
Novel Insight into the Mechanisms of the Bidirectional Relationship between Diabetes and Periodontitis



{kind=link}
{kind=link}
Abstract
:1. Periodontitis
2. Diabetes Mellitus
3. Bidirectional Relationship between Periodontitis and DM
4. Pathogenesis of Periodontitis
5. Mechanisms Linking Periodontitis and DM
5.1. Periodontitis—Type 2 DM Direction
5.1.1. Dissemination of Periodontal Bacteria
5.1.2. Inflammation
5.1.3. Oral-Gut Axis
5.2. DM-Periodontitis Direction
5.2.1. Changes in the Microbiota
5.2.2. Inflammatory Host Response
5.2.3. Immune Host Response
5.2.4. Periodontal Tissue Destruction
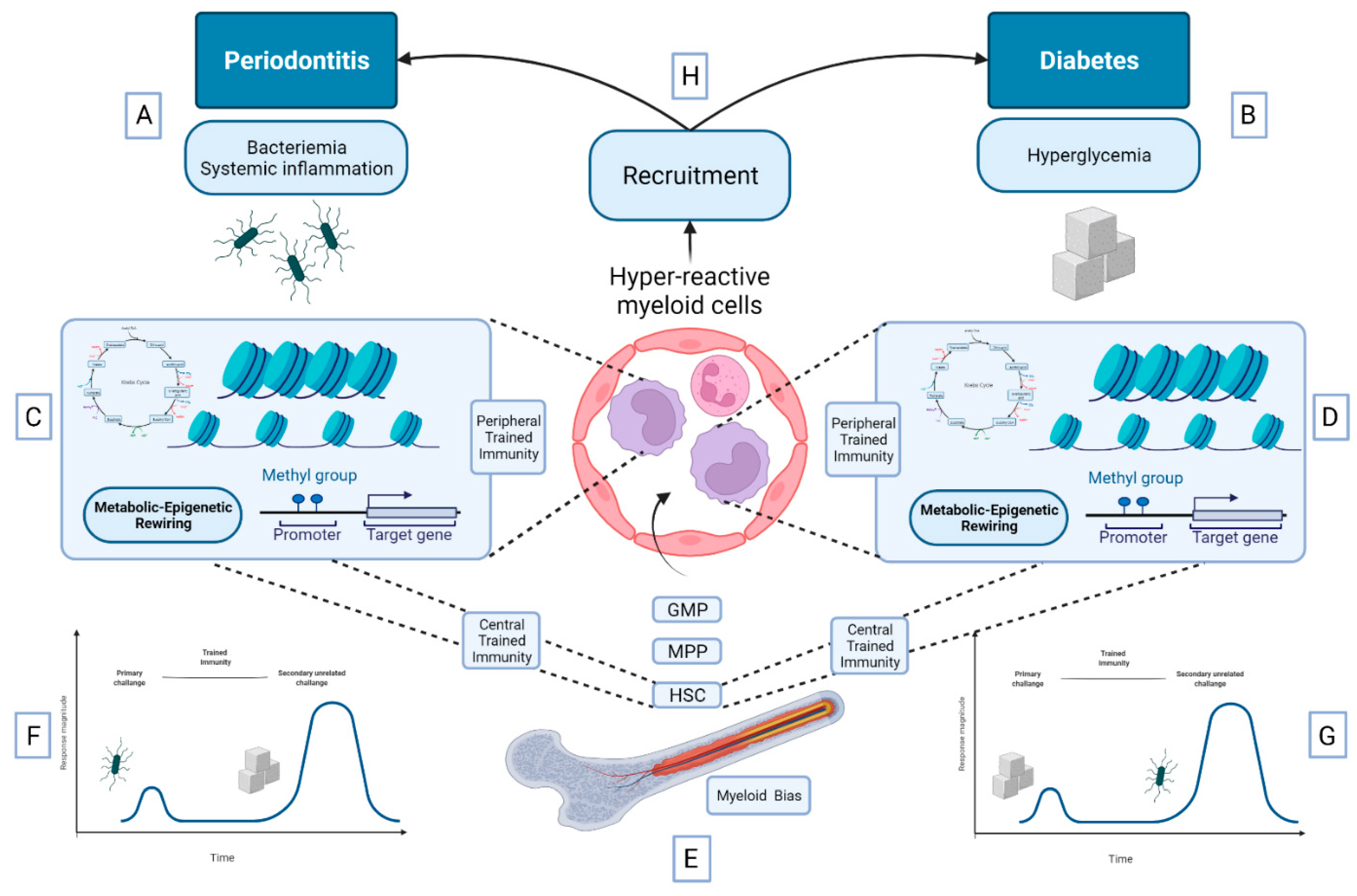
5.3. Bidirectional Relationship between DM and Periodontitis: A Role for Trained Immunity?
6. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Eke, P.I.; Thornton-Evans, G.O.; Wei, L.; Borgnakke, W.S.; Dye, B.A.; Genco, R.J. Periodontitis in US Adults: National Health and Nutrition Examination Survey 2009–2014. J. Am. Dent. Assoc. 2018, 149, 576–588.e6. [Google Scholar] [CrossRef] [PubMed]
- Kassebaum, N.J.; Bernabé, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.L.; Marcenes, W. Global Burden of Severe Periodontitis in 1990–2010: A Systematic Review and Meta-Regression. J. Dent. Res. 2014, 93, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus Report of Workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. 1), S173–S182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepsen, S.; Suvan, J.; Deschner, J. The Association of Periodontal Diseases with Metabolic Syndrome and Obesity. Periodontology 2000 2020, 83, 125–153. [Google Scholar] [CrossRef]
- Charupinijkul, A.; Arunyanak, S.; Rattanasiri, S.; Vathesatogkit, P.; Thienpramuk, L.; Lertpimonchai, A. The Effect of Obesity on Periodontitis Progression: The 10-Year Retrospective Cohort Study. Clin. Oral. Investig. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gobin, R.; Tian, D.; Liu, Q.; Wang, J. Periodontal Diseases and the Risk of Metabolic Syndrome: An Updated Systematic Review and Meta-Analysis. Front. Endocrinol. 2020, 11, 336. [Google Scholar] [CrossRef]
- Sanz, M.; Marco Del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases: Consensus Report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef]
- IDF Diabetes Atlas, Tenth Edition. Available online: https://diabetesatlas.org/ (accessed on 19 December 2021).
