
RICTOR Affects Melanoma Tumorigenesis and Its Resistance to Targeted Therapy





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Reagents
2.3. RNA Purification and Quantitative RT-PCR
2.4. Western Blotting and Antibodies
2.5. Human Melanoma Samples
2.6. Proximity Ligation Assay
2.7. Statistical Analysis
3. Results
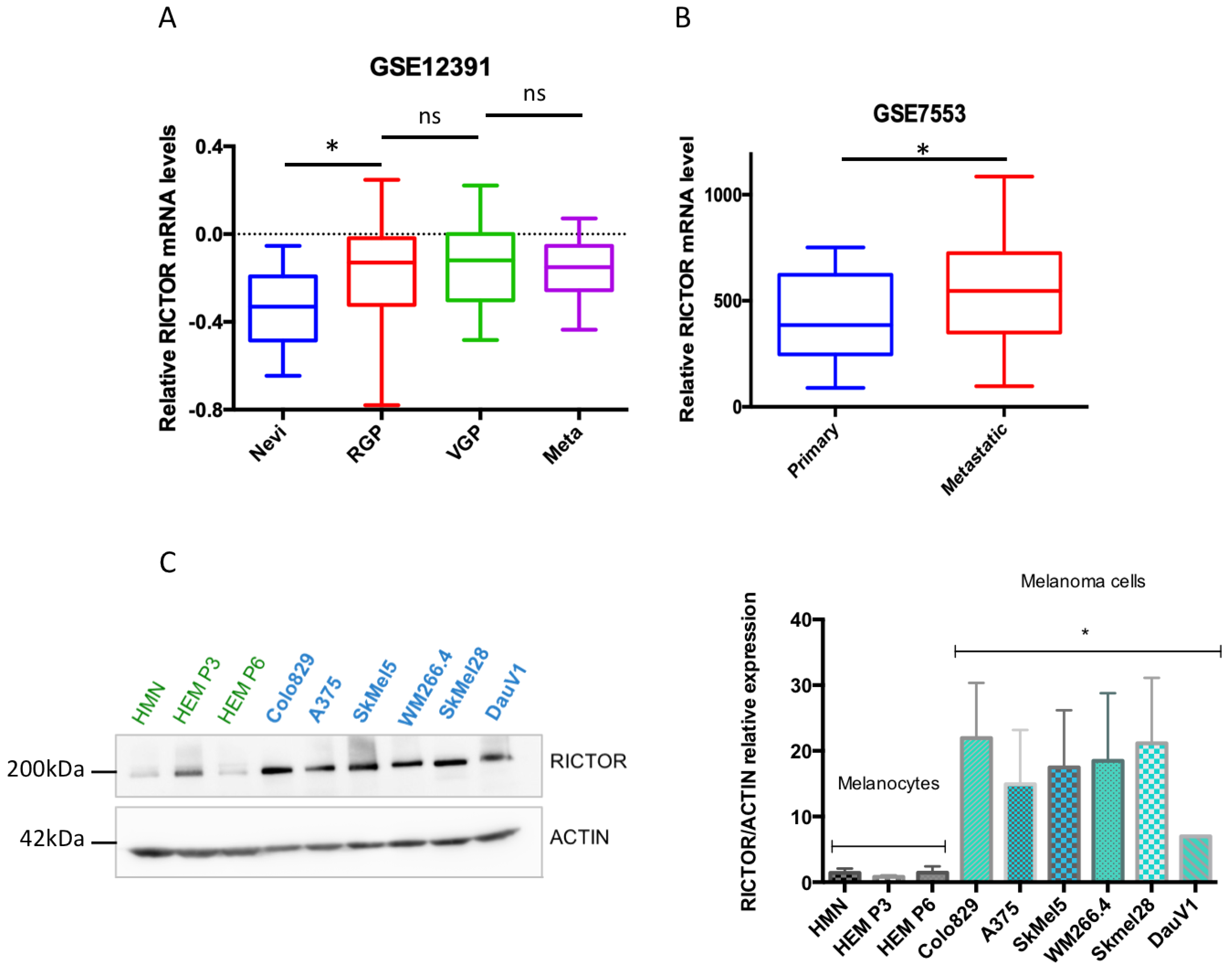
3.1. RICTOR Expression in Human Melanocytes and Melanoma Cells and Tissues
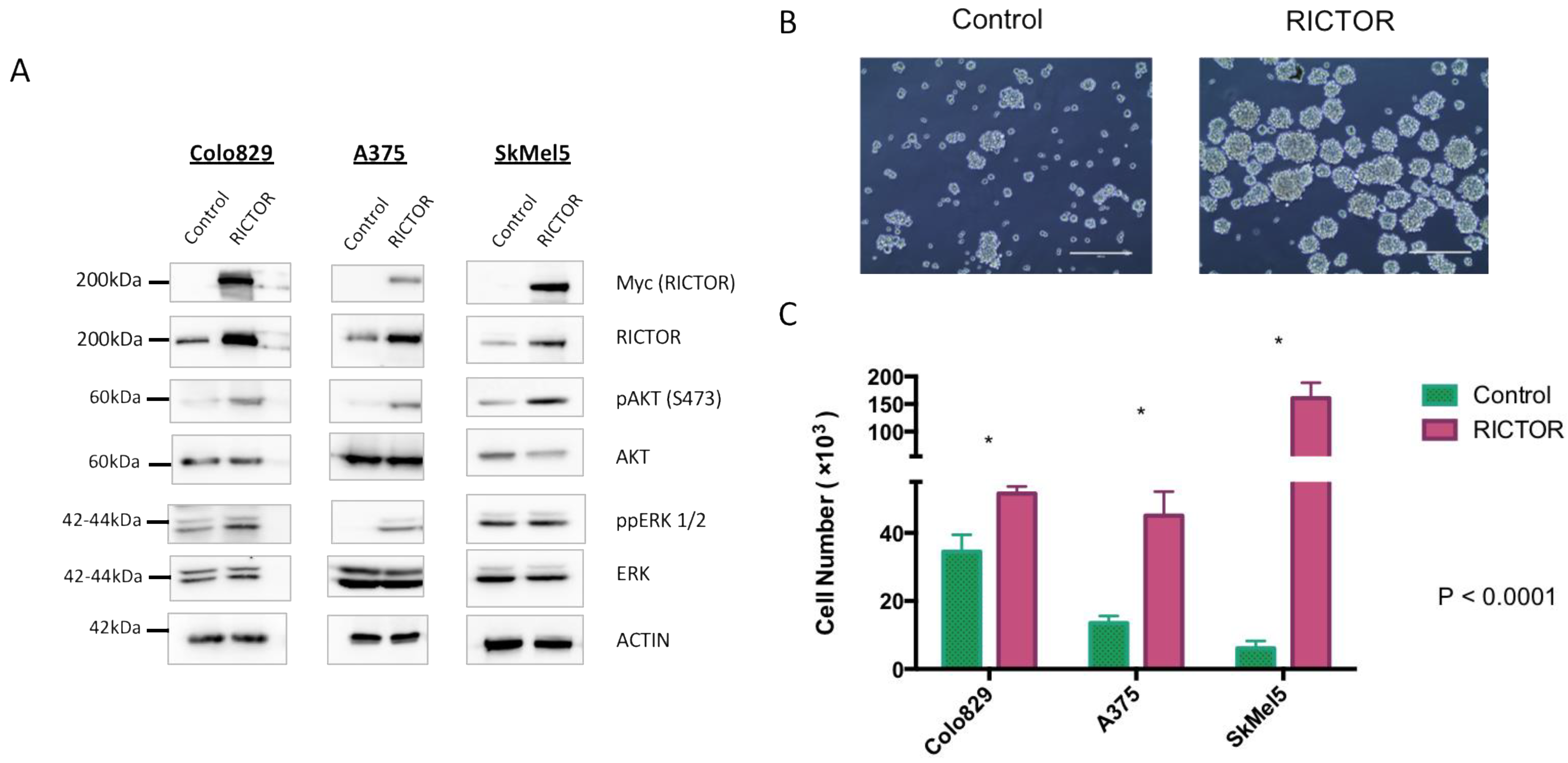
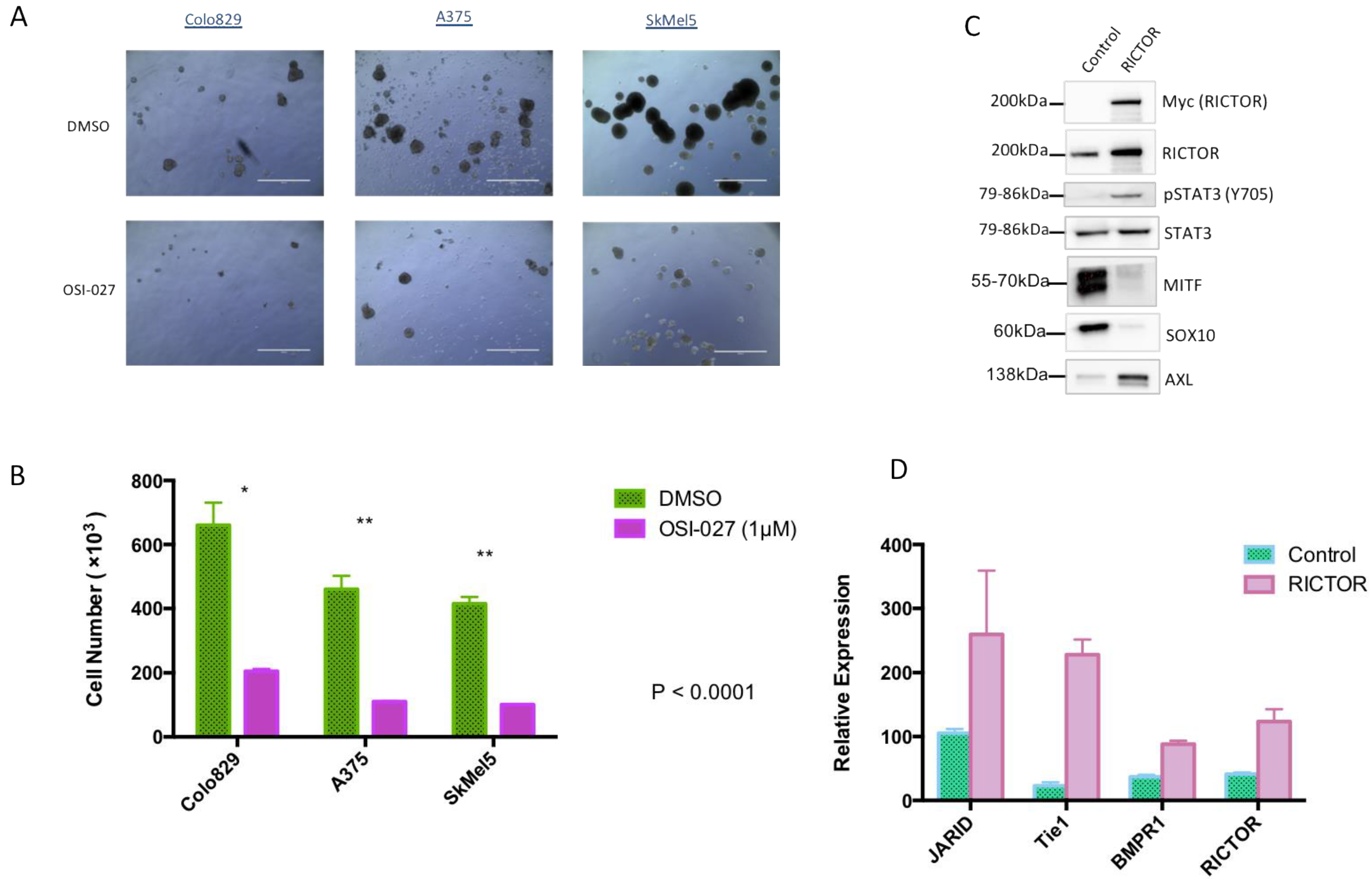
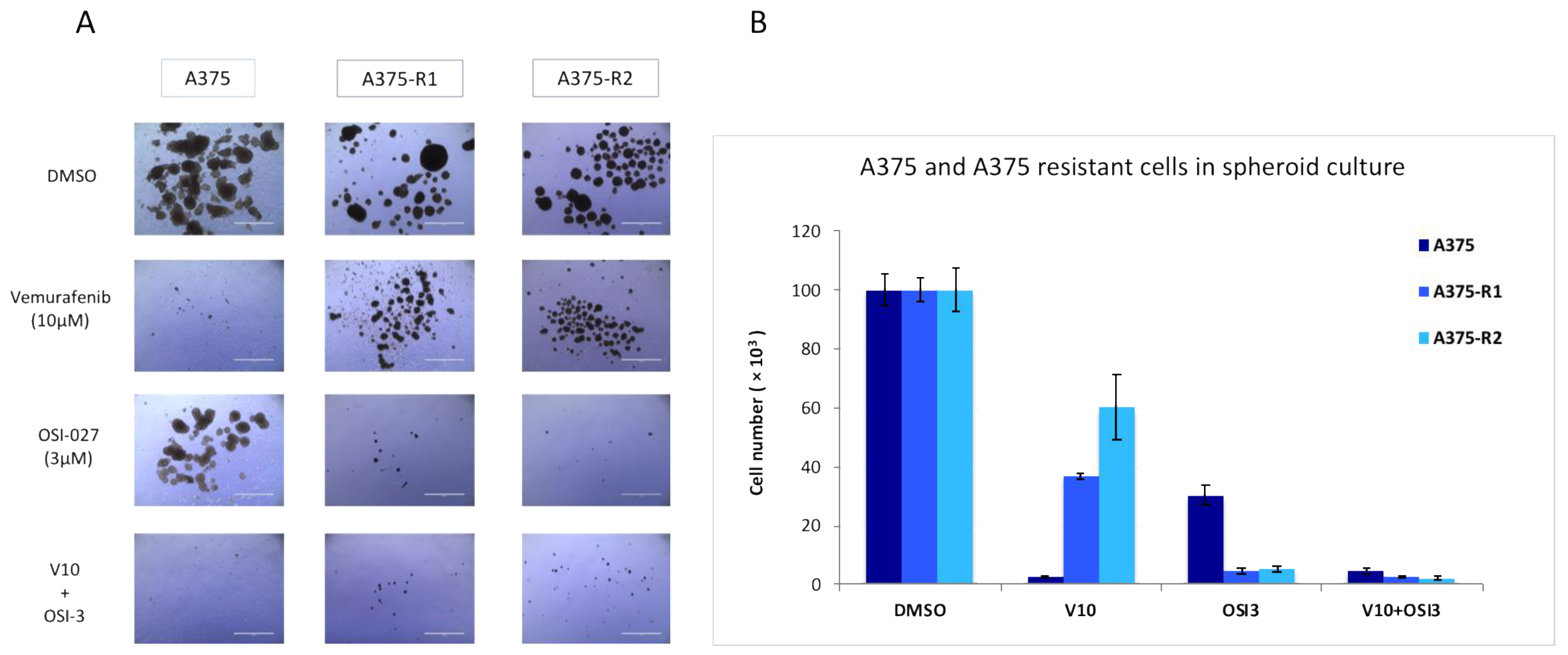
3.2. RICTOR Overexpression Promotes Cell Growth in Spheroid Culture
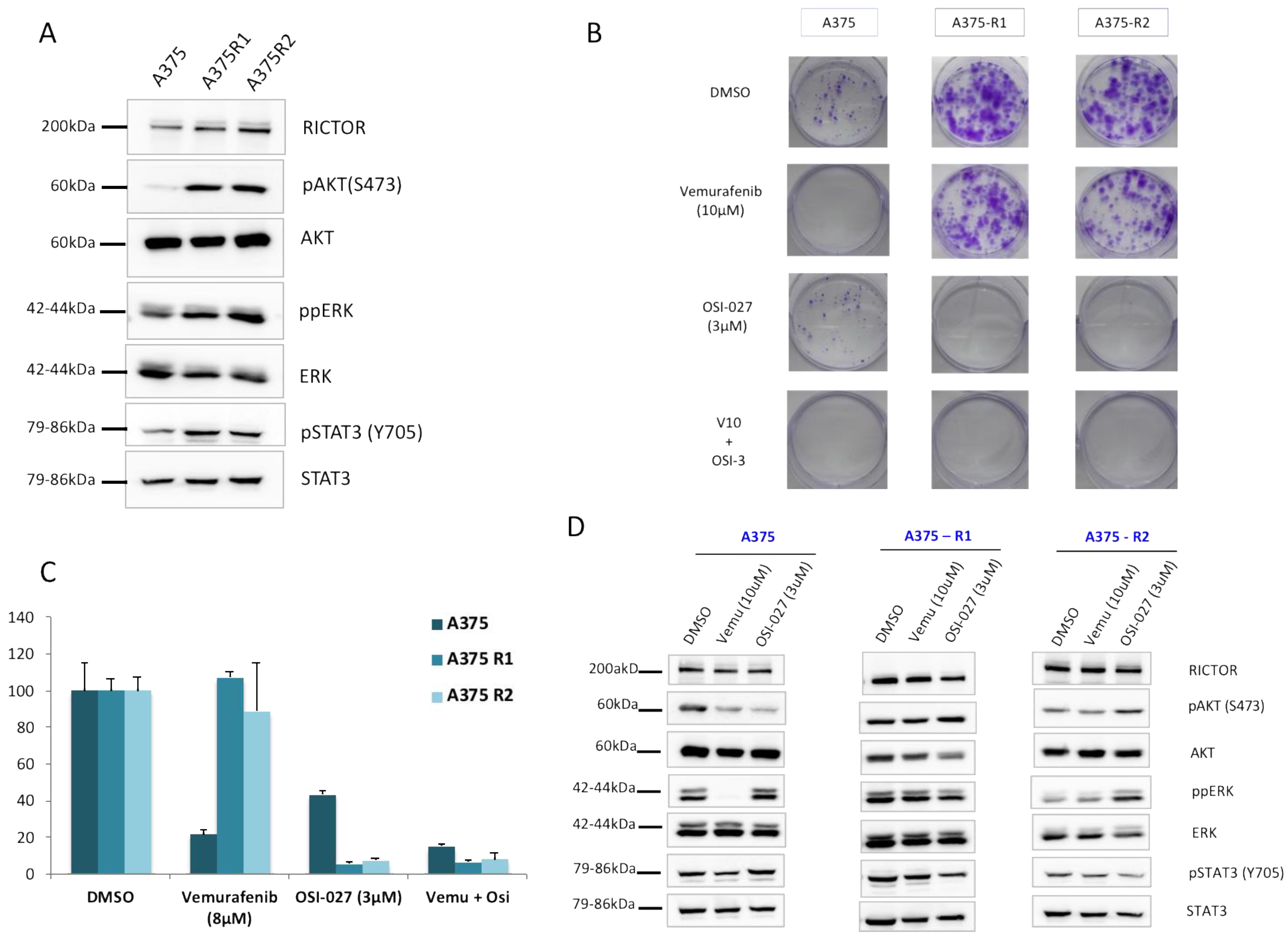
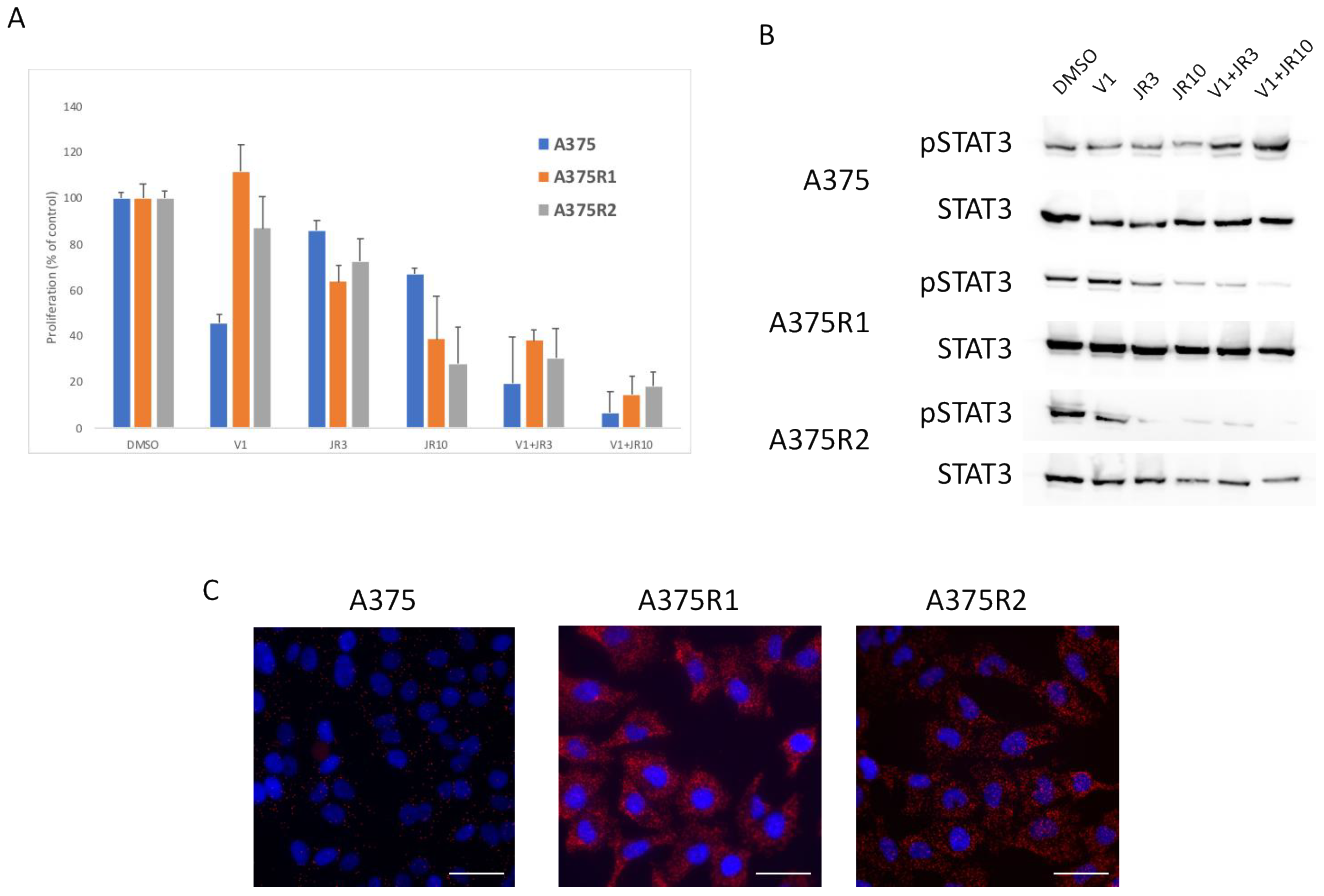
3.3. RICTOR Overexpression Contributes to Melanoma Targeted Therapy Resistance
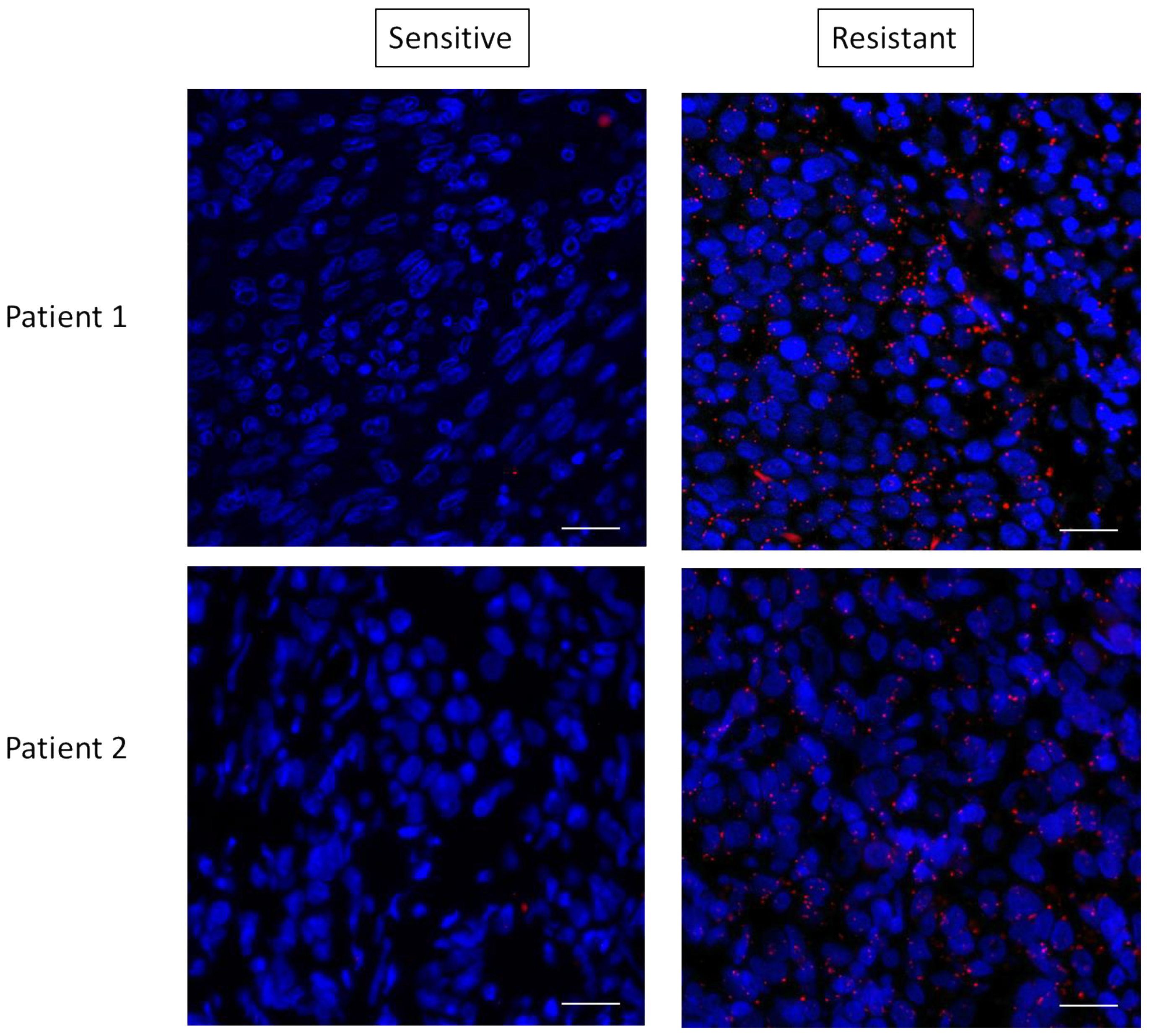
3.4. RAS–mTORC2 Interaction Is Involved in Melanoma Resistance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.G.M.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Goel, V.K.; Lazar, A.; Warneke, C.L.; Redston, M.S.; Haluska, F.G. Examination of Mutations in BRAF, NRAS, and PTEN in Primary Cutaneous Melanoma. J. Investig. Dermatol. 