
Approaching Sites of Action of Temozolomide for Pharmacological and Clinical Studies in Glioblastoma

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Understanding and Monitoring Temozolomide Fate and Biochemical Action
2.1. Absorption, Distribution, Metabolism, and Excretion of Temozolomide
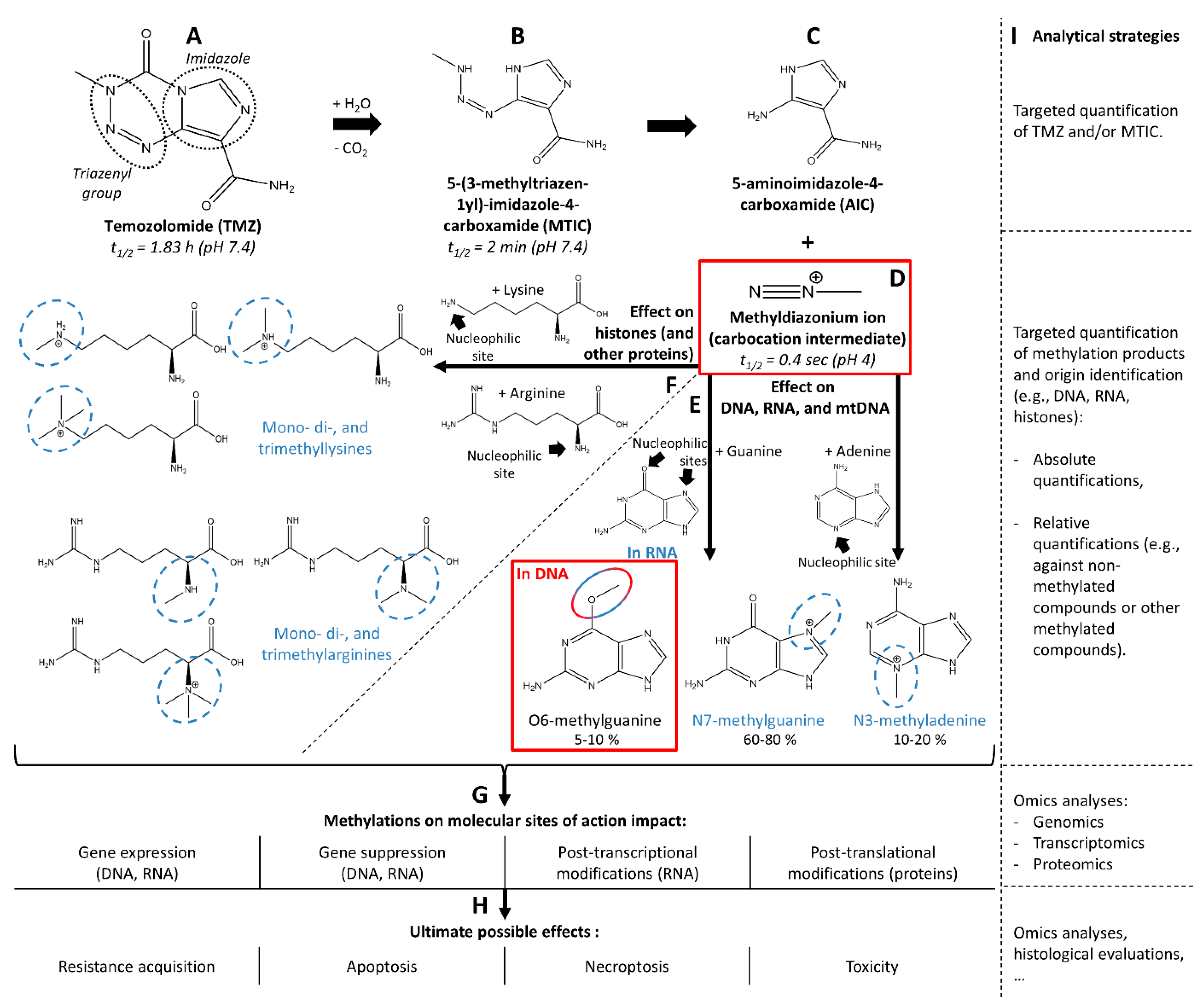
2.2. Chemical Action of Temozolomide
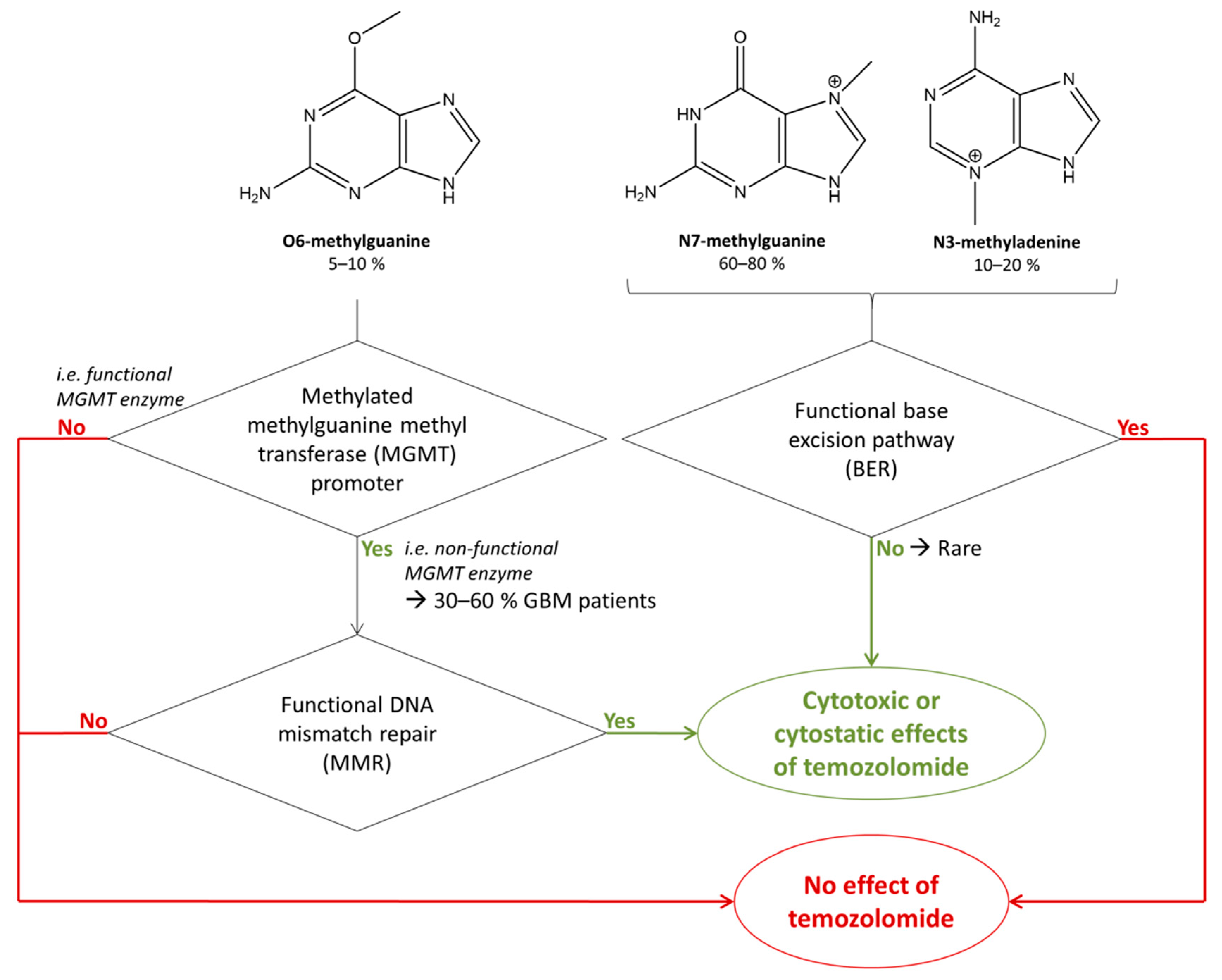
2.3. Biological Action of Temozolomide
2.4. Retrieval of the Intended Anatomical Site of Action of Temozolomide
3. Analytical Workflows to Quantify the Intended Chemical Action of Temozolomide in Glioblastoma
3.1. Sampling of the Intended Cellular Site of Action
3.2. Sample Processing for Intended Sites of Action of Temozolomide
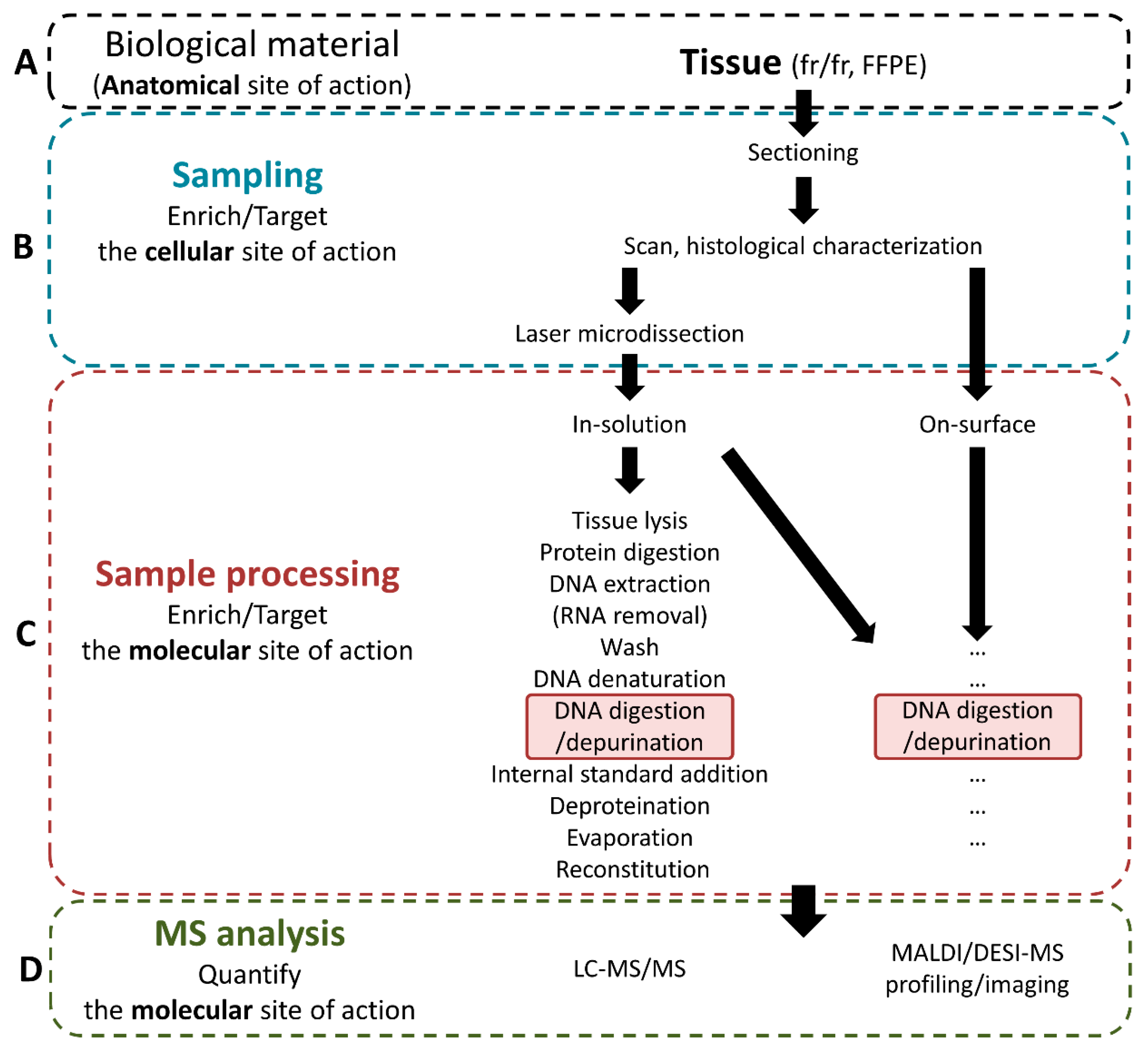
3.2.1. Retrieval of Unbound Temozolomide at the Intended Anatomical Site of Action
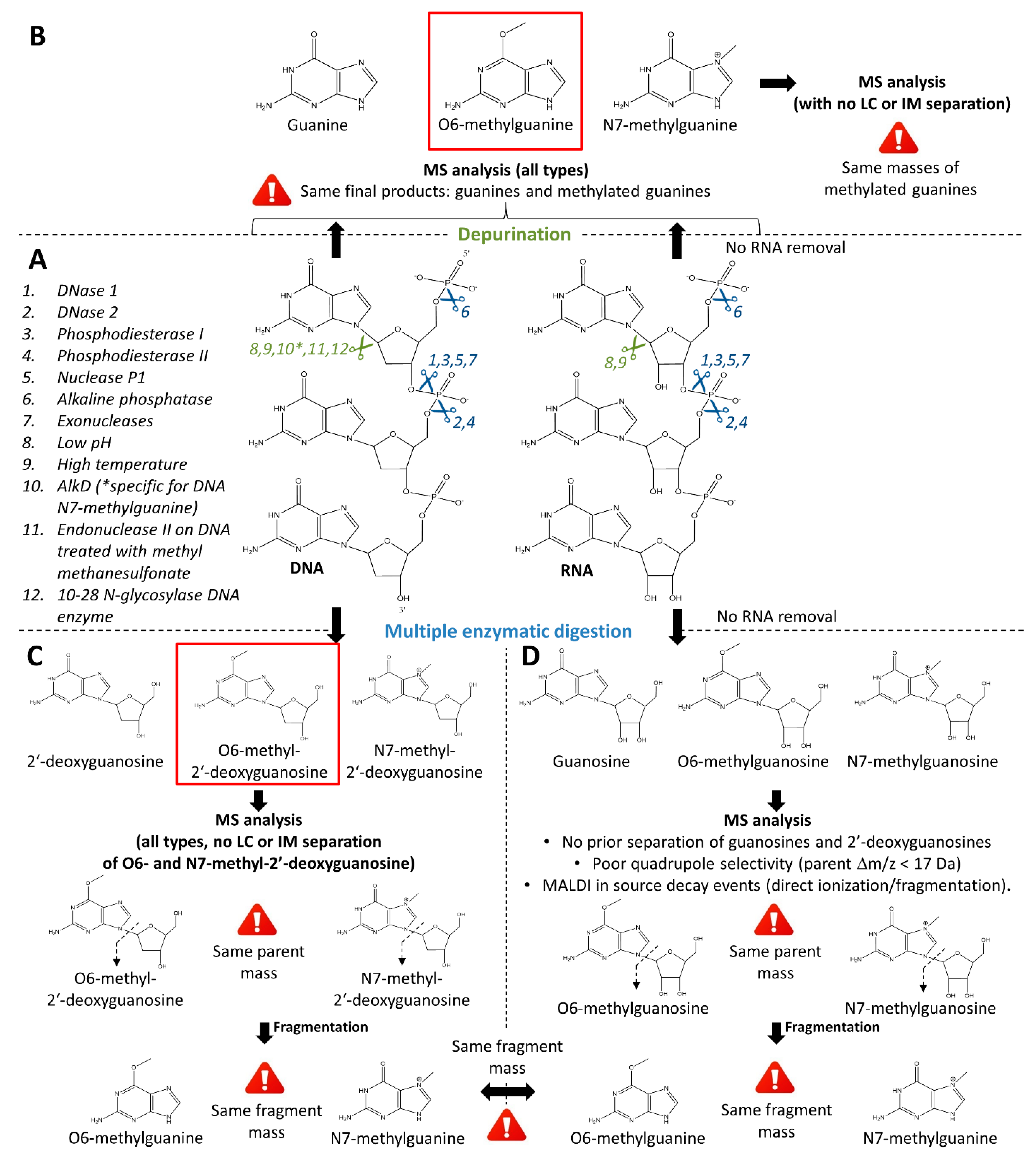
3.2.2. Retrieval of the Intended Modified Molecular Site of Action: Methylated Guanines
Retrieval of Deoxyribonucleosides
Retrieval of Purines
3.3. Analysis
3.3.1. LC-MS/MS
3.3.2. MALDI-MS
3.3.3. Quantification Approaches: Development and Validation
Quantification of Temozolomide
Quantification of Methylated Guanines and Guanosines
4. Biological Models to Study the Action of TMZ at SOAs
5. Considerations for the Analysis of Alternative Sites of Action
6. Measurement of the Biological Action of Temozolomide
6.1. Genomics and Transcriptomics
6.2. Proteomics
7. Applications
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; Westhoff, M.A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longuespee, R.; Theile, D.; Fresnais, M.; Burhenne, J.; Weiss, J.; Haefeli, W.E. Approaching sites of action of drugs in clinical pharmacology: New analytical options and their challenges. Br. J. Clin. Pharmacol. 2021, 87, 858–874. [Google Scholar] [CrossRef] [PubMed]
- Fresnais, M.; Burhenne, J.; Haefeli, W.E.; Longuespee, R. Desorption/Ionization-MS Methods for Drug Quantification in Biological Matrices and Their Validation Following Regulatory Guidance. Anal. Chem. 2021, 93, 7152–7163. [Google Scholar] [CrossRef]
- Beale, P.; Judson, I.; Moore, S.; Statkevich, P.; Marco, A.; Cutler, D.L.; Reidenberg, P.; Brada, M. Effect of gastric pH on the relative oral bioavailability and pharmacokinetics of temozolomide. Cancer Chemother. Pharmacol. 1999, 44, 389–394. [Google Scholar] [CrossRef]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [Green Version]
- Pitz, M.W.; Desai, A.; Grossman, S.A.; Blakeley, J.O. Tissue concentration of systemically administered antineoplastic agents in human brain tumors. J. Neurooncol. 2011, 104, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Guo, P.; Kruh, G.D.; Vicini, P.; Wang, X.; Gallo, J.M. Predicting human tumor drug concentrations from a preclinical pharmacokinetic model of temozolomide brain disposition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 4271–4279. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 309–317. [Google Scholar]
- Schreck, K.C.; Grossman, S.A. Role of Temozolomide in the Treatment of Cancers Involving the Central Nervous System. Oncology 2018, 32, 555–560. [Google Scholar] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers 2017, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wang, X.Q.; Zhou, B.; Zhang, L. The prognostic value of MGMT promoter methylation in Glioblastoma multiforme: A meta-analysis. Fam. Cancer 2013, 12, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef] [PubMed]
