

CNVbd: A Method for Copy Number Variation Detection and Boundary Search


Abstract
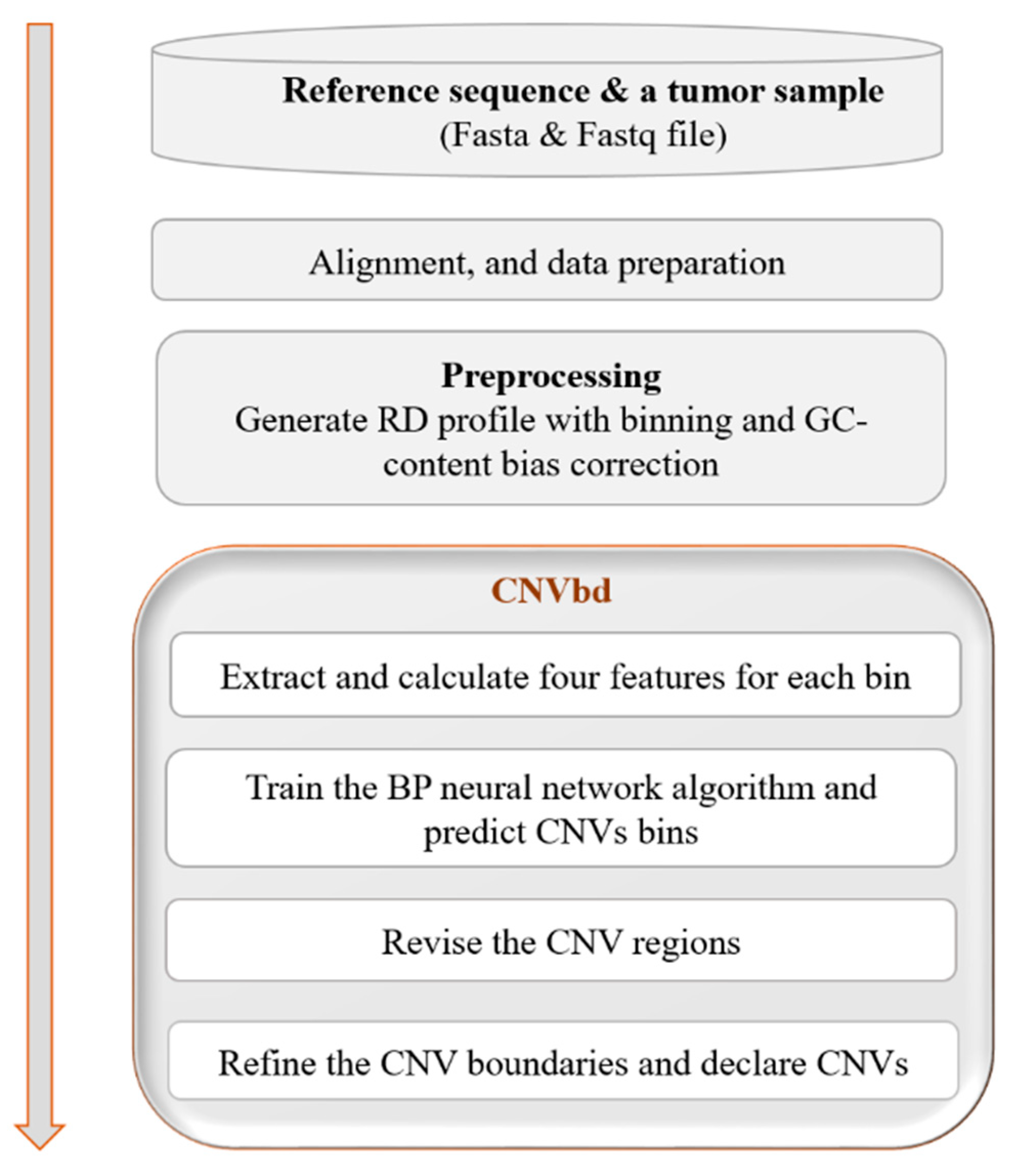
:1. Introduction
2. Methods
2.1. Overview of CNVbd
2.2. Preprocessing
2.3. Extract and Calculate CNV-Related Features
2.3.1. Calculate the Values of the Features LD and MD
2.3.2. Calculate the Value of the Feature ID
| Algorithm 1. Training an iTree. | |
| (1) | bins from all the bins; |
| (2) | Let Q be the list of RD values in X; |
| (3) | Randomly select a value as a root or sub-root and divide X into two subsets, the left side and the right side: where Q; |
| (4) | ; |
| (5) | . |
2.4. Predict the CNV Bins by Training the BP Neural Network Algorithm
2.5. Revise the CNV Regions
| Algorithm 2. Revise the normal bins on the left side of a CNV block. | |
| (1) | Calculate the CNV proportion in the region; |
| (2) | If , then calculate ; otherwise, keep bin as normal and quit. |
| (3) | If for a gain region (or for a loss region), then bin is revised to be a CNV gain (or loss); otherwise, keep bin as normal and quit. |
| (4) | Recalculate and let . |
| (5) | Repeat steps (2) to (4) until there is no normal bin left on the left side. |
- For the left gain block, the revision process starts at bin . (1) Calculate . Assume that and have been calculated. (2) Calculate and . It can be seen that , so the bin stays normal. In this case, the revision for the right side of the left gain block is over. Bin also stays normal.