2006, 126, 154–160. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Tortora, G.; Scarpa, A.; Bria, E.; Corbo, V. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018, 39, 971–980. [Google Scholar] [CrossRef]
- Madhunapantula, S.V.; Robertson, G.P. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2009, 22, 400–419. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. BrafV600E cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; McArthur, G.A. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, K.L.; Martin, A.-M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.-T.; et al. Tumor Genetic Analyses of Patients with Metastatic Melanoma Treated with the BRAF Inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kong, X.; Ribas, A.; Lo, R.S. Combinatorial Treatments That Overcome PDGFRβ-Driven Resistance of Melanoma Cells to V600EB-RAF Inhibition. Cancer Res. 2011, 71, 5067–5074. [Google Scholar] [CrossRef] [PubMed]
- Sabbatino, F.; Wang, Y.; Wang, X.; Flaherty, K.T.; Yu, L.; Pepin, D.; Scognamiglio, G.; Pepe, S.; Kirkwood, J.M.; Cooper, Z.A.; et al. PDGFRα up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget 2014, 5, 1926–1941. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef]
- Atefi, M.; Von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing Melanoma Cross-Resistance to BRAF and MEK Inhibitors by Co-Targeting the AKT/mTOR Pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; De Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Algazi, A.P.; Rotow, J.; Posch, C.; Ortiz-Urda, S.; Pelayo, A.; Munster, P.N.; Daud, A. A dual pathway inhibition strategy using BKM120 combined with vemurafenib is poorly tolerated in BRAF V600 E/K mutant advanced melanoma. Pigment Cell Melanoma Res. 2019, 32, 603–606. [Google Scholar] [CrossRef]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; Van Cutsem, E.; Pérez-García, J.; Stathis, A.; Britten, C.D.; et al. A Phase Ib Dose-Escalation Study of the Oral Pan-PI3K Inhibitor Buparlisib (BKM120) in Combination with the Oral MEK1/2 Inhibitor Trametinib (GSK1120212) in Patients with Selected Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar] [CrossRef]