- Houillier, C.; Wang, X.; Kaloshi, G.; Mokhtari, K.; Guillevin, R.; Laffaire, J.; Paris, S.; Boisselier, B.; Idbaih, A.; Laigle-Donadey, F.; et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 2010, 75, 1560–1566. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2001, 61, 5843–5849. [Google Scholar]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef]
- Aletta, J.M.; Cimato, T.R.; Ettinger, M.J. Protein methylation: A signal event in post-translational modification. Trends Biochem. Sci. 1998, 23, 89–91. [Google Scholar] [CrossRef]
- Wang, T.; Pickard, A.J.; Gallo, J.M. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Anticancer Res. 2016, 36, 3289–3299. [Google Scholar]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Rubens, U.; Hoyoux, R.; Vanosmael, L.; Ouras, M.; Tasset, M.; Hamilton, C.; Longuespee, R.; Maree, R. Cytomine: Toward an Open and Collaborative Software Platform for Digital Pathology Bridged to Molecular Investigations. Proteom. Clin. Appl. 2019, 13, e1800057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, M.P.; Maxwell, P.; Salto-Tellez, M. QuPath: The global impact of an open source digital pathology system. Comput. Struct. Biotechnol. J. 2021, 19, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Cizkova, K.; Foltynkova, T.; Gachechiladze, M.; Tauber, Z. Comparative Analysis of Immunohistochemical Staining Intensity Determined by Light Microscopy, ImageJ and QuPath in Placental Hofbauer Cells. Acta Histochem. Cytochem. 2021, 54, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Maree, R.; Rollus, L.; Stevens, B.; Hoyoux, R.; Louppe, G.; Vandaele, R.; Begon, J.M.; Kainz, P.; Geurts, P.; Wehenkel, L. Collaborative analysis of multi-gigapixel imaging data using Cytomine. Bioinformatics 2016, 32, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- von Eggeling, F.; Hoffmann, F. Microdissection-An Essential Prerequisite for Spatial Cancer Omics. Proteomics 2020, 20, e2000077. [Google Scholar] [CrossRef]
- Longuespee, R.; Casadonte, R.; Kriegsmann, M.; Pottier, C.; Picard de Muller, G.; Delvenne, P.; Kriegsmann, J.; De Pauw, E. MALDI mass spectrometry imaging: A cutting-edge tool for fundamental and clinical histopathology. Proteom. Clin. Appl. 2016, 10, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Chilakala, S.; Feng, Y.; Li, L.; Mahfouz, R.; Quteba, E.; Saunthararajah, Y.; Xu, Y. Tracking Decitabine Incorporation into Malignant Myeloid Cell DNA in vitro and in vivo by LC-MS/MS with Enzymatic Digestion. Sci. Rep. 2019, 9, 4558. [Google Scholar] [CrossRef]
- Ly, A.; Longuespee, R.; Casadonte, R.; Wandernoth, P.; Schwamborn, K.; Bollwein, C.; Marsching, C.; Kriegsmann, K.; Hopf, C.; Weichert, W.; et al. Site-to-Site Reproducibility and Spatial Resolution in MALDI-MSI of Peptides from Formalin-Fixed Paraffin-Embedded Samples. Proteom. Clin. Appl. 2019, 13, e1800029. [Google Scholar] [CrossRef] [Green Version]
- Alexovic, M.; Sabo, J.; Longuespee, R. Microproteomic sample preparation. Proteomics 2021, 21, e2000318. [Google Scholar] [CrossRef]
- Goldwirt, L.; Zahr, N.; Farinotti, R.; Fernandez, C. Development of a new UPLC-MSMS method for the determination of temozolomide in mice: Application to plasma pharmacokinetics and brain distribution study. Biomed. Chromatogr. BMC 2013, 27, 889–893. [Google Scholar] [CrossRef]
