
Using Machine Learning and Natural Language Processing for Unveiling Similarities between Microbial Data

Abstract
:1. Introduction
- The proposed microbial sequence data processing pipeline;
- The utilization of frequency analysis for microbial sequence data transformation into text vectors;
- The proposed hierarchical clustering method over microbial sequence text vectors;
- The evaluation of the proposed method on an animal feces dataset.
2. Background
3. Microbial Sequence Data Pipeline
4. Materials and Methods
4.1. Dataset
4.2. Preprocessing
4.3. Frequency Analysis
| Algorithm 1 General pseudocode of the FA sequence computation |
|
4.4. Clustering
- Data selection;
- Feature selection;
- Similarity measure;
- Clustering algorithm;
- Cluster evaluation;
- Cluster interpretation.
4.5. Proposed Hierarchical Clustering Method
| Algorithm 2 Hierarchical clustering algorithm |
|
| Algorithm 3 General pseudocode of the steps of the proposed method |
|
4.6. Visualization
5. Results and Discussion
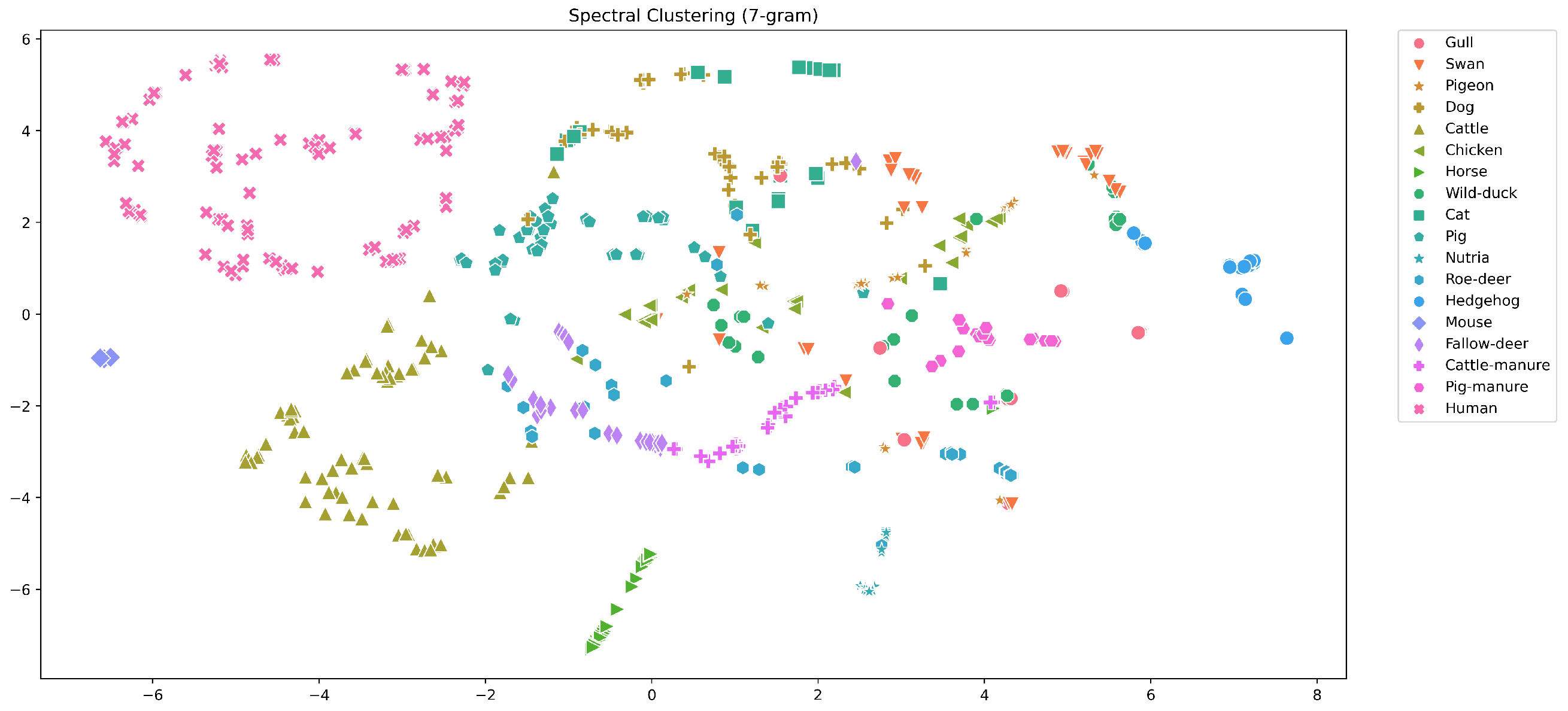
5.1. Analysis of the Groups
5.2. Analysis of the Beta Diversity
5.3. Quantitative Results Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sammut, C.; Webb, G.I. Encyclopedia of Machine Learning; Springer Science & Business Media: Berlin, Germany, 2011. [Google Scholar]
- Jordan, M.I.; Mitchell, T.M. Machine learning: Trends, perspectives, and prospects. Science 2015, 349, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Brezočnik, L.; Nalli, G.; De Leone, R.; Val, S.; Podgorelec, V.; Karakatič, S. Machine Learning Model for Student Drop-Out Prediction Based on Student Engagement. In Proceedings of the International Conference “New Technologies, Development and Applications”, Sarajevo, Bosnia and Herzegovina, 22–24 June 2023; Springer: Berlin, Germany, 2023; pp. 486–496. [Google Scholar]
- Podgorelec, V.; Kokol, P.; Stiglic, M.M.; Heričko, M.; Rozman, I. Knowledge discovery with classification rules in a cardiovascular dataset. Comput. Methods Programs Biomed. 2005, 80, S39–S49. [Google Scholar] [CrossRef] [PubMed]
- Nagarhalli, T.P.; Vaze, V.; Rana, N.K. Impact of Machine Learning in Natural Language Processing: A Review. In Proceedings of the 2021 Third International Conference on Intelligent Communication Technologies and Virtual Mobile Networks (ICICV), Tirunelveli, India, 4–6 February 2021; pp. 1529–1534. [Google Scholar] [CrossRef]
- Kameoka, S.; Motooka, D.; Watanabe, S.; Kubo, R.; Jung, N.; Midorikawa, Y.; Shinozaki, N.O.; Sawai, Y.; Takeda, A.K.; Nakamura, S. Benchmark of 16S rRNA gene amplicon sequencing using Japanese gut microbiome data from the V1–V2 and V3–V4 primer sets. BMC Genom. 2021, 22, 527. [Google Scholar] [CrossRef] [PubMed]
- Khurana, D.; Koli, A.; Khatter, K.; Singh, S. Natural language processing: State of the art, current trends and challenges. Multimed. Tools Appl. 2023, 82, 3713–3744. [Google Scholar] [CrossRef]
