
A Panel-Agnostic Strategy ‘HiPPo’ Improves Diagnostic Efficiency in the UK Genomic Medicine Service

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Recruitment and Patient Demographics
2.2. Research Exome Sequencing and Pipeline
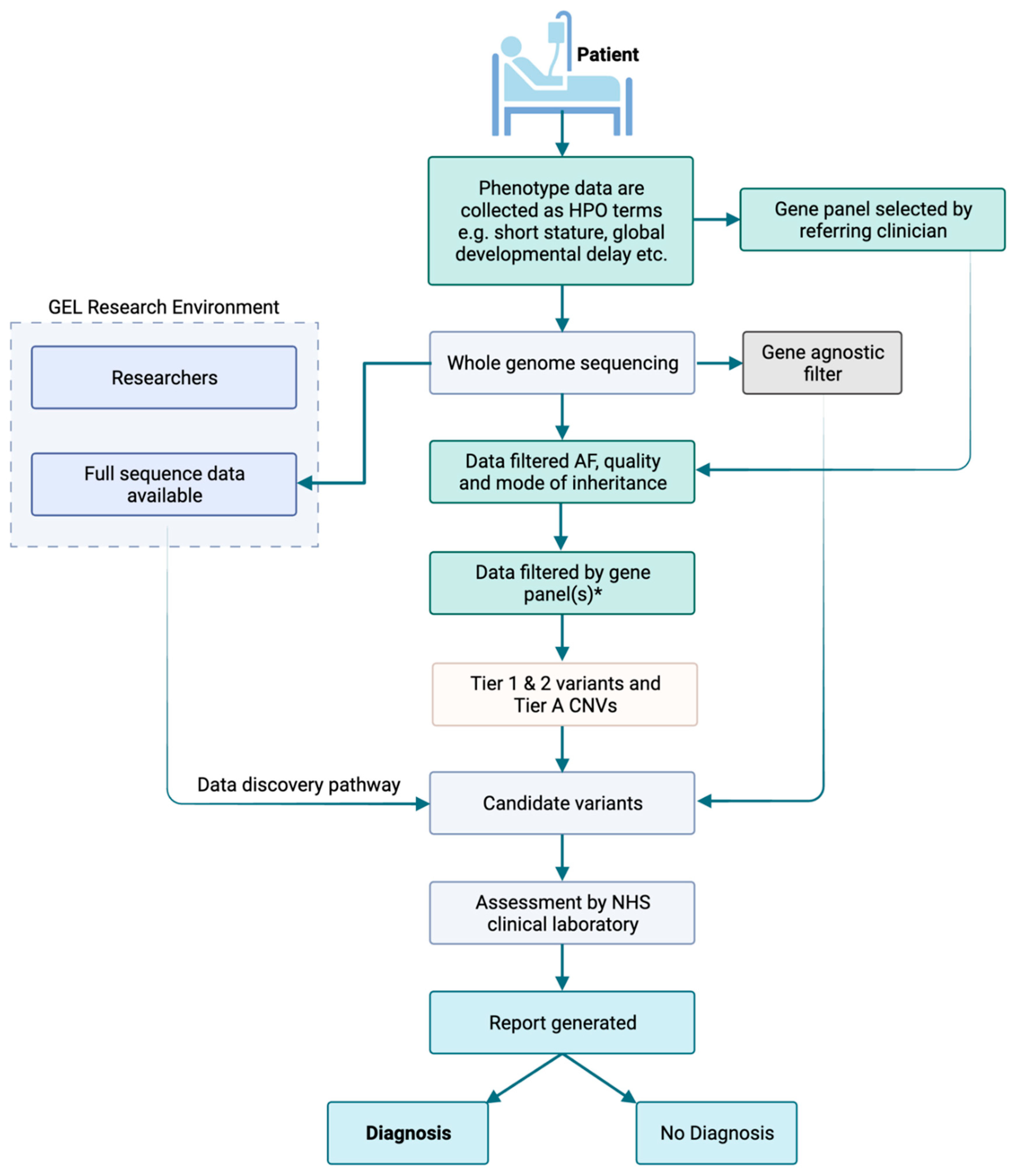
2.3. Genomic Medicine Service Pipeline
2.4. Data Analysis
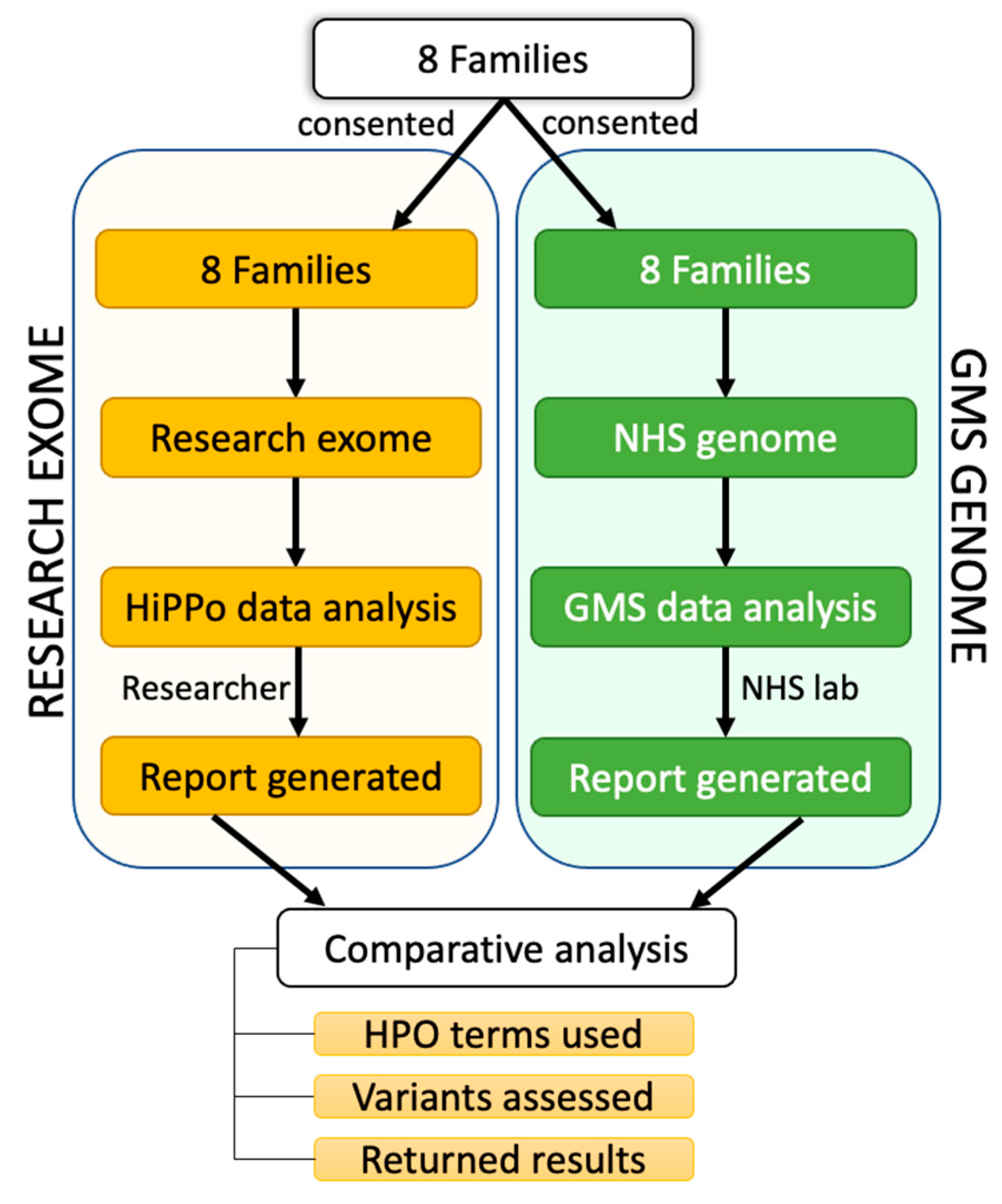
2.5. Research Exome Analysis
2.6. Reporting on Exome Variants
- 1.
- The variant was heterozygous in a known autosomal recessive disease gene, and no second hit (coding or noncoding) was identified;
- 2.
- 3.
- 4.
- 5.
- 6.
- The variant appeared artefactual upon visualisation of the read data in Integrative Genomics Viewer (IGV) [27].