- FDA. US Food and Drug Administration–Bioanalytical Method Validation Guidance for Industry, 2018th ed.; FDA: Silver Spring, MD, USA, 2018. [Google Scholar]
- EMA. European Medicines Agency–Guidelines on Bioanalytical Method Validation, 2011th ed.; EMA: Amsterdam, The Netherlands, 2011. [Google Scholar]
- ICH. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use–ICH Guideline M10 on Bioanalytical Method Validation, 2019th ed.; Draft version; ICH: Genevan, Switzerland, 2019. [Google Scholar]
- Yang, Y.; Nikolic, D.; Swanson, S.M.; van Breemen, R.B. Quantitative determination of N7-methyldeoxyguanosine and O6-methyldeoxyguanosine in DNA by LC-UV-MS-MS. Anal. Chem. 2002, 74, 5376–5382. [Google Scholar] [CrossRef]
- Zhang, F.; Bartels, M.J.; Pottenger, L.H.; Gollapudi, B.B.; Schisler, M.R. Simultaneous quantitation of 7-methyl- and O6-methylguanine adducts in DNA by liquid chromatography-positive electrospray tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 833, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.W.; Chen, C.M.; Ho, H.H.; Chao, M.R. Simultaneous quantification of methylated purines in DNA by isotope dilution LC-MS/MS coupled with automated solid-phase extraction. Anal. Bioanal. Chem. 2012, 402, 1199–1208. [Google Scholar] [CrossRef]
- Baskerville-Abraham, I.M.; Boysen, G.; Troutman, J.M.; Mutlu, E.; Collins, L.; Dekrafft, K.E.; Lin, W.; King, C.; Chaney, S.G.; Swenberg, J.A. Development of an ultraperformance liquid chromatography/mass spectrometry method to quantify cisplatin 1,2 intrastrand guanine-guanine adducts. Chem. Res. Toxicol. 2009, 22, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Hee, S.Q. Quantitation of normal and formaldehyde-modified deoxynucleosides by high-performance liquid chromatography/UV detection. Biomed. Chromatogr. BMC 2004, 18, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Beck, J.L.; Sheil, M.M.; Ralph, S.F. Identification of bifunctional GA and AG intrastrand crosslinks formed between cisplatin and DNA. J. Inorg. Biochem. 2005, 99, 552–559. [Google Scholar] [CrossRef]
- Da Pieve, C.; Sahgal, N.; Moore, S.A.; Velasco-Garcia, M.N. Development of a liquid chromatography/tandem mass spectrometry method to investigate the presence of biomarkers of DNA damage in urine related to red meat consumption and risk of colorectal cancer. Rapid Commun. Mass Spectrom. RCM 2013, 27, 2493–2503. [Google Scholar] [CrossRef]
- Donald, C.E.; Stokes, P.; O’Connor, G.; Woolford, A.J. A comparison of enzymatic digestion for the quantitation of an oligonucleotide by liquid chromatography-isotope dilution mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 817, 173–182. [Google Scholar] [CrossRef]
- Laukova, L.; Konecna, B.; Janovicova, L.; Vlkova, B.; Celec, P. Deoxyribonucleases and Their Applications in Biomedicine. Biomolecules 2020, 10, 1036. [Google Scholar] [CrossRef]
- Rittie, L.; Perbal, B. Enzymes used in molecular biology: A useful guide. J. Cell Commun. Signal. 2008, 2, 25–45. [Google Scholar] [CrossRef] [Green Version]
- Ekanayake, K.S.; Lebreton, P.R. Activation barriers for DNA alkylation by carcinogenic methane diazonium ions. J. Comput. Chem. 2006, 27, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- An, R.; Jia, Y.; Wan, B.; Zhang, Y.; Dong, P.; Li, J.; Liang, X. Non-enzymatic depurination of nucleic acids: Factors and mechanisms. PLoS ONE 2014, 9, e115950. [Google Scholar] [CrossRef] [Green Version]