- Algazi, A.P.; Esteve-Puig, R.; Nosrati, A.; Hinds, B.; Hobbs-Muthukumar, A.; Nandoskar, P.; Ortiz-Urda, S.; Chapman, P.B.; Daud, A. Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma. Pigment Cell Melanoma Res. 2018, 31, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Laugier, F.; Finet-Benyair, A.; André, J.; Rachakonda, S.; Kumar, R.; Bensussan, A.; Dumaz, N. RICTOR involvement in the PI3K/AKT pathway regulation in melanocytes and melanoma. Oncotarget 2015, 6, 28120–28131. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. Abstract 3287: An integrated TCGA pan-cancer clinical data resource to drive high quality survival outcome analytics. Bioinform. Syst. Biol. 2018, 78, 3287. [Google Scholar] [CrossRef]
- Schmidt, K.M.; Dietrich, P.; Hackl, C.; Guenzle, J.; Bronsert, P.; Wagner, C.; Fichtner-Feigl, S.; Schlitt, H.J.; Geissler, E.K.; Hellerbrand, C.; et al. Inhibition of mTORC2/RICTOR Impairs Melanoma Hepatic Metastasis. Neoplasia 2018, 20, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Sun, R.; Zhao, X.; Zhang, Y.; Gu, Q.; Dong, X.; Zhang, D.; Sun, J.; Sun, B. Rictor regulates the vasculogenic mimicry of melanoma via the AKT-MMP-2/9 pathway. J. Cell. Mol. Med. 2017, 21, 3579–3591. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A Tumorigenic Subpopulation with Stem Cell Properties in Melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Ramgolam, K.; Lauriol, J.; Lalou, C.; Lauden, L.; Michel, L.; de la Grange, P.; Khatib, A.-M.; Aoudjit, F.; Charron, D.; Alcaide-Loridan, C.; et al. Melanoma Spheroids Grown Under Neural Crest Cell Conditions Are Highly Plastic Migratory/Invasive Tumor Cells Endowed with Immunomodulator Function. PLoS ONE 2011, 6, e18784. [Google Scholar] [CrossRef]
- Benavides-Serrato, A.; Lee, J.; Holmes, B.; Landon, K.A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLoS ONE 2017, 12, e0176599. [Google Scholar] [CrossRef]
- Kovalski, J.R.; Bhaduri, A.; Zehnder, A.M.; Neela, P.H.; Che, Y.; Wozniak, G.G.; Khavari, P.A. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol. Cell 2019, 73, 830–844.e12. [Google Scholar] [CrossRef]