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44, S15–S33. [Google Scholar] [CrossRef] [PubMed]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The Aetiology and Molecular Landscape of Insulin Resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of Hepatic Glucose Metabolism in Health and Disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leto, D.; Saltiel, A.R. Regulation of Glucose Transport by Insulin: Traffic Control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bao, W.; Liu, J.; Ouyang, Y.-Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.-L.; Zhang, Y.; Yao, P.; et al. Inflammatory Markers and Risk of Type 2 Diabetes: A Systematic Review and Meta-Analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Kaminga, A.C.; Wen, S.W.; Liu, A. Chemokines in Prediabetes and Type 2 Diabetes: A Meta-Analysis. Front. Immunol. 2021, 12, 622438. [Google Scholar] [CrossRef] [PubMed]
- Vozarova, B.; Weyer, C.; Lindsay, R.S.; Pratley, R.E.; Bogardus, C.; Tataranni, P.A. High White Blood Cell Count Is Associated with a Worsening of Insulin Sensitivity and Predicts the Development of Type 2 Diabetes. Diabetes 2002, 51, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.W. Bidirectional Interrelationships between Diabetes and Periodontal Diseases: An Epidemiologic Perspective. Ann. Periodontol. 2001, 6, 99–112. [Google Scholar] [CrossRef]
- Löe, H. Periodontal disease: The sixth complication of diabetes mellitus. Diabetes Care 1993, 16, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, C.; Ali, A.; Shih, Y.A.; Xie, Q.; Guo, C. Prevalence of Periodontitis in People Clinically Diagnosed with Diabetes Mellitus: A Meta-Analysis of Epidemiologic Studies. Acta Diabetol. 2021, 58, 1307–1327. [Google Scholar] [CrossRef]
- Wu, C.-Z.; Yuan, Y.-H.; Liu, H.-H.; Li, S.-S.; Zhang, B.-W.; Chen, W.; An, Z.-J.; Chen, S.-Y.; Wu, Y.-Z.; Han, B.; et al. Epidemiologic Relationship between Periodontitis and Type 2 Diabetes Mellitus. BMC Oral. Health 2020, 20, 204. [Google Scholar] [CrossRef]
- Kocher, T.; König, J.; Borgnakke, W.S.; Pink, C.; Meisel, P. Periodontal Complications of Hyperglycemia/Diabetes Mellitus: Epidemiologic Complexity and Clinical Challenge. Periodontology 2000 2018, 78, 59–97. [Google Scholar] [CrossRef]
- Genco, R.J.; Borgnakke, W.S. Diabetes as a Potential Risk for Periodontitis: Association Studies. Periodontology 2000 2020, 83, 40–45. [Google Scholar] [CrossRef]
- Romano, F.; Perotto, S.; Mohamed, S.E.O.; Bernardi, S.; Giraudi, M.; Caropreso, P.; Mengozzi, G.; Baima, G.; Citterio, F.; Berta, G.N.; et al. Bidirectional Association between Metabolic Control in Type-2 Diabetes Mellitus and Periodontitis Inflammatory Burden: A Cross-Sectional Study in an Italian Population. J. Clin. Med. 2021, 10, 1787. [Google Scholar] [CrossRef]
- Tonetti, M.S.; Greenwell, H.; Kornman, K.S. Staging and Grading of Periodontitis: Framework and Proposal of a New Classification and Case Definition. J. Clin. Periodontol. 2018, 45 (Suppl. 20), S149–S161. [Google Scholar] [CrossRef] [Green Version]
- Borgnakke, W.S.; Poudel, P. Diabetes and Oral Health: Summary of Current Scientific Evidence for Why Transdisciplinary Collaboration Is Needed. Front. Dent. Med. 2021, 2, 50. [Google Scholar] [CrossRef]
- Romano, F.; Perotto, S.; Mohamed, S.E.O.; Giraudi, M.; Bernardi, S.; Durazzo, M.; Gruden, G.; Aimetti, M. Type 2 Diabetes Mellitus and Periodontitis: Are Diabetic Patients Aware about This Bidirectional Association? Acta Diabetol. 2021, 58, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Ziukaite, L.; Slot, D.E.; Van der Weijden, F.A. Prevalence of Diabetes Mellitus in People Clinically Diagnosed with Periodontitis: A Systematic Review and Meta-Analysis of Epidemiologic Studies. J. Clin. Periodontol. 2018, 45, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Lee, J.S.; Lee, K.-J.; Woo, H.G.; Song, T.-J. Improved Oral Hygiene Is Associated with Decreased Risk of New-Onset Diabetes: A Nationwide Population-Based Cohort Study. Diabetologia 2020, 63, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Borgnakke, W.S.; Ylöstalo, P.V.; Taylor, G.W.; Genco, R.J. Effect of Periodontal Disease on Diabetes: Systematic Review of Epidemiologic Observational Evidence. J. Clin. Periodontol. 2013, 40 (Suppl. 14), S135–S152. [Google Scholar] [CrossRef]
- Graziani, F.; Gennai, S.; Solini, A.; Petrini, M. A Systematic Review and Meta-Analysis of Epidemiologic Observational Evidence on the Effect of Periodontitis on Diabetes An Update of the EFP-AAP Review. J. Clin. Periodontol. 2018, 45, 167–187. [Google Scholar] [CrossRef]
- Song, T.-J.; Jeon, J.; Kim, J. Cardiovascular Risks of Periodontitis and Oral Hygiene Indicators in Patients with Diabetes Mellitus. Diabetes Metab. 2021, 47, 101252. [Google Scholar] [CrossRef]