- Glavin, D.P.; Schubert, M.; Bada, J.L. Direct isolation of purines and pyrimidines from nucleic acids using sublimation. Anal. Chem. 2002, 74, 6408–6412. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Y. Mass spectrometry for the assessment of the occurrence and biological consequences of DNA adducts. Chem. Soc. Rev. 2015, 44, 7829–7854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T.; Karlstrom, O. Heat-induced depyrimidination of deoxyribonucleic acid in neutral solution. Biochemistry 1973, 12, 5151–5154. [Google Scholar] [CrossRef] [PubMed]
- Rubinson, E.H.; Christov, P.P.; Eichman, B.F. Depurination of N7-methylguanine by DNA glycosylase AlkD is dependent on the DNA backbone. Biochemistry 2013, 52, 7363–7365. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kirtikar, D.M.; Goldthwait, D.A. The enzymatic release of O6-methylguanine and 3-methyladenine from DNA reacted with the carcinogen N-methyl-N-nitrosourea. Proc. Natl. Acad. Sci. USA 1974, 71, 2022–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, T.L.; Ordoukhanian, P.; Joyce, G.F. A DNA enzyme with N-glycosylase activity. Proc. Natl. Acad. Sci. USA 2000, 97, 7802–7807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresnais, M.; Muck, A.; Majewsky, M.; Statz, B.; Krausert, S.; Benzel, J.; Castel, D.; Le Dret, L.; Pfister, S.; Haefeli, W.E.; et al. Rapid and Sensitive Drug Quantification in Tissue Sections Using Matrix Assisted Laser Desorption Ionization-Ion Mobility-Mass Spectrometry Profiling. J. Am. Soc. Mass Spectrom. 2020, 31, 742–751. [Google Scholar] [CrossRef]
- Guo, C.; Chen, Q.; Chen, J.; Yu, J.; Hu, Y.; Zhang, S.; Zheng, S. 8-Hydroxyguanosine as a possible RNA oxidative modification marker in urine from colorectal cancer patients: Evaluation by ultra performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1136, 121931. [Google Scholar] [CrossRef]
- Pabst, M.; Grass, J.; Fischl, R.; Leonard, R.; Jin, C.; Hinterkorner, G.; Borth, N.; Altmann, F. Nucleotide and nucleotide sugar analysis by liquid chromatography-electrospray ionization-mass spectrometry on surface-conditioned porous graphitic carbon. Anal. Chem. 2010, 82, 9782–9788. [Google Scholar] [CrossRef]
- Barnes, J.; Tian, L.; Loftis, J.; Hiznay, J.; Comhair, S.; Lauer, M.; Dweik, R. Isolation and analysis of sugar nucleotides using solid phase extraction and fluorophore assisted carbohydrate electrophoresis. MethodsX 2016, 3, 251–260. [Google Scholar] [CrossRef]
- Godoy, A.T.; Eberlin, M.N.; Simionato, A.V.C. Targeted metabolomics: Liquid chromatography coupled to mass spectrometry method development and validation for the identification and quantitation of modified nucleosides as putative cancer biomarkers. Talanta 2020, 210, 120640. [Google Scholar] [CrossRef]
- Valle-Algarra, F.M.; Medina, A.; Gimeno-Adelantado, J.V.; Llorens, A.; Jimenez, M.; Mateo, R. Comparative assessment of solid-phase extraction clean-up procedures, GC columns and perfluoroacylation reagents for determination of type B trichothecenes in wheat by GC-ECD. Talanta 2005, 66, 194–201. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalo, E.; Herrero-Herrero, L.; Garcia-Gomez, D. Development, validation and application of a fast analytical methodology for the simultaneous determination of DNA- and RNA-derived urinary nucleosides by liquid chromatography coupled to tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1019, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Cervinkova, B.; Krcmova, L.K.; Sestakova, V.; Solichova, D.; Solich, P. A fully validated bioanalytical method using an UHPLC-MS/MS system for quantification of DNA and RNA oxidative stress biomarkers. Anal. Bioanal. Chem. 2017, 409, 3611–3621. [Google Scholar] [CrossRef]