- Asnicar, F.; Thomas, A.M.; Passerini, A.; Waldron, L.; Segata, N. Machine learning for microbiologists. Nat. Rev. Microbiol. 2024, 22, 191–205. [Google Scholar] [CrossRef]
- Walsh, C.; Stallard-Olivera, E.; Fierer, N. Nine (not so simple) steps: A practical guide to using machine learning in microbial ecology. Mbio 2024, 15, e02050-23. [Google Scholar] [CrossRef]
- Gihawi, A.; Ge, Y.; Lu, J.; Puiu, D.; Xu, A.; Cooper, C.S.; Brewer, D.S.; Pertea, M.; Salzberg, S.L. Major data analysis errors invalidate cancer microbiome findings. MBio 2023, 14, e01607-23. [Google Scholar] [CrossRef]
- Mohanty, S.; Behera, A.; Mishra, S.; Alkhayyat, A.; Gupta, D.; Sharma, V. Resumate: A Prototype to Enhance Recruitment Process with NLP based Resume Parsing. In Proceedings of the 2023 4th International Conference on Intelligent Engineering and Management (ICIEM), London, UK, 9–11 May 2023; pp. 1–6. [Google Scholar] [CrossRef]
- Ismail, S.S.; Mansour, R.F.; Abd El-Aziz, R.M.; Taloba, A.I. Efficient E-mail spam detection strategy using genetic decision tree processing with NLP features. Comput. Intell. Neurosci. 2022, 2022, 7710005. [Google Scholar] [CrossRef]
- Chen, L.; Gu, Y.; Ji, X.; Sun, Z.; Li, H.; Gao, Y.; Huang, Y. Extracting medications and associated adverse drug events using a natural language processing system combining knowledge base and deep learning. J. Am. Med. Inform. Assoc. 2020, 27, 56–64. [Google Scholar] [CrossRef]
- Afzal, M.; Hussain, M.; Malik, K.M.; Lee, S. Impact of automatic query generation and quality recognition using deep learning to curate evidence from biomedical literature: Empirical study. JMIR Med. Inform. 2019, 7, e13430. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, W.; Kim, S.; Kim, D.; Kim, S.; So, C.H.; Kang, J. BioBERT: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020, 36, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Ngiam, K.Y. How data science and AI-based technologies impact genomics. Singap. Med. J. 2023, 64, 59–66. [Google Scholar] [CrossRef]
- Yang, M.Q.; Wang, Z.J.; Zhai, C.B.; Chen, L.Q. Research progress on the application of 16S rRNA gene sequencing and machine learning in forensic microbiome individual identification. Front. Microbiol. 2024, 15, 1360457. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.J.; Rawson, N.; Bailey, B.A.; Fernandez-Guerra, A.; Sisk-Hackworth, L.; Kelley, S.T. Meta-SourceTracker: Application of Bayesian source tracking to shotgun metagenomics. PeerJ 2020, 8, e8783. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Ng, S.K.; Sung, J.J.; Goh, W.W.B.; Wong, S.H. Data pre-processing for analyzing microbiome data–A mini review. Comput. Struct. Biotechnol. J. 2023, 21, 4804–4815. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef]