2.7. Taking Novel Exome Candidates forward
2.8. GMS Clinical Genome Analysis Pipeline
2.9. Reporting of GMS Genome Variants
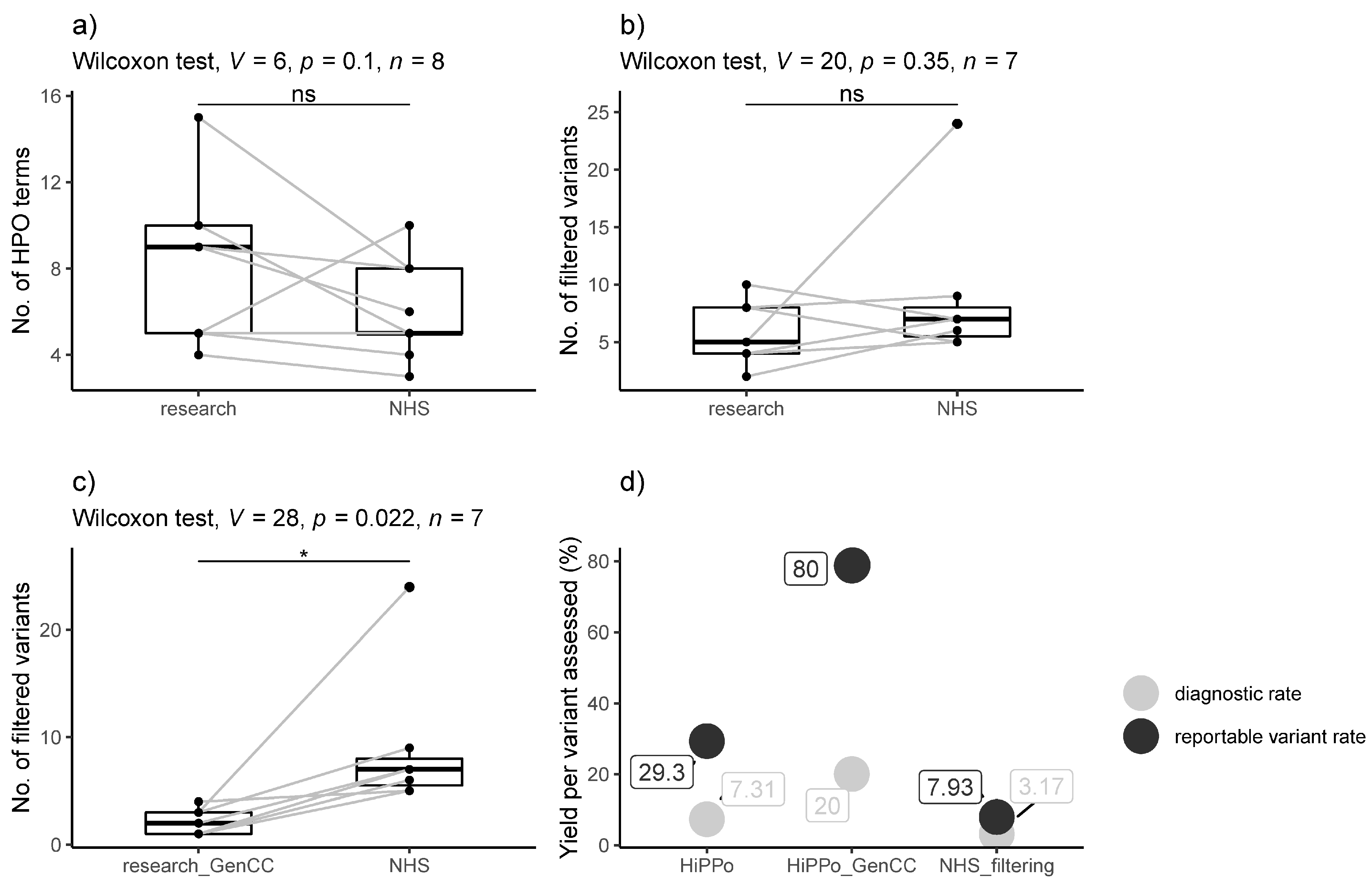
2.10. Comparison of Two Filtering Approaches
3. Results
3.1. GMS Clinical Genome Analysis Strategy
3.2. Research Exome HiPPo Strategy
3.3. Comparison between Exome Study and GMS Clinical Genome Results
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- 100,000 Genomes Project Pilot Investigators. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar] [CrossRef]
- Seaby, E.G.; Ennis, S. Challenges in the diagnosis and discovery of rare genetic disorders using contemporary sequencing technologies. Brief. Funct. Genom. 2020, 19, 243–258. [Google Scholar] [CrossRef]
- Seaby, E.G.; Smedley, D.; Taylor Tavares, A.L.; Brittain, H.; van Jaarsveld, R.H.; Baralle, D.; Rehm, H.L.; O’Donnell-Luria, A.; Ennis, S.; Genomics England Research Consortium. A gene-to-patient approach uplifts novel disease gene discovery and identifies 18 putative novel disease genes. Genet. Med. 2022, 24, 1697–1707. [Google Scholar] [CrossRef]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.U.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef]
- Turnbull, C.; Scott, R.H.; Thomas, E.; Jones, L.; Murugaesu, N.; Pretty, F.B.; Halai, D.; Baple, E.; Craig, C.; Hamblin, A.; et al. The 100,000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ 2018, 361, k1687. [Google Scholar] [CrossRef]
- Smedley, D.; Jacobsen, J.O.; Jäger, M.; Köhler, S.; Holtgrewe, M.; Schubach, M.; Siragusa, E.; Zemojtel, T.; Buske, O.J.; Washington, N.L.; et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 2015, 10, 2004–2015. [Google Scholar] [CrossRef]
- Lelieveld, S.H.; Spielmann, M.; Mundlos, S.; Veltman, J.A.; Gilissen, C. Comparison of exome and genome sequencing technologies for the complete capture of protein-coding regions. Hum. Mutat. 2015, 36, 815–822. [Google Scholar] [CrossRef]
- Rehm, H.L. Time to make rare disease diagnosis accessible to all. Nat. Med. 2022, 28, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Shendure, J.A.; Valle, D.; Hamosh, A.; Lupski, J.R.; Gibbs, R.A.; Boerwinkle, E.; Lifton, R.P.; Gerstein, M.; Gunel, M.; et al. The Centers for Mendelian Genomics: A new large-scale initiative to identify the genes underlying rare Mendelian conditions. Am. J. Med. Genet. Part A 2012, 158, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Pais, L.S.; Snow, H.; Weisburd, B.; Zhang, S.; Baxter, S.M.; DiTroia, S.; O’Heir, E.; England, E.; Chao, K.R.; Lemire, G.; et al. seqr: A web-based analysis and collaboration tool for rare disease genomics. Hum. Mutat. 2022, 43, 698–707. [Google Scholar] [CrossRef]
- DiStefano, M.T.; Goehringer, S.; Babb, L.; Alkuraya, F.S.; Amberger, J.; Amin, M.; Austin-Tse, C.; Balzotti, M.; Berg, J.S.; Birney, E.; et al. The Gene Curation Coalition: A global effort to harmonize gene-disease evidence resources. Genet. Med. 2022, 24, 1732–1742. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Azzariti, D.R.; Hamosh, A. Genomic Data Sharing for Novel Mendelian Disease Gene Discovery: The Matchmaker Exchange. Annu. Rev. Genom. Hum. Genet. 2020, 21, 305–326. [Google Scholar] [CrossRef]