- Petru, K.; Siroka, J.; Bydzovska, L.; Krcmova, L.; Polasek, M. Assay of urinary 8-hydroxy-2’-deoxyguanosine by capillary electrophoresis with spectrophotometric detection using a high-sensitivity detection cell and solid-phase extraction. Electrophoresis 2014, 35, 2546–2549. [Google Scholar] [CrossRef] [PubMed]
- Garaguso, I.; Halter, R.; Krzeminski, J.; Amin, S.; Borlak, J. Method for the rapid detection and molecular characterization of DNA alkylating agents by MALDI-TOF mass spectrometry. Anal. Chem. 2010, 82, 8573–8582. [Google Scholar] [CrossRef]
- Li, Y.; Yu, H.; Zhao, W.; Xu, X.; Zhou, J.; Xu, M.; Gao, W.; Yuan, G. Analysis of urinary methylated nucleosides of patients with coronary artery disease by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2014, 28, 2054–2058. [Google Scholar] [CrossRef]
- Machon, C.; Jordheim, L.P.; Puy, J.Y.; Lefebvre, I.; Dumontet, C.; Guitton, J. Fully validated assay for the quantification of endogenous nucleoside mono- and triphosphates using online extraction coupled with liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 2925–2941. [Google Scholar] [CrossRef]
- Liebich, H.M.; Muller-Hagedorn, S.; Klaus, F.; Meziane, K.; Kim, K.R.; Frickenschmidt, A.; Kammerer, B. Chromatographic, capillary electrophoretic and matrix-assisted laser desorption ionization time-of-flight mass spectrometry analysis of urinary modified nucleosides as tumor markers. J. Chromatogr. A 2005, 1071, 271–275. [Google Scholar] [CrossRef]
- Kammerer, B.; Frickenschmidt, A.; Gleiter, C.H.; Laufer, S.; Liebich, H. MALDI-TOF MS analysis of urinary nucleosides. J. Am. Soc. Mass Spectrom. 2005, 16, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Lai, Y.; Chen, G.; Cai, Z. Matrix interference-free method for the analysis of small molecules by using negative ion laser desorption/ionization on graphene flakes. Anal. Chem. 2011, 83, 3161–3169. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, L.; Wang, J.; Hou, J.; He, Q.; Liu, J.; Wang, J.; Xiong, S.; Yang, G.; Nie, Z. 2,3,4,5-Tetrakis(3’,4’-dihydroxylphenyl)thiophene: A new matrix for the selective analysis of low molecular weight amines and direct determination of creatinine in urine by MALDI-TOF MS. Anal. Chem. 2012, 84, 10291–10297. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Tan, B.H.; Sugrue, R.J.; Tang, K. MALDI mass spectrometry for nucleic acid analysis. Top. Curr. Chem. 2013, 331, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Fujisaka, A.; Obika, S. Nucleobase derivatives induce in-source decay of oligonucleotides as new matrix-assisted laser desorption/ionization matrices. Rapid Commun. Mass Spectrom. 2020, 34, e8620. [Google Scholar] [CrossRef]
- Ciccimaro, E.; Blair, I.A. Stable-isotope dilution LC-MS for quantitative biomarker analysis. Bioanalysis 2010, 2, 311–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresnais, M.; Yildirim, E.; Karabulut, S.; Jager, D.; Zornig, I.; Benzel, J.; Pajtler, K.W.; Pfister, S.M.; Burhenne, J.; Haefeli, W.E.; et al. Rapid MALDI-MS Assays for Drug Quantification in Biological Matrices: Lessons Learned, New Developments, and Future Perspectives. Molecules 2021, 26, 1281. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Comincini, S.; Schinelli, S. In Vitro Glioblastoma Models: A Journey into the Third Dimension. Cancers 2021, 13, 2449. [Google Scholar] [CrossRef] [PubMed]