- Van Dyke, T.E.; Kholy, K.E.; Ishai, A.; Takx, R.A.P.; Mezue, K.; Abohashem, S.M.; Ali, A.; Yuan, N.; Hsue, P.; Osborne, M.T.; et al. Inflammation of the Periodontium Associates with Risk of Future Cardiovascular Events. J. Periodontol. 2021, 92, 348–358. [Google Scholar] [CrossRef]
- Nguyen, A.T.M.; Akhter, R.; Garde, S.; Scott, C.; Twigg, S.M.; Colagiuri, S.; Ajwani, S.; Eberhard, J. The Association of Periodontal Disease with the Complications of Diabetes Mellitus. A Systematic Review. Diabetes Res. Clin. Pract. 2020, 165, 108244. [Google Scholar] [CrossRef] [PubMed]
- Borgnakke, W.S.; Anderson, P.F.; Shannon, C.; Jivanescu, A. Is There a Relationship between Oral Health and Diabetic Neuropathy? Curr. Diabetes Rep. 2015, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Khawaja, A.T.; Jin, L.; Chan, K.W.; Tonetti, M.; Tang, S.C.W.; Pelekos, G. Effect of Non-Surgical Periodontal Therapy on Renal Function in Chronic Kidney Disease Patients with Periodontitis: A Systematic Review and Meta-Analysis of Interventional Studies. Clin. Oral. Investig. 2020, 24, 1607–1618. [Google Scholar] [CrossRef]
- Alvarenga, M.O.P.; Miranda, G.H.N.; Ferreira, R.O.; Saito, M.T.; Fagundes, N.C.F.; Maia, L.C.; Lima, R.R. Association Between Diabetic Retinopathy and Periodontitis—A Systematic Review. Front. Public Health 2020, 8, 550614. [Google Scholar] [CrossRef] [PubMed]
- Genco, R.J.; Graziani, F.; Hasturk, H. Effects of Periodontal Disease on Glycemic Control, Complications, and Incidence of Diabetes Mellitus. Periodontology 2000 2020, 83, 59–65. [Google Scholar] [CrossRef]
- Sharma, P.; Dietrich, T.; Ferro, C.J.; Cockwell, P.; Chapple, I.L.C. Association between Periodontitis and Mortality in Stages 3-5 Chronic Kidney Disease: NHANES III and Linked Mortality Study. J. Clin. Periodontol. 2016, 43, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stöhr, J.; Barbaresko, J.; Neuenschwander, M.; Schlesinger, S. Bidirectional Association between Periodontal Disease and Diabetes Mellitus: A Systematic Review and Meta-Analysis of Cohort Studies. Sci. Rep. 2021, 11, 13686. [Google Scholar] [CrossRef] [PubMed]
- Teshome, A.; Yitayeh, A. The Effect of Periodontal Therapy on Glycemic Control and Fasting Plasma Glucose Level in Type 2 Diabetic Patients: Systematic Review and Meta-Analysis. BMC Oral. Health 2016, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Zhan, Q.; Wu, C.-Z.; Yuan, Y.-H.; Chen, W.; Yu, F.-Y.; Li, Y.; Li, L.-J. Baseline HbA1c Level Influences the Effect of Periodontal Therapy on Glycemic Control in People with Type 2 Diabetes and Periodontitis: A Systematic Review on Randomized Controlled Trails. Diabetes Ther. 2021, 12, 1249–1278. [Google Scholar] [CrossRef]
- Cao, R.; Li, Q.; Wu, Q.; Yao, M.; Chen, Y.; Zhou, H. Effect of Non-Surgical Periodontal Therapy on Glycemic Control of Type 2 Diabetes Mellitus: A Systematic Review and Bayesian Network Meta-Analysis. BMC Oral. Health 2019, 19, 176. [Google Scholar] [CrossRef]
- Baeza, M.; Morales, A.; Cisterna, C.; Cavalla, F.; Jara, G.; Isamitt, Y.; Pino, P.; Gamonal, J. Effect of Periodontal Treatment in Patients with Periodontitis and Diabetes: Systematic Review and Meta-Analysis. J. Appl. Oral. Sci. 2020, 28, e20190248. [Google Scholar] [CrossRef]
- Montero, E.; López, M.; Vidal, H.; Martínez, M.; Virto, L.; Marrero, J.; Herrera, D.; Zapatero, A.; Sanz, M. Impact of Periodontal Therapy on Systemic Markers of Inflammation in Patients with Metabolic Syndrome: A Randomized Clinical Trial. Diabetes Obes. Metab. 2020, 22, 2120–2132. [Google Scholar] [CrossRef]
- D’Aiuto, F.; Gkranias, N.; Bhowruth, D.; Khan, T.; Orlandi, M.; Suvan, J.; Masi, S.; Tsakos, G.; Hurel, S.; Hingorani, A.D.; et al. Systemic Effects of Periodontitis Treatment in Patients with Type 2 Diabetes: A 12 Month, Single-Centre, Investigator-Masked, Randomised Trial. Lancet Diabetes Endocrinol. 2018, 6, 954–965. [Google Scholar] [CrossRef]
- de Araújo, A.A.; de Morais, H.B.; de Medeiros, C.A.C.X.; de Castro Brito, G.A.; Guedes, P.M.M.; Hiyari, S.; Pirih, F.Q.; de Araújo Júnior, R.F. Gliclazide Reduced Oxidative Stress, Inflammation, and Bone Loss in an Experimental Periodontal Disease Model. J. Appl. Oral. Sci. 2019, 27, e20180211. [Google Scholar] [CrossRef]
- Kawahara, Y.; Kaneko, T.; Yoshinaga, Y.; Arita, Y.; Nakamura, K.; Koga, C.; Yoshimura, A.; Sakagami, R. Effects of Sulfonylureas on Periodontopathic Bacteria-Induced Inflammation. J. Dent. Res. 2020, 99, 830–838. [Google Scholar] [CrossRef]