- Philippakis, A.A.; Azzariti, D.R.; Beltran, S.; Brookes, A.J.; Brownstein, C.A.; Brudno, M.; Brunner, H.G.; Buske, O.J.; Carey, K.; Doll, C.; et al. The Matchmaker Exchange: A platform for rare disease gene discovery. Hum. Mutat. 2015, 36, 915–921. [Google Scholar] [CrossRef]
- Buske, O.J.; Schiettecatte, F.; Hutton, B.; Dumitriu, S.; Misyura, A.; Huang, L.; Hartley, T.; Girdea, M.; Sobreira, N.; Mungall, C.; et al. The Matchmaker Exchange API: Automating patient matching through the exchange of structured phenotypic and genotypic profiles. Hum. Mutat. 2015, 36, 922–927. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Cummings, B.B.; Karczewski, K.J.; Kosmicki, J.A.; Seaby, E.G.; Watts, N.A.; Singer-Berk, M.; Mudge, J.M.; Karjalainen, J.; Kyle Satterstrom, F.; O’Donnell-Luria, A.; et al. Transcript expression-aware annotation improves rare variant discovery and interpretation. bioRxiv 2019. bioRxiv:554444. [Google Scholar]
- Mungall, C.J.; McMurry, J.A.; Köhler, S.; Balhoff, J.P.; Borromeo, C.; Brush, M.; Carbon, S.; Conlin, T.; Dunn, N.; Engelstad, M.; et al. The Monarch Initiative: An integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 2017, 45, D712–D722. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Ellard, S.; Baple, E.L.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; Deans, Z.C. ACGS best practice guidelines for variant classification 2019. ACGS Guidel. 2019, 1, 32. [Google Scholar]
- Robinson, P.N.; Köhler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The clinical genome resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.D.; Hernandez, E.J.; Hobbs, C.; Jenkins, S.M.; Moore, M.B.; Juarez, E.R.; Zoucha, S.; Sanford, E.; Bainbridge, M.N.; Frise, E.; et al. Automated Prioritization of Sick Newborns for Rapid Whole Genome Sequencing Using Clinical Natural Language Processing and Machine Learning. Genome Med. 2022, 15, 18. [Google Scholar] [CrossRef]
- Uguen, K.; Krysiak, K.; Audebert-Bellanger, S.; Redon, S.; Benech, C.; Viora-Dupont, E.; Tran Mau-Them, F.; Rondeau, S.; Elsharkawi, I.; Granadillo, J.L.; et al. Heterozygous HMGB1 loss-of-function variants are associated with developmental delay and microcephaly. Clin. Genet. 2021, 100, 386–395. [Google Scholar] [CrossRef]
- Purcell, R.H.; Toro, C.; Gahl, W.A.; Hall, R.A. A disease-associated mutation in the adhesion GPCR BAI2 (ADGRB2) increases receptor signaling activity. Hum. Mutat. 2017, 38, 1751–1760. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Clinical Data | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Fam_ID | Pro_ID | Pat_ID | Mat_ID | Sib_ID | Age | Sex | WES Phenotype—HPO Terms Identified from Clinical Notes | WGS Phenotype—HPO Terms Identified by Clinician | WGS—Gene Panel Applied |
| FAM_1 | 1 | 2 | 3 | 0–5 | M | Gastroesophageal reflux, Myopia, Delayed eruption of primary teeth, Triangular face, Prominent forehead, Cow milk allergy, Egg allergy, Nut food product allergy, Sacral dimple, Clinodactyly of the 5th finger, Short 5th toe, 2–3 toe syndactyly, Mild global developmental delay, Delayed speech and language development, Oligohydramnios | Global developmental delay, Delayed speech and language development, Triangular face, Prominent forehead, Feeding difficulties, Delayed gross motor development, Oligohydramnios, Delayed eruption of primary teeth. | Intellectual disability (R.29.4), Congenital malformation and dysmorphism syndromes (R27.3), Skeletal dysplasia (R104.3), Likely inborn error of metabolism (R98.2) | |