- Rybin, M.J.; Ivan, M.E.; Ayad, N.G.; Zeier, Z. Organoid Models of Glioblastoma and Their Role in Drug Discovery. Front. Cell Neurosci. 2021, 15, 605255. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Bergmann, S.; Lawler, S.E.; Qu, Y.; Fadzen, C.M.; Wolfe, J.M.; Regan, M.S.; Pentelute, B.L.; Agar, N.Y.R.; Cho, C.F. Blood-brain-barrier organoids for investigating the permeability of CNS therapeutics. Nat. Protoc. 2018, 13, 2827–2843. [Google Scholar] [CrossRef] [Green Version]
- Waghule, T.; Narayan Saha, R.; Singhvi, G. UV spectroscopic method for estimation of temozolomide: Application in stability studies in simulated plasma pH, degradation rate kinetics, formulation design, and selection of dissolution media. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 258, 119848. [Google Scholar] [CrossRef]
- Stoccoro, A.; Coppede, F. Mitochondrial DNA Methylation and Human Diseases. Int. J. Mol. Sci. 2021, 22, 4594. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Kalxdorf, M.; Longuespee, R.; Kazdal, D.N.; Stenzinger, A.; Krijgsveld, J. Automated sample preparation with SP3 for low-input clinical proteomics. Mol. Syst. Biol. 2020, 16, e9111. [Google Scholar] [CrossRef]
- Liu, N.Q.; Braakman, R.B.; Stingl, C.; Luider, T.M.; Martens, J.W.; Foekens, J.A.; Umar, A. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J. Mammary Gland. Biol. Neoplasia 2012, 17, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.Z.; Xiang, W.; Feng, W.Y.; Chen, Z.Y.; Li, Y.M.; Deng, S.Z.; Guo, M.L.; Zhao, L.; Sun, X.G.; He, M.Y.; et al. Identification of Key Candidate Proteins and Pathways Associated with Temozolomide Resistance in Glioblastoma Based on Subcellular Proteomics and Bioinformatical Analysis. Biomed. Res. Int. 2018, 2018, 5238760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.; Li, Y.; Chan, A.; Ng, S.; Loong, H.; Chan, D.; Wong, G.; Poon, W.-S. A multifaceted review of temozolomide resistance mechanisms in glioblastoma beyond O-6-methylguanine-DNA methyltransferase. Glioma 2019, 2, 68–82. [Google Scholar] [CrossRef]
- Noberini, R.; Longuespee, R.; Richichi, C.; Pruneri, G.; Kriegsmann, M.; Pelicci, G.; Bonaldi, T. PAT-H-MS coupled with laser microdissection to study histone post-translational modifications in selected cell populations from pathology samples. Clin Epigenet. 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leutert, M.; Entwisle, S.W.; Villen, J. Decoding Post-Translational Modification Crosstalk With Proteomics. Mol. Cell Proteom. 2021, 20, 100129. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Param. | Description |
|---|---|
| P1 | Dose of therapeutic compound |
| P2 | Unbound drug concentration in plasma (drug free fraction) |
| P3 | Localization of the drug’s cellular SOA (e.g., intravascular or interstitial compartment) |
| P4 | Expression and activity of transporters at the cellular SOA |
| P5 | Unbound and bound drug concentration on or in the cellular SOA |
| P6 | Abundance of the molecular SOA and number of available specific binding sites |
| P7 | Chemical interaction between the drug and its molecular SOA |
| P8 | Alternative (off-)site binding |
| P9 | Concentration of drug metabolites in the cellular SOA |
| P10 | Expression and function of drug metabolizing enzymes and other clearance mechanisms |