- Love, C.J.; Gubert, C.; Kodikara, S.; Kong, G.; Lê Cao, K.A.; Hannan, A.J. Microbiota DNA isolation, 16S rRNA amplicon sequencing, and bioinformatic analysis for bacterial microbiome profiling of rodent fecal samples. STAR Protoc. 2022, 3, 101772. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; Collaboration, I.N.S.D. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef]
- Topçuoğlu, B.D.; Lesniak, N.A.; Ruffin IV, M.T.; Wiens, J.; Schloss, P.D. A framework for effective application of machine learning to microbiome-based classification problems. MBio 2020, 11, 10-1128. [Google Scholar] [CrossRef]
- Su, X.; Jing, G.; Sun, Z.; Liu, L.; Xu, Z.; McDonald, D.; Wang, Z.; Wang, H.; Gonzalez, A.; Zhang, Y.; et al. Multiple-disease detection and classification across cohorts via microbiome search. Msystems 2020, 5, 10-1128. [Google Scholar] [CrossRef]
- Hu, Y.; Satten, G.A.; Hu, Y.J. LOCOM: A logistic regression model for testing differential abundance in compositional microbiome data with false discovery rate control. Proc. Natl. Acad. Sci. USA 2022, 119, e2122788119. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; van Es, H.M.; Buckley, D.H. Predicting measures of soil health using the microbiome and supervised machine learning. Soil Biol. Biochem. 2022, 164, 108472. [Google Scholar] [CrossRef]
- Han, J.; Kamber, M.; Pei, J. 2-Getting to Know Your Data. In Data Mining, 3rd ed.; Han, J., Kamber, M., Pei, J., Eds.; The Morgan Kaufmann Series in Data Management Systems; Morgan Kaufmann: Boston, MA, USA, 2012; pp. 39–82. [Google Scholar] [CrossRef]
- Zou, H. Clustering algorithm and its application in data mining. Wirel. Pers. Commun. 2020, 110, 21–30. [Google Scholar] [CrossRef]
- Žlender, T.; Brezočnik, L.; Podgorelec, V.; Rupnik, M. Uncovering cattle-associated markers of faecal pollution through 16s rRNA gene analysis. In Proceedings of the 13th International Gut Microbiology Symposium, Aberdeen, Scotland, 13–15 June 2023; P&J LIVE: Aberdeen, Scotland; Hong Kong, China, 2023; p. 87. [Google Scholar]
- Žlender, T.; Brezočnik, L.; Podgorelec, V.; Rupnik, M. Identifying Markers of Cattle Fecal Pollution Using Comparative Analysis of the 16S rRNA Gene. In Proceedings of the Power of Microbes in Industry and Environment: Book of Abstracts, Poreč, Croatia, 15–18 May 2023; Croatian Microbiological Society: Zagreb, Croatia, 2023; p. 119. [Google Scholar]
- López-Aladid, R.; Fernández-Barat, L.; Alcaraz-Serrano, V.; Bueno-Freire, L.; Vázquez, N.; Pastor-Ibáñez, R.; Palomeque, A.; Oscanoa, P.; Torres, A. Determining the most accurate 16S rRNA hypervariable region for taxonomic identification from respiratory samples. Sci. Rep. 2023, 13, 3974. [Google Scholar] [CrossRef]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Flisar, J.; Podgorelec, V. Improving short text classification using information from DBpedia ontology. Fundam. Inform. 2020, 172, 261–297. [Google Scholar] [CrossRef]
- Müllner, D. Modern hierarchical, agglomerative clustering algorithms. arXiv 2011, arXiv:1109.2378. [Google Scholar]