- Sawada, N.; Adachi, K.; Nakamura, N.; Miyabe, M.; Ito, M.; Kobayashi, S.; Miyajima, S.-I.; Suzuki, Y.; Kikuchi, T.; Mizutani, M.; et al. Glucagon-Like Peptide-1 Receptor Agonist Liraglutide Ameliorates the Development of Periodontitis. J. Diabetes Res. 2020, 2020, 8843310. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Wang, P.; Xiang, S.; Xu, D.; Jin, C.; Jiang, Z.; Hu, N. Effects of Type 2 Diabetes and Metformin on Salivary Microbiota in Patients with Chronic Periodontitis. Microb. Pathog. 2021, 161, 105277. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, M.; Xia, L.; Fang, Z.; Yu, S.; Gao, J.; Feng, Q.; Yang, P. Alteration of Salivary Microbiome in Periodontitis with or without Type-2 Diabetes Mellitus and Metformin Treatment. Sci. Rep. 2020, 10, 15363. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, P.; Wang, Q.; Ji, N.; Xia, S.; Ding, Y.; Wang, Q. Metformin Ameliorates Experimental Diabetic Periodontitis Independently of Mammalian Target of Rapamycin (MTOR) Inhibition by Reducing NIMA-Related Kinase 7 (Nek7) Expression. J. Periodontol. 2019, 90, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, X.; Wu, Y.; Pei, M.; Yang, M.; Wu, X.; Pang, Y.; Wang, J. Liraglutide Regulates Bone Destruction and Exhibits Anti-Inflammatory Effects in Periodontitis in Vitro and in Vivo. J. Dent. 2020, 94, 103310. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, A.R.; Rao, N.S.; Naik, S.B.; Kumari, M. Efficacy of Varying Concentrations of Subgingivally Delivered Metformin in the Treatment of Chronic Periodontitis: A Randomized Controlled Clinical Trial. J. Periodontol. 2013, 84, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.C.; Grisa, T.A.; Muniz, F.W.M.G.; Rösing, C.K.; Cavagni, J. Effect of Adjuvant Use of Metformin on Periodontal Treatment: A Systematic Review and Meta-Analysis. Clin. Oral. Investig. 2019, 23, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Akram, Z.; Vohra, F.; Javed, F. Locally Delivered Metformin as Adjunct to Scaling and Root Planing in the Treatment of Periodontal Defects: A Systematic Review and Meta-Analysis. J. Periodontal Res. 2018, 53, 941–949. [Google Scholar] [CrossRef]
- Van Dyke, T.E.; Bartold, P.M.; Reynolds, E.C. The Nexus Between Periodontal Inflammation and Dysbiosis. Front. Immunol. 2020, 11, 511. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The Oral Microbiota: Dynamic Communities and Host Interactions. Nat. Rev. MicroBiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. Local and Systemic Mechanisms Linking Periodontal Disease and Inflammatory Comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Dewhirst, F.E.; Borisy, G.G. Biogeography of the Oral Microbiome: The Site-Specialist Hypothesis. Annu. Rev. MicroBiol. 2019, 73, 335–358. [Google Scholar] [CrossRef]
- Curtis, M.A.; Diaz, P.I.; Van Dyke, T.E. The Role of the Microbiota in Periodontal Disease. Periodontology 2000 2020, 83, 14–25. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. Periodontal Microbial Ecology. Periodontology 2000 2005, 38, 135–187. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Beyond the Red Complex and into More Complexity: The Polymicrobial Synergy and Dysbiosis (PSD) Model of Periodontal Disease Etiology. Mol. Oral. MicroBiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Popadiak, K.; Potempa, J.; Riesbeck, K.; Blom, A.M. Biphasic Effect of Gingipains from Porphyromonas Gingivalis on the Human Complement System. J. Immunol. 2007, 178, 7242–7250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potempa, M.; Potempa, J.; Kantyka, T.; Nguyen, K.-A.; Wawrzonek, K.; Manandhar, S.P.; Popadiak, K.; Riesbeck, K.; Eick, S.; Blom, A.M. Interpain A, a Cysteine Proteinase from Prevotella Intermedia, Inhibits Complement by Degrading Complement Factor C3. PLoS Pathog. 2009, 5, e1000316. [Google Scholar] [CrossRef]
- Wang, M.; Krauss, J.L.; Domon, H.; Hosur, K.B.; Liang, S.; Magotti, P.; Triantafilou, M.; Triantafilou, K.; Lambris, J.D.; Hajishengallis, G. Microbial Hijacking of Complement-Toll-like Receptor Crosstalk. Sci. Signal 2010, 3, ra11. [Google Scholar] [CrossRef] [Green Version]
- Jusko, M.; Potempa, J.; Karim, A.Y.; Ksiazek, M.; Riesbeck, K.; Garred, P.; Eick, S.; Blom, A.M. A Metalloproteinase Karilysin Present in the Majority of Tannerella Forsythia Isolates Inhibits All Pathways of the Complement System. J. Immunol. 2012, 188, 2338–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas Gingivalis Manipulates Complement and TLR Signaling to Uncouple Bacterial Clearance from Inflammation and Promote Dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taxman, D.J.; Swanson, K.V.; Broglie, P.M.; Wen, H.; Holley-Guthrie, E.; Huang, M.T.