| Sample Type | Targeted Compound | Sample Preparation | Enzymatic Digestion (Specificity Indication) | Analytical Method | Quantification Strategy | Quality Parameters | Ref. |
|---|---|---|---|---|---|---|---|
| Tissue (mice dosed with TMZ) | TMZ | Low pH conditions/Tissue homogeneization/Protein precipitation | n.a. | LC-MS/MS 4 min LC gradient MRM (QqQ) | Tissue homogenate spiked with TMZ and theophylline (IS) for CALs and QCs | Partial validation (assay precision, accuracy, recovery, linearity, specificty, and matrix effect) [7,46,47,48] | [45] |
| Cells (cell lines and patient cells exposed with decitabine) | Decitabine (5-aza-2′-deoxycyti dine) incorporated in DNA | Cell lysis/Protein digestion (proteinase K)/DNA extraction/RNA removal (RNase)/DNA denaturing and hydrolysis/IS spiking/Deproteination/ Evaporation/Reconstitu tion | DNA hydrolysis: DNase I (DNA phosphodiester bonds) NPI (RNA and ssDNA phosphodiester bonds) PDE I (phosphodiester bonds) ALP (removal of 5′-phosphate) | LC-MS/MS MRM (Q-Trap) |
| Partial validation (assay precision, accuracy, recovery, linearity, and matrix effect) [7,46,47,48] | [42] |
| DNA (from calf thymus) Liver tissues (from rat exposed to MNU) | N7- and O6-methyl-2′-deoxy guanosine | DNA: DNA methylation by MNU in buffer/DNA precipitation. Tissues: Homogeneization and cell lysis/RNA digestion/Protein digestion (protease K)/DNA precipitation. All samples: DNA hydrolysis/ultrafiltration (<30,000 g/mol) | All samples (DNA hydrolysis): NP1 ALP | LC-UV-MS/MS 23-min gradient MRM (QqQ) | LC mobile phase spiked with N7- and O6-methyl-2′-deoxyguanosines, and [2H3]-N7- and [2H3]-O6-methyl-2′-deoxyguanosines (IS) for CALs | Stability experiments at −20 °C, room temperature, and 37 °C using N7- and O6-methyl-2′-deoxyguanosine solutions in Tris buffer. Recovery: DNA hydrolysates spiked with standards at different concentrations before and after ultrafiltration compared to standards in water without processing. | [49] |
| DNA (from salmon testis) exposed to MNU and MMS | N7- and O6-methylguanines | DNA methylation by MNU or MMS in buffer/DNA precipitation and isolation/DNA depurination by simultaneous heat-induced hydrolysis (90% FA, 85 °C for 60 min) | n.a. | LC-MS/MS 8-min gradient MRM (QqQ) | Water with 5% FA spiked with N7- and O6-methylguanines, and [2H3]-N7- and [2H3]-O6-methylguanine (IS) for CALs and QCs. | Partial validation (assay precision, accuracy, linearity) [7,46,47,48] Stability experiments: Freeze-and-thaw and 45-day stabilities at −20 °C. Recovery: control DNA acidic hydrolytes spiked with QC concentrations of N7- and O6-methylguanines and reated IS, compared to QC samples (standards and IS spiked in water with 5% FA). | [50] |
| DNA (from calf thymus) exposed to MMS | N7- and O6-methylguanines, and N3-methyladenines | DNA methylation by MMS in buffer/DNA precipitation/DNA depurination by simultaneous heat-induced and acidic hydrolysis (0.1 M HCl, 80 °C for 30 min) | n.a. | LC-MS/MS 4-min gradient MRM (QqQ) | Water/MeOH/TFA 97:3:0.1 (v/v/v) spiked with N7- and O6-methylguanines, and N3-methyladenine, and 15N5-N7-methylguanine (IS), d3-O6-methylguanine (IS), and d3- N3- methyladenine (IS) for CALs and QCs. | Partial validation (assay accuracy and linearity) [7,46,47,48] Intra- and inter-day precision calculated between replicates of untreated DNA spiked with unlabeled standard mixture of fixed concentrations. Recovery: untreated DNA spiked with unlabeled standard mixtures of three different concentrations. Matrix effect: comparison of IS areas between CALs and DNA samples. | [51] |