- For the right gain block, the revision process starts at bin . (1) Calculate and . It can be seen that , so bin is revised to be a gain. (2) For bin i, recalculate and . Since , bin stays normal.
2.6. Refine the CNV Boundary and Declare CNVs
| Algorithm 3. The left boundary search for a gain block. | |
| (1) | Divide and in half into four sub-bins: , , , and , from right to left. |
| (2) | Calculate and the deviations , , , and of the four sub-bins. |
| (3) | Choose two adjacent ones from the four sub-bins from right to left: take the first pair of sub-bins whose deviations change from non-negative to negative, if they exist; or take the leftmost two sub-bins, and , if ; otherwise, take the rightmost two bins, and . |
| (4) | Replace and with the two sub-bins chosen in step (3), recalculate and , and repeat steps (1) to (3) until the lengths of the two sub-bins reduce to a single base. |
3. Results
3.1. Simulation Studies
3.2. Application to Real Blood Samples
4. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Coe, B.P.; Girirajan, S.; Eichler, E.E. The genetic variability and commonality of neurodevelopmental disease. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160, 118–129. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef]
- Yuan, X.-G.; Zhao, Y.; Guo, Y.; Ge, L.-M.; Liu, W.; Wen, S.-Y.; Li, Q.; Wan, Z.-B.; Zheng, P.-N.; Guo, T.; et al. COSINE: A web server for clonal and subclonal structure inference and evolution in cancer genomics. Zool. Res. 2022, 43, 75–77. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef]
- Yuan, X.; Yu, G.; Hou, X.; Shih Ie, M.; Clarke, R.; Zhang, J.; Hoffman, E.P.; Wang, R.R.; Zhang, Z.; Wang, Y. Genome-wide identification of significant aberrations in cancer genome. BMC Genom. 2012, 13, 342. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Stranger, B.E. The impact of human copy number variation on gene expression. Brief. Funct. Genom. 2015, 14, 352–357. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef]
- Tan, R.; Wang, Y.; Kleinstein, S.E.; Liu, Y.; Zhu, X.; Guo, H.; Jiang, Q.; Allen, A.S.; Zhu, M. An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum. Mutat. 2014, 35, 899–907. [Google Scholar] [CrossRef]
- Zare, F.; Dow, M.; Monteleone, N.; Hosny, A.; Nabavi, S. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinform. 2017, 18, 286. [Google Scholar] [CrossRef]
- Teo, S.M.; Pawitan, Y.; Ku, C.S.; Chia, K.S.; Salim, A. Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics 2012, 28, 2711–2718. [Google Scholar] [CrossRef]
- Yoon, S.; Xuan, Z.; Makarov, V.; Ye, K.; Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009, 19, 1586–1592. [Google Scholar] [CrossRef]
- Dharanipragada, P.; Parekh, N. Copy number variation detection workflow using next generation sequencing data. In Proceedings of the 2016 International Conference on Bioinformatics and Systems Biology, Allahabad, India, 4–6 March 2016; pp. 1–5. [Google Scholar]
- Chiang, D.Y.; Getz, G.; Jaffe, D.B.; O’kelly, M.J.; Zhao, X.; Carter, S.L.; Russ, C.; Nusbaum, C.; Meyerson, M.; Lander, E.S. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods 2009, 6, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Hampton, O.; Coarfa, C.; Milosavljevic, A. ReadDepth: A parallel R package for detecting copy number alterations from short sequencing reads. PLoS ONE 2011, 6, e16327. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Zinovyev, A.; Bleakley, K.; Vert, J.-P.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics 2010, 27, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Dharanipragada, P.; Vogeti, S.; Parekh, N. iCopyDAV: Integrated platform for copy number variations-Detection, annotation and visualization. PLoS ONE 2018, 13, e0195334. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Velds, A.; Kemper, K.; Ranzani, M.; Bombardelli, L.; Hoogstraat, M.; Nevedomskaya, E.; Xu, G.; de Ruiter, J.; Lolkema, M.P.; et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.; Kawash, J.K.; Grigoriev, A. GROM-RD: Resolving genomic biases to improve read depth detection of copy number variants. PeerJ 2015, 3, e836. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, L.; Wang, Y.; Cao, M.; Gelowani, V.; Xu, M.C.; Agrawal, S.A.; Li, Y.M.; Daiger, S.P.; Gibbs, R.; et al. SeqCNV: A novel method for identification of copy number variations in targeted next-generation sequencing data. BMC Bioinform. 2017, 18, 147. [Google Scholar] [CrossRef]