| FAM_1 | 2 | 41–45 | M | unaffected | unaffected | ||||
| FAM_1 | 3 | 41–45 | F | unaffected | unaffected | ||||
| FAM_2 | 4 | 5 | 6 | 6–10 | M | Simple ear, Astigmatism, Obesity, Patchy hypo- and hyperpigmentation, 2–3 toe syndactyly, Short finger, Specific learning disability, Global developmental delay, Intellectual disability, Delayed speech and language development | Chronic otitis media, Obesity, Severe intellectual disability, Autistic behaviour, Global developmental delay | Intellectual disability (R.29.4), severe early-onset obesity (R149.1) | |
| FAM_2 | 5 | 31–35 | M | unaffected | unaffected | ||||
| FAM_2 | 6 | 31–35 | F | unaffected | unaffected | ||||
| FAM_3 | 7 | 8 | 9 | 6–10 | F | Low-set ears, Hypermetropia, Abnormality of the palmar creases, Broad distal phalanges of all fingers, Shallow orbits, Cranial asymmetry, Plagiocephaly, Mild global developmental delay, Intellectual disability | Thin upper lip vermillion, Long philtrum, Downslanted palpebral fissures, Deep palmar crease, Intellectual disability, Plagiocephaly | Intellectual disability (R29.4) | |
| FAM_3 | 8 | 61–65 | M | unaffected | unaffected | ||||
| FAM_3 | 9 | 46–50 | F | unaffected | unaffected | ||||
| FAM_4 | 10 | 11 | 12 | 13 | 46–50 | F | Delayed ability to walk, Delayed speech and language development, Spastic paraparesis, Global developmental delay | Global developmental delay, Intellectual disability, and Spastic paraparesis | Intellectual disability (R29.4) |
| FAM_4 | 11 | 76–80 | M | unaffected | unaffected | ||||
| FAM_4 | 12 | 76–80 | F | unaffected | unaffected | ||||
| FAM_4 | 13 | 46–50 | F | Delayed ability to walk, Delayed speech and language development, Seizure, Spastic paraparesis, Global developmental delay | Developmental delay, Intellectual disability, Spastic paraparesis, and Seizure | Intellectual disability (R29.4) | |||
| FAM_5 | 14 | 15 | 16 | 0–5 | F | Prominent forehead, Low hanging columella, Prominent fingertip pads, Preauricular pit, Hypopigmented macule, Frontal bossing, Flat occiput, Joint hypermobility, Confluent hyperintensity of cerebral white matter on MRI, Mild global developmental delay, Polydipsia | Prominent forehead, Moderate global developmental delay, Relative macrocephaly, Anxiety, Low hanging columella | Intellectual disability (R29.4) | |
| FAM_5 | 15 | 26–30 | M | unaffected | unaffected | ||||
| FAM_5 | 16 | 21–25 | F | unaffected | unaffected | ||||
| FAM_6 | 17 | 18 | 19 | 0–5 | F | Epicanthic folds, Joint hypermobility, Global developmental delay, Intellectual disability, Increased nuchal translucency | Global developmental delay, Increased prenatal nuchal translucency, Short toenails, Epicanthic folds | Intellectual disability (R29.4) | |
| FAM_6 | 18 | 31–35 | M | unaffected | unaffected | ||||
| FAM_6 | 19 | 31–35 | F | unaffected | unaffected | ||||
| FAM_7 | 20 | 21 | 22 | 6–10 | M | Hypertelorism, Bilateral polymicrogyria, Global developmental delay, Delayed speech and language development, Delayed fine motor development, Delayed gross motor development, Focal seizures, Generalised seizures, Intellectual disability | Focal seizures, Generalised seizures, Infantile encephalopathy, Polymicrogyria, Delayed speech and language development, Severe intellectual disability, Global developmental delay | Early-onset or syndromic epilepsy (R59.3), Cerebral malformation (R87.3) | |