-H.; Callaway, J.B.; Eitas, T.K.; Duncan, J.A.; Ting, J.P.Y. Porphyromonas Gingivalis Mediates Inflammasome Repression in Polymicrobial CultuRes. through a Novel Mechanism Involving Reduced Endocytosis. J. Biol. Chem. 2012, 287, 32791–32799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; McIntosh, M.L.; Alsam, A.; Kirkwood, K.L.; Lambris, J.D.; et al. Low-Abundance Biofilm Species Orchestrates Inflammatory Periodontal Disease through the Commensal Microbiota and Complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G. The Inflammophilic Character of the Periodontitis-Associated Microbiota. Mol. Oral. MicroBiol. 2014, 29, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, D.M.; Shelef, K.M.; Gonzalez, A.; Davis, C.L.; Dethlefsen, L.; Burns, A.R.; Loomer, P.M.; Armitage, G.C.; Ryder, M.I.; Millman, M.E.; et al. Microbial Biogeography and Ecology of the Mouth and Implications for Periodontal Diseases. Periodontology 2000 2020, 82, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Stabholz, A.; Soskolne, W.A.; Shapira, L. Genetic and Environmental Risk Factors for Chronic Periodontitis and Aggressive Periodontitis. Periodontology 2000 2010, 53, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, E.; Hajishengallis, G. Neutrophil Homeostasis and Periodontal Health in Children and Adults. J. Dent. Res. 2014, 93, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutsopoulos, N.M.; Konkel, J.; Sarmadi, M.; Eskan, M.A.; Wild, T.; Dutzan, N.; Abusleme, L.; Zenobia, C.; Hosur, K.B.; Abe, T.; et al. Defective Neutrophil Recruitment in Leukocyte Adhesion Deficiency Type I Disease Causes Local IL-17-Driven Inflammatory Bone Loss. Sci. Transl. Med. 2014, 6, 229ra40. [Google Scholar] [CrossRef] [Green Version]
- Eskan, M.A.; Jotwani, R.; Abe, T.; Chmelar, J.; Lim, J.-H.; Liang, S.; Ciero, P.A.; Krauss, J.L.; Li, F.; Rauner, M.; et al. The Leukocyte Integrin Antagonist Del-1 Inhibits IL-17-Mediated Inflammatory Bone Loss. Nat. Immunol. 2012, 13, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryder, M.I. Comparison of Neutrophil Functions in Aggressive and Chronic Periodontitis. Periodontology 2000 2010, 53, 124–137. [Google Scholar] [CrossRef]
- Hajishengallis, G. Immunomicrobial Pathogenesis of Periodontitis: Keystones, Pathobionts, and Host Response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, F.; Rothhammer, V.; Claussen, M.C.; Haas, J.D.; Blanco, L.R.; Heink, S.; Prinz, I.; Hemmer, B.; Kuchroo, V.K.; Oukka, M.; et al. Γδ T Cells Enhance Autoimmunity by Restraining Regulatory T Cell Responses via an Interleukin-23-DepenDent. Mechanism. Immunity 2010, 33, 351–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a Cross-Talk between Human Neutrophils and Th17 Cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef]
- Zhang, S.; Gang, X.; Yang, S.; Cui, M.; Sun, L.; Li, Z.; Wang, G. The Alterations in and the Role of the Th17/Treg Balance in Metabolic Diseases. Front. Immunol. 2021, 12, 678355. [Google Scholar] [CrossRef]
- Huang, Z.; Pei, X.; Graves, D.T. The Interrelationship Between Diabetes, IL-17 and Bone Loss. Curr. Osteoporos Rep. 2020, 18, 23–31. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [Green Version]
- Graves, D.T.; Ding, Z.; Yang, Y. The Impact of Diabetes on Periodontal Diseases—PubMed. Periodontology 2000 2020, 82, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, C.; Zheng, X.; Jia, X.; Peng, X.; Yang, R.; Zhou, X.; Xu, X. Porphyromonas Gingivalis Induces Insulin Resistance by Increasing BCAA Levels in Mice. J. Dent. Res. 2020, 99, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Komazaki, R.; Katagiri, S.; Takahashi, H.; Maekawa, S.; Shiba, T.; Takeuchi, Y.; Kitajima, Y.; Ohtsu, A.; Udagawa, S.; Sasaki, N.; et al. Periodontal Pathogenic Bacteria, Aggregatibacter Actinomycetemcomitans Affect Non-Alcoholic Fatty Liver Disease by Altering Gut Microbiota and Glucose Metabolism. Sci. Rep. 2017, 7, 13950. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Petro, B.J.; Shlimon, A.E.; Unterman, T.G. Effect of Periodontitis on Insulin Resistance and the Onset of Type 2 Diabetes Mellitus in Zucker Diabetic Fatty Rats. J. Periodontol. 2008, 79, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Baque, V.; Garidou, L.; Pomié, C.; Escoula, Q.; Loubieres, P.; Le Gall-David, S.; Lemaitre, M.; Nicolas, S.; Klopp, P.; Waget, A.; et al. Periodontitis Induced by Porphyromonas Gingivalis Drives Periodontal Microbiota Dysbiosis and Insulin Resistance via an Impaired Adaptive Immune Response. Gut 2017, 66, 872–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkein, H.A.; Papapanou, P.N.; Genco, R.; Sanz, M. Mechanisms Underlying the Association between Periodontitis and Atherosclerotic Disease. Periodontology 2000 2020, 83, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Horliana, A.C.R.T.; Chambrone, L.; Foz, A.M.; Artese, H.P.C.; de Sousa Rabelo, M.; Pannuti, C.M.; Romito, G.A. Dissemination of Periodontal Pathogens in the Bloodstream after Periodontal Procedures: A Systematic Review. PLoS ONE 2014, 9, e98271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of Bacteremia after Chewing, Tooth Brushing and Scaling in Individuals with Periodontal Inflammation. J. Clin. Periodontol. 2006, 33, 401–407. [Google Scholar] [CrossRef]