- Theus, M. High-dimensional Data Visualization. In Handbook of Data Visualization; Springer: Berlin/Heidelberg, Germany, 2008; pp. 151–178. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Penington, J.S.; Penno, M.A.; Ngui, K.M.; Ajami, N.J.; Roth-Schulze, A.J.; Wilcox, S.A.; Bandala-Sanchez, E.; Wentworth, J.M.; Barry, S.C.; Brown, C.Y.; et al. Influence of fecal collection conditions and 16S rRNA gene sequencing at two centers on human gut microbiota analysis. Sci. Rep. 2018, 8, 4386. [Google Scholar] [CrossRef]
- Lloyd, S. Least squares quantization in PCM. IEEE Trans. Inf. Theory 1982, 28, 129–137. [Google Scholar] [CrossRef]
- Shi, J.; Malik, J. Normalized cuts and image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2000, 22, 888–905. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Caliński, T.; Harabasz, J. A dendrite method for cluster analysis. Commun.-Stat.-Theory Methods 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Davies, D.L.; Bouldin, D.W. A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. 1979, PAMI-1, 224–227. [Google Scholar] [CrossRef]
- Ekemeyong Awong, L.E.; Zielinska, T. Comparative Analysis of the Clustering Quality in Self-Organizing Maps for Human Posture Classification. Sensors 2023, 23, 7925. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Samples | Number of ZOTUs | |
|---|---|---|---|
| 1 | Cat | 33 | 11.310 |
| 2 | Cattle | 79 | 28.328 |
| 3 | Cattle manure | 31 | 34.668 |
| 4 | Chicken | 35 | 19.861 |
| 5 | Dog | 43 | 12.506 |
| 6 | Fallow deer | 34 | 28.978 |
| 7 | Gull | 21 | 9.460 |
| 8 | Hedgehog | 28 | 6.164 |
| 9 | Horse | 36 | 28.835 |
| 10 | Human | 185 | 6.635 |
| 11 | Mouse | 26 | 6.660 |
| 12 | Nutria | 23 | 10.025 |
| 13 | Pig | 45 | 21.777 |
| 14 | Pig manure | 21 | 21.593 |
| 15 | Pigeon | 24 | 13.924 |
| 16 | Roe deer | 34 | 28.685 |
| 17 | Swan | 41 | 17.986 |
| 18 | Wild duck | 26 | 16.507 |
| AA | AC | AG | … | TT | |
|---|---|---|---|---|---|
| Sample 1 | 0.07351 | 0.08578 | 0.05976 | … | 0.08654 |
| Sample 2 | 0.07612 | 0.04586 | 0.05228 | … | 0.04661 |
| … | … | … | … | … | … |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brezočnik, L.; Žlender, T.; Rupnik, M.; Podgorelec, V. Using Machine Learning and Natural Language Processing for Unveiling Similarities between Microbial Data. Mathematics 2024, 12, 2717. https://doi.org/10.3390/math12172717
Brezočnik L, Žlender T, Rupnik M, Podgorelec V. Using Machine Learning and Natural Language Processing for Unveiling Similarities between Microbial Data. Mathematics. 2024; 12(17):2717. https://doi.org/10.3390/math12172717
Chicago/Turabian StyleBrezočnik, Lucija, Tanja Žlender, Maja Rupnik, and Vili Podgorelec. 2024. "Using Machine Learning and Natural Language Processing for Unveiling Similarities between Microbial Data" Mathematics 12, no. 17: 2717. https://doi.org/10.3390/math12172717