| FAM_7 | 21 | 36–40 | M | unaffected | unaffected | ||||
| FAM_7 | 22 | 36–40 | F | unaffected | unaffected | ||||
| FAM_8 | 23 | 24 | 25 | 0–5 * | F | Microphthalmia, Cataract, Retinal dystrophy, Congenital nephrotic syndrome, Microcephaly | Intrauterine growth restriction, Microcephaly, Congenital nephrotic syndrome, Renal failure, Bilateral congenital cataract, Cerebellopontine hypoplasia, Retinal dysfunction, Thrombocytopaenia, Giant platelets, Howell–Jolly bodies | Congenital malformation and dysmorphic syndromes (R27), Structural eye disease (R36), Unexplained paediatric onset end-stage renal disease (R257), Cerebellar anomalies (R84), Severe microcephaly (R88), Proteinuric renal disease (R195) | |
| FAM_8 | 24 | 36–40 | F | unaffected | unaffected | ||||
| FAM_8 | 25 | 41–45 | M | unaffected | unaffected | ||||
| Research Exome HiPPo Strategy | NHS Genome Panel-Based Strategy | |||
|---|---|---|---|---|
| Dominant | Recessive | Dominant | Recessive | |
| Inheritance | De novo/dominant search | Recessive search & | De novo/dominant search | Recessive search & |
| AF (gnomAD exomes, gnomAD genomes, TOPMED *, ExAC, 1000 g) | <0.001 | <0.05 | <0.001 | <0.01 |
| Cohort^ AF | <0.01 | <0.01 | No filter applied | No filter applied |
| Variant type | All coding +/− 20 bp, excluding synonymous, on any transcript | All coding +/− 20 bp, excluding synonymous, on any transcript | All coding +/− 8 bp on any transcript, excluding synonymous | All coding +/− 8 bp on any transcript, excluding synonymous |
| SpliceAI (for splicing variants) | >0.2 | >0.2 | No filter applied | No filter applied |
| CADD (all variants) | >15 | >15 | No filter applied | No filter applied |
| ClinVar | Remove benign/likely benign | Remove benign/likely benign | No filter applied | No filter applied |
| Genes | All genes and later restricted to GenCC definitive and strong genes | All genes and later restricted to GenCC definitive and strong genes | Green in PanelApp Panel(s) | Green in PanelApp Panel(s) |
| Allele balance | >0.2 | >0.2 | N/A | N/A |
| Genotype Quality | >40 | >40 | >30 | >30 |
| QC | all variants | all variants | pass | pass |
| Other | Pathogenic variants in ClinVar retained even if in unaffected parents | N/A | In any gene: Exomiser top 3 rank variant (coding) with score of ≥0.95 or any de novo (coding) | In any gene: Exomiser top 3 rank variant (coding) with score of ≥0.95 or any de novo (coding) |
| SV/CNV | Not assessed | Not assessed | CNV (>10 KB) overlaps a ClinGen curated pathogenic region relevant to a panel applied, or the CNV overlaps with a green gene in the panel applied. | CNV (>10 KB) overlaps a ClinGen curated pathogenic region relevant to a panel applied, or the CNV overlaps with a green gene in the panel applied. |
| Samples | Research Exome | GMS Genome | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FamID | ProID | Reported Variants | Status | No. HiPPo Variants | No. HiPPo Variants in GenCC Genes | Reported Variants | No. of Variants Passing Filtering | De Novo | Exomiser | Additional HiPPo Variants | GMS Interpretation of HiPPo Variants |