- Rafferty, B.; Jönsson, D.; Kalachikov, S.; Demmer, R.T.; Nowygrod, R.; Elkind, M.S.V.; Bush, H.; Kozarov, E. Impact of Monocytic Cells on Recovery of Uncultivable Bacteria from Atherosclerotic Lesions. J. Intern Med. 2011, 270, 273–280. [Google Scholar] [CrossRef]
- Benedyk, M.; Mydel, P.M.; Delaleu, N.; Płaza, K.; Gawron, K.; Milewska, A.; Maresz, K.; Koziel, J.; Pyrc, K.; Potempa, J. Gingipains: Critical Factors in the Development of Aspiration Pneumonia Caused by Porphyromonas Gingivalis. J. Innate Immun. 2016, 8, 185–198. [Google Scholar] [CrossRef]
- Gnanasekaran, J.; Binder Gallimidi, A.; Saba, E.; Pandi, K.; Eli Berchoer, L.; Hermano, E.; Angabo, S.; Makkawi, H.A.; Khashan, A.; Daoud, A.; et al. Intracellular Porphyromonas Gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells. Cancers 2020, 12, 2331. [Google Scholar] [CrossRef] [PubMed]
- Rangé, H.; Labreuche, J.; Louedec, L.; Rondeau, P.; Planesse, C.; Sebbag, U.; Bourdon, E.; Michel, J.-B.; Bouchard, P.; Meilhac, O. Periodontal Bacteria in Human Carotid Atherothrombosis as a Potential Trigger for Neutrophil Activation. Atherosclerosis 2014, 236, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Belstrøm, D.; Holmstrup, P.; Damgaard, C.; Borch, T.S.; Skjødt, M.-O.; Bendtzen, K.; Nielsen, C.H. The Atherogenic Bacterium Porphyromonas Gingivalis Evades Circulating Phagocytes by Adhering to Erythrocytes. Infect. Immun. 2011, 79, 1559–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Shakhatreh, M.-A.K.; James, D.; Liang, S.; Nishiyama, S.-I.; Yoshimura, F.; Demuth, D.R.; Hajishengallis, G. Fimbrial Proteins of Porphyromonas Gingivalis Mediate in Vivo Virulence and Exploit TLR2 and Complement Receptor 3 to Persist in Macrophages. J. Immunol. 2007, 179, 2349–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slocum, C.; Coats, S.R.; Hua, N.; Kramer, C.; Papadopoulos, G.; Weinberg, E.O.; Gudino, C.V.; Hamilton, J.A.; Darveau, R.P.; Genco, C.A. Distinct Lipid a Moieties Contribute to Pathogen-Induced Site-Specific Vascular Inflammation. PLoS Pathog. 2014, 10, e1004215. [Google Scholar] [CrossRef] [PubMed]
- Carey, I.M.; Critchley, J.A.; DeWilde, S.; Harris, T.; Hosking, F.J.; Cook, D.G. Risk of Infection in Type 1 and Type 2 Diabetes Compared With the General Population: A Matched Cohort Study. Diabetes Care 2018, 41, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Toniolo, A.; Cassani, G.; Puggioni, A.; Rossi, A.; Colombo, A.; Onodera, T.; Ferrannini, E. The Diabetes Pandemic and Associated Infections: Suggestions for Clinical Microbiology. Rev. Med. MicroBiol. 2019, 30, 1–17. [Google Scholar] [CrossRef]
- Abu-Ashour, W.; Twells, L.K.; Valcour, J.E.; Gamble, J.-M. Diabetes and the Occurrence of Infection in Primary Care: A Matched Cohort Study. BMC Infect. Dis. 2018, 18, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafar, N.; Edriss, H.; Nugent, K. The Effect of Short-Term Hyperglycemia on the Innate Immune System. Am. J. Med. Sci. 2016, 351, 201–211. [Google Scholar] [CrossRef]
- Ghosh, P.; Vaidya, A.; Sahoo, R.; Goldfine, A.; Herring, N.; Bry, L.; Chorev, M.; Halperin, J.A. Glycation of the Complement Regulatory Protein CD59 Is a Novel Biomarker for Glucose Handling in Humans. J. Clin. Endocrinol. Metab. 2014, 99, E999–E1006. [Google Scholar] [CrossRef]
- Hu, R.; Xia, C.-Q.; Butfiloski, E.; Clare-Salzler, M. Effect of High Glucose on Cytokine Production by Human Peripheral Blood Immune Cells and Type I Interferon Signaling in Monocytes: Implications for the Role of Hyperglycemia in the Diabetes Inflammatory Process and Host Defense against Infection. Clin. Immunol. 2018, 195, 139–148. [Google Scholar] [CrossRef]
- Wang, X.; Ota, N.; Manzanillo, P.; Kates, L.; Zavala-Solorio, J.; Eidenschenk, C.; Zhang, J.; Lesch, J.; Lee, W.P.; Ross, J.; et al. Interleukin-22 Alleviates Metabolic Disorders and RestoRes. Mucosal Immunity in Diabetes. Nature 2014, 514, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Gronke, K.; Diefenbach, A. A Catch-22: Interleukin-22 and Cancer. Eur. J. Immunol. 2018, 48, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya-Rosales, A.; Castro-Garcia, P.; Torres-Juarez, F.; Enciso-Moreno, J.A.; Rivas-Santiago, B. Glucose Levels Affect LL-37 Expression in Monocyte-Derived Macrophages Altering the Mycobacterium Tuberculosis Intracellular Growth Control. Microb. Pathog. 2016, 97, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Lee, K.O.; Low, K.C.; Gamage, A.M.; Liu, Y.; Tan, G.-Y.G.; Koh, H.Q.V.; Alonso, S.; Gan, Y.-H. Glutathione Deficiency in Type 2 Diabetes Impairs Cytokine Responses and Control of Intracellular Bacteria. J. Clin. Invest. 2012, 122, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Delamaire, M.; Maugendre, D.; Moreno, M.; Le Goff, M.C.; Allannic, H.; Genetet, B. Impaired Leucocyte Functions in Diabetic Patients. Diabet Med. 1997, 14, 29–34. [Google Scholar] [CrossRef]