- Johnson, D.B.; Sosman, J.A. Therapeutic Advances and Treatment Options in Metastatic Melanoma. JAMA Oncol. 2015, 1, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Jebali, A.; Dumaz, N. The role of RICTOR downstream of receptor tyrosine kinase in cancers. Mol. Cancer 2018, 17, 1–10. [Google Scholar] [CrossRef]
- Cheli, Y.; Guiliano, S.; Botton, T.; Rocchi, S.; Hofman, V.; Hofman, P.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 2011, 30, 2307–2318. [Google Scholar] [CrossRef]
- Lee, I.; Fox, P.S.; Ferguson, S.D.; Bassett, J.R.L.; Kong, L.-Y.; Schacherer, C.W.; Gershenwald, J.E.; Grimm, E.A.; Fuller, G.; Heimberger, A.B. The Expression of p-STAT3 in Stage IV Melanoma: Risk of CNS Metastasis and Survival. Oncotarget 2012, 3, 336–344. [Google Scholar] [CrossRef][Green Version]
- Vultur, A.; Villanueva, J.; Krepler, C.; Rajan, G.; Chen, Q.; Xiao, M.; Li, L.; Gimotty, P.A.; Wilson, M.; Hayden, J.; et al. MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene 2014, 33, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.-J.; Settleman, J. Drug Resistance via Feedback Activation of Stat3 in Oncogene-Addicted Cancer Cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Won, J.-K.; Yang, H.W.; Shin, S.-Y.; Lee, J.H.; Heo, W.D.; Cho, K.-H. The crossregulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. J. Mol. Cell Biol. 2012, 4, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Louveau, B.; Delyon, J.; De Moura, C.R.; Battistella, M.; Jouenne, F.; Golmard, L.; Sadoux, A.; Podgorniak, M.-P.; Chami, I.; Marco, O.; et al. A targeted genomic alteration analysis predicts survival of melanoma patients under BRAF inhibitors. Oncotarget 2019, 10, 1669–1687. [Google Scholar] [CrossRef]
- Micevic, G.; Muthusamy, V.; Damsky, W.; Theodosakis, N.; Liu, X.; Meeth, K.; Wingrove, E.; Santhanakrishnan, M.; Bosenberg, M. DNMT3b Modulates Melanoma Growth by Controlling Levels of mTORC2 Component RICTOR. Cell Rep. 2016, 14, 2180–2192. [Google Scholar] [CrossRef] [PubMed]