| FAM_1 | 1 | VUS: HMGB1: 13:30462666:CT:C; c.342del; p.Gly115GlufsTer37 (frameshift, de novo). | Potential new disease gene, submitted to MME. Variant also detected by NHS. | 8 | 3 | VUS: HMGB1: 13:30462666:CT:C; c.342del; p.Gly115GlufsTer37 | 9 | REST HMGB1 | 1. HMGB1 2. KDM4 3. ROBO1 | None | N/A |
| FAM_2 | 4 | VUS: INTS1: 7:1480876:G:C; c.3908C > G; p.Thr1303Ser (missense). | Phenotype partially fitting with disease gene—undergoing clinical review. | 4 | 4 | No variants reported | 5 | None | 1. KDM5A 2. RPS3A 3. COL16A1 | INTS1—VUS × 2 | INTS1 (Tier 2) discounted as weak evidence |
| VUS: INTS1: 7:1497193:C:G; c.1547G > C; p.Cys516Ser (missense). | |||||||||||
| (Variants in trans) | |||||||||||
| FAM_3 | 7 | Pathogenic: PPP1CB: 2:28776944:C:G; c.146C > G; p.Pro49Arg (missense, de novo). | Confirmed diagnosis (also detected by NHS). | 4 | 2 | Pathogenic: PPP1CB: 2:28776944:C:G; c.146C > G; p.Pro49Arg | 7 | MYO7B PPP1CB EXOC7 | 1. PPP1CB 2. SELENBP1 3. EFCAB11 | None | N/A |
| FAM_4 | 10 | VUS: ADGRB2: 1:31731030:G:A; c.4150C > T; p.Arg1384Ter (de novo, nonsense). | Potential new disease gene. Confirmed de novo by Sanger sequencing and in identical twin (FAM_4_13). Functional work underway. | 68 | 23 | No variants reported | 14 | ADGRB2 CRNN PCDHB7 NFYB PIEZO1 | 1. FBXO46 2. CEP290 3. NFYB | ADGRB2—VUS × 2 | De novo (ADGRB2) variant discounted as in novel gene |
| FAM_4 | 13 | VUS: ADGRB2: 1:31731030:G:A; c.4150C > T; p.Arg1384Ter (de novo, stop gained). | The same variant is present in identical twin (FAM_4_10) | No variants reported | |||||||
| FAM_5 | 14 | Pathogenic: ABCC8: 11:17413408:G:A; c.2464C > T; p.Gln822Ter (nonsense, inherited from parent) | Clinically agreed as partial diagnosis. | 8 | 1 | No variants reported | 5 | GOLGA8T | 1. PTPRF 2. NPHP4 3. PRKDC | ABCC8—Pathogenic | ABCC8 not analysed as untiered and gene absent from R29 panel |
| FAM_6 | 17 | Pathogenic: CHAMP1: 13:114325034:C:T; c.1192C > T; p.Arg398Ter (de novo, nonsense). | Confirmed diagnosis (also detected by NHS). | 2 | 1 | Pathogenic: CHAMP1: 13:114325034:C:T; c.1192C > T; p.Arg398Ter | 6 | KRTAP5-5 | 1. CHAMP1 2. MDK 3. CRAC2RA | None | N/A |
| FAM_7 | 20 | VUS: FOXB2: 9:77020700:A:G; c.1046A > G; p.Lys349Arg (missense, de novo). VUS: PKD1L3: 16:71951734:T:G; c.3020A > C; p.Glu1007Ala (missense). | Both FOXB2 and PFK1L3 are potential novel disease genes and have been submitted to MME. | 10 | 1 | No variants reported | 7 | FOXB2 RP1L1 | 1. IGFN1 2. ZXDA 3. CADNA1F | FOXB2—VUS PKD1L3—VUS × 2 | FOXB2 de novo variant—Discounted PKD1L3—Not analysed—Tier 3; Exomiser rank 33 |
| VUS: PKD1L3: 16:71951734:T:G; c.3020A > C; p.Glu1007Ala (missense). PFK1L3 variants are in trans. | |||||||||||
| FAM_8 | 23 | VUS: ZNF91: 19:23361341:G:C; c.1638C > G; p.Tyr546Ter (de novo, nonsense). | ZNF91 is a novel disease gene. A group is working on this gene, and we have joined their case series. The SDCCAG8 variants are in trans but are not felt to fit with the clinical phenotype. | 5 | 3 | VUS: SDCCAG8: 14:92449109:A > C, c.1552A > G, p.Arg518Gly (missense). Pathogenic: SDCCAG8: 1:243341070:TG>T, c.1255del, p.Glu419ArgfsTer43 (frameshift). | 24 | ZNF91 ZNF91 | 1. RIN3 2. ERAP2 3. ZNF91 | None | ZNF91 variant discounted as no established disease association |
| VUS: SDCCAG8: 14:92449109:A > C, c.1552A > G, p.Arg518Gly (missense). | |||||||||||