- Yuan, X.; Li, J.; Bai, J.; Xi, J. A local outlier factor-based detection of copy number variations from NGS data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1811–1820. [Google Scholar] [CrossRef]
- Haque, A.A.; Xie, K.; Liu, K.; Zhao, H.; Yang, X.; Yuan, X. Detection of copy number variations from NGS data by using an adaptive kernel density estimation-based outlier factor. Digit. Signal Process. 2022, 126, 103524. [Google Scholar] [CrossRef]
- Xie, K.; Liu, K.; Alvi, H.A.K.; Ji, W.; Wang, S.; Chang, L.; Yuan, X. IhybCNV: An intra-hybrid approach for CNV detection from next-generation sequencing data. Digit. Signal Process. 2022, 121, 103304. [Google Scholar] [CrossRef]
- Hu, T.; Chen, S.; Ullah, A.; Xue, H. AluScanCNV2: An R package for copy number variation calling and cancer risk prediction with next-generation sequencing data. Genes Dis. 2019, 6, 43–46. [Google Scholar] [CrossRef]
- Yuan, X.; Yu, J.; Xi, J.; Yang, L.; Shang, J.; Li, Z.; Duan, J. CNV_IFTV: An isolation forest and total variation-based detection of CNVs from short-read sequencing data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 539–549. [Google Scholar] [CrossRef]
- Onsongo, G.; Baughn, L.B.; Bower, M.; Henzler, C.; Schomaker, M.; Silverstein, K.A.T.; Thyagarajan, B. CNV-RF is a random forest-based copy number variation detection method using next-generation sequencing. J. Mol. Diagn. 2016, 18, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Tian, Y.; Yuan, X. A density peak-based method to detect copy number variations from next-generation sequencing data. Front. Genet. 2021, 11, 632311. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Laio, A. Clustering by fast search and find of density peaks. Science 2014, 344, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning Internal Representations by Error Propagation; MIT Press: Cambridge, MA, USA, 1985. [Google Scholar]
- Hahnloser, R.H.; Sarpeshkar, R.; Mahowald, M.A.; Douglas, R.J.; Seung, H.S. Digital selection and analogue amplification coexist in a cortex-inspired silicon circuit. Nature 2000, 405, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, J.; Yang, L.; Bai, J.; Fan, P. Detection of significant copy number variations from multiple samples in next-generation sequencing data. IEEE Trans. Nanobiosci. 2018, 17, 12–20. [Google Scholar] [CrossRef]
- Yuan, X.; Bai, J.; Zhang, J.; Yang, L.; Duan, J.; Li, Y.; Gao, M. CONDEL: Detecting copy number variation and genotyping deletion Zygosity from single tumor samples using sequence data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1141–1153. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, J.; Yang, L. IntSIM: An integrated simulator of next-generation sequencing data. IEEE Trans. Biomed. Eng. 2017, 64, 441–451. [Google Scholar] [CrossRef]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Description |
|---|---|
| Read-depth (RD), | The corrected RD for each bin |
| Local density (LD), | The number of points that are closer than the cutoff distance to point |
| Minimum distance (MD), | The minimum distance between the point and any other point with higher density |
| IFTV deep (ID), hi | The isolation forest deep, i.e., the average depth of in all iTrees |
| Sample | CNVbd | CNV-IFTV | FREEC | CNVnator | dpCNV |
|---|---|---|---|---|---|
| LC | 294.138 | 239.989 | 33.868 | 19.553 | 206.781 |
| 2053_1 | 293.321 | 96.898 | 329.671 | 43.774 | 4.0 |
| 2561_1 | 249.202 | 91.875 | 319.158 | 18.718 | 60.295 |
| Average | 278.887 | 142.921 | 227.566 | 27.348 | 90.359 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, J.; Liao, Z.; Haque, A.K.A.; Yu, Q.; Xie, K.; Guo, Y. CNVbd: A Method for Copy Number Variation Detection and Boundary Search. Mathematics 2024, 12, 420. https://doi.org/10.3390/math12030420
Lan J, Liao Z, Haque AKA, Yu Q, Xie K, Guo Y. CNVbd: A Method for Copy Number Variation Detection and Boundary Search. Mathematics. 2024; 12(3):420. https://doi.org/10.3390/math12030420
Chicago/Turabian StyleLan, Jingfen, Ziheng Liao, A. K. Alvi Haque, Qiang Yu, Kun Xie, and Yang Guo. 2024. "CNVbd: A Method for Copy Number Variation Detection and Boundary Search" Mathematics 12, no. 3: 420. https://doi.org/10.3390/math12030420