- Zykova, S.N.; Jenssen, T.G.; Berdal, M.; Olsen, R.; Myklebust, R.; Seljelid, R. Altered Cytokine and Nitric Oxide Secretion in Vitro by Macrophages from Diabetic Type II-like Db/Db Mice. Diabetes 2000, 49, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Llorente, L.; De La Fuente, H.; Richaud-Patin, Y.; Alvarado-De La Barrera, C.; Diaz-Borjón, A.; López-Ponce, A.; Lerman-Garber, I.; Jakez-Ocampo, J. Innate Immune Response Mechanisms in Non-Insulin DepenDent. Diabetes Mellitus Patients Assessed by Flow Cytoenzymology. Immunol. Lett. 2000, 74, 239–244. [Google Scholar] [CrossRef]
- Thomsen, R.W.; Hundborg, H.H.; Lervang, H.-H.; Johnsen, S.P.; Schønheyder, H.C.; Sørensen, H.T. Diabetes Mellitus as a Risk and Prognostic Factor for Community-Acquired Bacteremia Due to Enterobacteria: A 10-Year, Population-Based Study among Adults. Clin. Infect. Dis. 2005, 40, 628–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, A.; Nakagawa, I.; Okahashi, N.; Hamada, N. Variations of Porphyromonas Gingivalis Fimbriae in Relation to Microbial Pathogenesis. J. Periodontal Res. 2004, 39, 136–142. [Google Scholar] [CrossRef]
- Ojima, M.; Takeda, M.; Yoshioka, H.; Nomura, M.; Tanaka, N.; Kato, T.; Shizukuishi, S.; Amano, A. Relationship of Periodontal Bacterium Genotypic Variations with Periodontitis in Type 2 Diabetic Patients. Diabetes Care 2005, 28, 433–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, H.; Yoshida, K.; Okamura, H.; Fujiwara, N.; Ozaki, K. Porphyromonas Gingivalis Attenuates the Insulin-Induced Phosphorylation and Translocation of Forkhead Box Protein O1 in Human Hepatocytes. Arch. Oral. Biol. 2016, 69, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y.; Aburaya, S.; Sugiyama, N.; Narukawa, Y.; Sakamoto, Y.; Takahashi, M.; Uemura, H.; Yamashita, R.; Tominaga, S.; Hayashi, S.; et al. Porphyromonas Gingivalis Induces Entero-Hepatic Metabolic Derangements with Alteration of Gut Microbiota in a Type 2 Diabetes Mouse Model. Sci. Rep. 2021, 11, 18398. [Google Scholar] [CrossRef] [PubMed]
- Seyama, M.; Yoshida, K.; Yoshida, K.; Fujiwara, N.; Ono, K.; Eguchi, T.; Kawai, H.; Guo, J.; Weng, Y.; Haoze, Y.; et al. Outer Membrane Vesicles of Porphyromonas Gingivalis Attenuate Insulin Sensitivity by Delivering Gingipains to the Liver. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165731. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Yoshida, K.; Okamura, H.; Ochiai, K.; Takamura, H.; Fujiwara, N.; Ozaki, K. Oral Porphyromonas Gingivalis Translocates to the Liver and Regulates Hepatic Glycogen Synthesis through the Akt/GSK-3β Signaling Pathway. Biochim. Biophys. Acta 2013, 1832, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.-S. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients 2016, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- White, P.J.; McGarrah, R.W.; Herman, M.A.; Bain, J.R.; Shah, S.H.; Newgard, C.B. Insulin Action, Type 2 Diabetes, and Branched-Chain Amino Acids: A Two-Way Street. Mol. Metab. 2021, 52, 101261. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A Branched-Chain Amino Acid-Related Metabolic Signature That Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.B.; Kastenmüller, G.; Shin, S.-Y.; Petersen, A.-K.; Hyde, C.; Psatha, M.; Ward, K.J.; et al. Biomarkers for Type 2 Diabetes and Impaired Fasting Glucose Using a Nontargeted Metabolomics Approach. Diabetes 2013, 62, 4270–4276. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, S.S.; Siddiqui, K. The Emerging Role of Branch Chain Amino Acids in the Prediction of Diabetes: A Brief Review. Curr. Diabetes Rev. 2020, 16, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.H.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human Gut MicrObes. Impact Host Serum Metabolome and Insulin Sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Ilievski, V.; Bhat, U.G.; Suleiman-Ata, S.; Bauer, B.A.; Toth, P.T.; Olson, S.T.; Unterman, T.G.; Watanabe, K. Oral Application of a Periodontal Pathogen Impacts SerpinE1 Expression and Pancreatic Islet Architecture in Prediabetes. J. Periodontal Res. 2017, 52, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Ilievski, V.; Toth, P.T.; Valyi-Nagy, K.; Valyi-Nagy, T.; Green, S.J.; Marattil, R.S.; Aljewari, H.W.; Wicksteed, B.; O’Brien-Simpson, N.M.; Reynolds, E.C.; et al. Identification of a Periodontal Pathogen and Bihormonal Cells in Pancreatic Islets of Humans and a Mouse Model of Periodontitis. Sci. Rep. 2020, 10, 9976. [Google Scholar] [CrossRef] [PubMed]