- Werzowa, J.; Cejka, D.; Fuereder, T.; Dekrout, B.; Thallinger, C.; Pehamberger, H.; Wacheck, V.; Pratscher, B. Suppression of mTOR complex 2-dependent AKT phosphorylation in melanoma cells by combined treatment with rapamycin and LY294002. Br. J. Dermatol. 2009, 160, 955–964. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jebali, A.; Battistella, M.; Lebbé, C.; Dumaz, N. RICTOR Affects Melanoma Tumorigenesis and Its Resistance to Targeted Therapy. Biomedicines 2021, 9, 1498. https://doi.org/10.3390/biomedicines9101498
Jebali A, Battistella M, Lebbé C, Dumaz N. RICTOR Affects Melanoma Tumorigenesis and Its Resistance to Targeted Therapy. Biomedicines. 2021; 9(10):1498. https://doi.org/10.3390/biomedicines9101498
Chicago/Turabian StyleJebali, Ahlem, Maxime Battistella, Céleste Lebbé, and Nicolas Dumaz. 2021. "RICTOR Affects Melanoma Tumorigenesis and Its Resistance to Targeted Therapy" Biomedicines 9, no. 10: 1498. https://doi.org/10.3390/biomedicines9101498
APA StyleJebali, A., Battistella, M., Lebbé, C., & Dumaz, N. (2021). RICTOR Affects Melanoma Tumorigenesis and Its Resistance to Targeted Therapy. Biomedicines, 9(10), 1498. https://doi.org/10.3390/biomedicines9101498