivariate Analysis. Biometrika 1966, 53, 325–338. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate Analyses in Microbial Ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [Green Version]
- Cortopassi, K.A.; Bradbury, J.W. The Comparison of Harmonically Rich Sounds Using Spectrographic Cross-Correlation and Principal Coordinates Analysis. Bioacoustics 2000, 11, 89–127. [Google Scholar] [CrossRef]
- Jones, M.C.; Rice, J.A. Displaying the Important Features of Large Collections of Similar Curves. Am. Stat. 1992, 46, 140–145. [Google Scholar] [CrossRef]
- Wangersky, P.J. Lotka-Volterra Population Models Lotka-Volterra*: 4140 Population Models. Source Annu. Rev. Ecol. Syst. Ann. Rev. Ecol. Syst 1978, 9, 189–218. [Google Scholar] [CrossRef]
- Lotka, A.J. Contribution to the Theory of Periodic Reactions. J. Phys. Chem. 1909, 14, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Volterra, V. Variazioni e Fluttuazioni Del Numero d’individui in Specie Animali Conviventi; Atti dell’ Accademia Nazionale dei Lincei: Lincei, Italy, 1926; Volume 62, pp. 31–113. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step Pattern | Recursion Formula |
|---|---|
| symmetric1 | |
| symmetric2 | |
| asymmetric | |
| Rabiner-Juang |
| a | b | C | d | |
|---|---|---|---|---|
| PCoA1 | 0.14 | −0.09 | 0.62 | −0.64 |
| PCoA2 | 0.03 | −0.36 | −0.12 | 0.14 |
| a | b | c | d | |
|---|---|---|---|---|
| PCoA1 | 0.54 | −0.59 | 0.12 | 0.23 |
| PCoA2 | −0.33 | −0.05 | −0.37 | 0.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimenko, A.I.; Vorobeva, D.A.; Lashin, S.A. A New Visualization and Analysis Method for a Convolved Representation of Mass Computational Experiments with Biological Models. Mathematics 2023, 11, 2783. https://doi.org/10.3390/math11122783
Klimenko AI, Vorobeva DA, Lashin SA. A New Visualization and Analysis Method for a Convolved Representation of Mass Computational Experiments with Biological Models. Mathematics. 2023; 11(12):2783. https://doi.org/10.3390/math11122783
Chicago/Turabian StyleKlimenko, Alexandra I., Diana A. Vorobeva, and Sergey A. Lashin. 2023. "A New Visualization and Analysis Method for a Convolved Representation of Mass Computational Experiments with Biological Models" Mathematics 11, no. 12: 2783. https://doi.org/10.3390/math11122783
APA StyleKlimenko, A. I., Vorobeva, D. A., & Lashin, S. A. (2023). A New Visualization and Analysis Method for a Convolved Representation of Mass Computational Experiments with Biological Models. Mathematics, 11(12), 2783. https://doi.org/10.3390/math11122783