| Pathogenic: SDCCAG8: 1:243341070:TG > T, c.1255del, p.Glu419ArgfsTer43 (frameshift). | |||||||||||
| Variant | Gene | Consequence | Gnomad | Cadd | Revel | Hgvsc | Hgvsp | ClinVar | ACMG | FamID | ProbandID | P_AC | Sample_2 | S2_AC | Sample_3 | S3_AC | Returned by GMS? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13:30462666:CT:C | HMGB1 | frameshift | 0 | 38 | ENST00000341423.9:c.342del | p.Gly115GlufsTer37 | VUS | FAM_1 | 1 | 1 | 2 | 0 | 3 | 0 | Yes | ||
| 7:1480876:G:C | INTS1 | missense | 5.56−4 | 23.5 | 0.243 | ENST00000404767.7:c.3908C > G | p.Thr1303Ser | VUS | FAM_2 | 4 | 1 | 5 | 1 | 6 | 0 | No | |
| 7:1497193:C:G | INTS1 | missense | 7.76−5 | 24 | 0.315 | ENST00000404767.7:c.1547G > C | p.Cys516Ser | VUS | FAM_2 | 4 | 1 | 5 | 0 | 6 | 1 | No | |
| 2:28776944:C:G | PPP1CB | missense | 0 | 26.7 | 0.438 | ENST00000395366.2:c.146C > G | p.Pro49Arg | P | P | FAM_3 | 7 | 1 | 8 | 0 | 9 | 0 | Yes |
| 1:31731030:G:A | ADGRB2 | stop_gained | 0 | 38 | ENST00000373655.6:c.4150C > T | p.Arg1384Ter | VUS | FAM_4 | 13 | 1 | 11 | 0 | 13 | 1 | No | ||
| 1:31731030:G:A | ADGRB2 | stop_gained | 0 | 38 | ENST00000373655.6:c.4150C > T | p.Arg1384Ter | VUS | FAM_4 | 10 | 1 | 11 | 0 | 13 | 1 | No | ||
| 13:114325034:C:T | CHAMP1 | stop_gained | 0 | 35 | ENST00000643483.1:c.1192C > T | p.Arg398Ter | P | P | FAM_6 | 17 | 1 | 18 | 0 | 19 | 0 | Yes | |
| 11:17413408:G:A | ABCC8 | stop_gained | 0 | 43 | ENST00000302539.9:c.2464C > T | p.Gln822Ter | LP | FAM_5 | 14 | 1 | 15 | 0 | 19 | 1 | No | ||
| 16:71951734:T:G | PKD1L3 | missense | 5.09−4 | 23.1 | ENST00000620267.1:c.3020A > C | p.Glu1007Ala | VUS | FAM_7 | 20 | 1 | 21 | 1 | 22 | 0 | No | ||
| 16:71973386:C:T | PKD1L3 | missense | 1.02−4 | 22 | ENST00000620267.1:c.1891G > A | p.Ala631Thr | VUS | FAM_7 | 20 | 1 | 21 | 0 | 22 | 1 | No | ||
| 9:77020700:A:G | FOXB2 | missense | 0 | 25.3 | 0.534 | ENST00000376708.1:c.1046A > G | p.Lys349Arg | VUS | FAM_7 | 20 | 1 | 21 | 0 | 22 | 0 | No | |
| 19:23361341:G:C | ZNF91 | stop_gained | 0 | 32 | ENST00000300619.11:c.1638C > G | p.Tyr546Ter | VUS | FAM_8 | 23 | 1 | 24 | 0 | 25 | 0 | No | ||
| 1:243341070:TG:T | SDCCAG8 | frameshift | 0 | 26 | ENST00000366541.7:c.1255del | p.Glu419ArgfsTer43 | P | FAM_8 | 23 | 1 | 24 | 1 | 25 | 0 | Yes | ||
| 1:243378799:A:G | SDCCAG8 | missense | 9.55−5 | 22.2 | 0.195 | ENST00000366541.7:c.1552A > G | p.Arg518Gly | VUS | VUS | FAM_8 | 23 | 1 | 24 | 0 | 25 | 1 | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seaby, E.G.; Thomas, N.S.; Hunt, D.; Baralle, D.; Rehm, H.L.; O’Donnell-Luria, A.; Ennis, S. A Panel-Agnostic Strategy ‘HiPPo’ Improves Diagnostic Efficiency in the UK Genomic Medicine Service. Healthcare 2023, 11, 3179. https://doi.org/10.3390/healthcare11243179
Seaby EG, Thomas NS, Hunt D, Baralle D, Rehm HL, O’Donnell-Luria A, Ennis S. A Panel-Agnostic Strategy ‘HiPPo’ Improves Diagnostic Efficiency in the UK Genomic Medicine Service. Healthcare. 2023; 11(24):3179. https://doi.org/10.3390/healthcare11243179
Chicago/Turabian StyleSeaby, Eleanor G., N. Simon Thomas, David Hunt, Diana Baralle, Heidi L. Rehm, Anne O’Donnell-Luria, and Sarah Ennis. 2023. "A Panel-Agnostic Strategy ‘HiPPo’ Improves Diagnostic Efficiency in the UK Genomic Medicine Service" Healthcare 11, no. 24: 3179. https://doi.org/10.3390/healthcare11243179