| DNA (from calf thymus) exposed to cisplatin | 1,2 guanine-guanine intrastrand cisplatin adducts (CP-d(GpG)) | DNA hydrolysis/SPE (SCX and C18) or HPLC clean-up | DNAase I NP1 ALP | LC-MS/MS 22-min gradient SRM (QqQ) | Preparation of a CP-d(GpG) analyte standard in 10 mM ammonium acetate and of a 15N10-CP-d(GpG) IS in 10 mM ammonium acetate with 0.1% glacial acetic acid for CALs. | Linearity: one calibration curve. Interday- and interpreparation-precisions: four replicates of CP-d(GpG) at different concentrations. Recovery: analyte standard processed with or without clean-up method. | [52] |
| DNA (from human placenta) exposed to FA | Hydroxymethyl deoxynuclosides | DNA incubation with FA/DNA precipitation/DNA hydrolysis | DNAse I PDE ALP | LC-UV | Creation of standard hydroxymethyldeoxydeoxynuclosides by exposition of deoxynucleosides with FA. Calibration curves built without IS normalization. | n.a | [53] |
| Oligonucleotides (synthetic) exposed to cisplatin | Guanine-guanine (GG), guanine-adenine (GA), and adenine-guanine (AG) adducts with cisplatin adducts (cis-Pt(NH3)2) | Oligonucleotide incubation with cysplatin/Separation of unreacted and cisplatin derived oligonucleotides/DNA hydrolysis | PDE I or PDE II | LC-UV | n.a. | n.a. | [54] |
| Urine (from human) | O6-carboxymethyl guanine, O6-carboxymethyl-2′-deoxyguanosine, O6-methylguanine and O6-methyl-2′-deoxy guanosine | DNA depurination by simultaneous heat-induced and acidic hydrolysis (0.1 M FA, 70 °C for 1 h)/SPE (C18) clean-up | n.a. | LC-MS/MS 23-min gradient SRM (QqQ) | Synthetic urine spiked with serial dilutions of O6-carboxymethylgua nine, O6-carboxymethyl-2′-deoxyguano sine, O6-methylguanine (control) and O6-methyl-2′-deoxyguanosine (control) and fixed concentrations of tubercidin (IS) for CALs and QCs. | Partial validation (assay linearity, intra-and inter-day accuracy and precision) [7,46,47,48] | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fresnais, M.; Turcan, S.; Theile, D.; Ungermann, J.; Abou Zeed, Y.; Lindner, J.R.; Breitkopf, M.; Burhenne, J.; Haefeli, W.E.; Longuespée, R. Approaching Sites of Action of Temozolomide for Pharmacological and Clinical Studies in Glioblastoma. Biomedicines 2022, 10, 1. https://doi.org/10.3390/biomedicines10010001
Fresnais M, Turcan S, Theile D, Ungermann J, Abou Zeed Y, Lindner JR, Breitkopf M, Burhenne J, Haefeli WE, Longuespée R. Approaching Sites of Action of Temozolomide for Pharmacological and Clinical Studies in Glioblastoma. Biomedicines. 2022; 10(1):1. https://doi.org/10.3390/biomedicines10010001
Chicago/Turabian StyleFresnais, Margaux, Sevin Turcan, Dirk Theile, Johannes Ungermann, Yasmin Abou Zeed, Joshua Raoul Lindner, Marius Breitkopf, Jürgen Burhenne, Walter E. Haefeli, and Rémi Longuespée. 2022. "Approaching Sites of Action of Temozolomide for Pharmacological and Clinical Studies in Glioblastoma" Biomedicines 10, no. 1: 1. https://doi.org/10.3390/biomedicines10010001
APA StyleFresnais, M., Turcan, S., Theile, D., Ungermann, J., Abou Zeed, Y., Lindner, J. R., Breitkopf, M., Burhenne, J., Haefeli, W. E., & Longuespée, R. (2022). Approaching Sites of Action of Temozolomide for Pharmacological and Clinical Studies in Glioblastoma. Biomedicines, 10(1), 1. https://doi.org/10.3390/biomedicines10010001