- Diomede, F.; Thangavelu, S.R.; Merciaro, I.; D’Orazio, M.; Bramanti, P.; Mazzon, E.; Trubiani, O. Porphyromonas Gingivalis Lipopolysaccharide Stimulation in Human Periodontal Ligament Stem Cells: Role of Epigenetic Modifications to the Inflammation. Eur. J. HistoChem. 2017, 61, 2826. [Google Scholar] [CrossRef] [Green Version]
- Solini, A.; Suvan, J.; Santini, E.; Gennai, S.; Seghieri, M.; Masi, S.; Petrini, M.; D’Aiuto, F.; Graziani, F. Periodontitis Affects Glucoregulatory Hormones in Severely Obese Individuals. Int. J. Obes. 2019, 43, 1125–1129. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, Y.; Konishi, K.; Gomi, T.; Yagishita, H.; Yajima, A.; Yoshikawa, M. Enzymatic Properties of Dipeptidyl Aminopeptidase IV Produced by the Periodontal Pathogen Porphyromonas Gingivalis and Its Participation in Virulence. Infect. Immun. 2000, 68, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Ohara-Nemoto, Y.; Nakasato, M.; Shimoyama, Y.; Baba, T.T.; Kobayakawa, T.; Ono, T.; Yaegashi, T.; Kimura, S.; Nemoto, T.K. Degradation of Incretins and Modulation of Blood Glucose Levels by Periodontopathic Bacterial Dipeptidyl Peptidase 4. Infect. Immun. 2017, 85, e00277-17. [Google Scholar] [CrossRef] [Green Version]
- Konkel, J.E.; O’Boyle, C.; Krishnan, S. Distal Consequences of Oral Inflammation. Front. Immunol. 2019, 10, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loos, B.G.; Craandijk, J.; Hoek, F.J.; Wertheim-van Dillen, P.M.; van der Velden, U. Elevation of Systemic Markers Related to Cardiovascular Diseases in the Peripheral Blood of Periodontitis Patients. J. Periodontol. 2000, 71, 1528–1534. [Google Scholar] [CrossRef]
- Roca-Millan, E.; González-Navarro, B.; Sabater-Recolons, M.-M.; Marí-Roig, A.; Jané-Salas, E.; López-López, J. Periodontal Treatment on Patients with Cardiovascular Disease: Systematic Review and Meta-Analysis. Med. Oral. Patol. Oral. Cir. Bucal 2018, 23, e681–e690. [Google Scholar] [CrossRef]
- Türer, Ç.C.; Durmuş, D.; Balli, U.; Güven, B. Effect of Non-Surgical Periodontal Treatment on Gingival Crevicular Fluid and Serum Endocan, Vascular Endothelial Growth Factor-A, and Tumor Necrosis Factor-Alpha Levels. J. Periodontol. 2017, 88, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Kato, T.; Takahashi, N.; Nakajima, M.; Arimatsu, K.; Minagawa, T.; Sato, K.; Ohno, H.; Yamazaki, K. Ligature-Induced Periodontitis in Mice Induces Elevated Levels of Circulating Interleukin-6 but Shows Only Weak Effects on Adipose and Liver Tissues. J. Periodontal Res. 2016, 51, 639–646. [Google Scholar] [CrossRef]
- O’Boyle, C.; Haley, M.J.; Lemarchand, E.; Smith, C.J.; Allan, S.M.; Konkel, J.E.; Lawrence, C.B. Ligature-Induced Periodontitis Induces Systemic Inflammation but Does Not Alter Acute Outcome after Stroke in Mice. Int. J. Stroke 2020, 15, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Katagiri, S.; Komazaki, R.; Watanabe, K.; Maekawa, S.; Shiba, T.; Udagawa, S.; Takeuchi, Y.; Ohtsu, A.; Kohda, T.; et al. Endotoxemia by Porphyromonas Gingivalis Injection Aggravates Non-Alcoholic Fatty Liver Disease, Disrupts Glucose/Lipid Metabolism, and Alters Gut Microbiota in Mice. Front. MicroBiol. 2018, 9, 2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, F.; Xie, M.; Huang, X.; Long, Y.; Lu, X.; Wang, X.; Chen, L. Porphyromonas Gingivalis and Its Systemic Impact: Current Status. Pathogens 2020, 9, 944. [Google Scholar] [CrossRef] [PubMed]
- Arimatsu, K.; Yamada, H.; Miyazawa, H.; Minagawa, T.; Nakajima, M.; Ryder, M.I.; Gotoh, K.; Motooka, D.; Nakamura, S.; Iida, T.; et al. Oral Pathobiont Induces Systemic Inflammation and Metabolic Changes Associated with Alteration of Gut Microbiota. Sci. Rep. 2014, 4, 4828. [Google Scholar] [CrossRef] [Green Version]
- Hatasa, M.; Ohsugi, Y.; Katagiri, S.; Yoshida, S.; Niimi, H.; Morita, K.; Tsuchiya, Y.; Shimohira, T.; Sasaki, N.; Maekawa, S.; et al. Endotoxemia by Porphyromonas Gingivalis Alters Endocrine Functions in Brown Adipose Tissue. Front. Cell Infect. MicroBiol. 2020, 10, 580577. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Q. Periodontitis Aggravated Pancreatic β-Cell Dysfunction in Diabetic Mice through Interleukin-12 Regulation on Klotho. J. Diabetes Investig. 2016, 7, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Utsugi, T.; Ohno, T.; Ohyama, Y.; Uchiyama, T.; Saito, Y.; Matsumura, Y.; Aizawa, H.; Itoh, H.; Kurabayashi, M.; Kawazu, S.; et al. Decreased Insulin Production and Increased Insulin Sensitivity in the Klotho Mutant Mouse, a Novel Animal Model for Human Aging. Metabolism 2000, 49, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.; Bergsten, E.; et al. Extensive Transmission of MicrObes. along the Gastrointestinal Tract. Elife 2019, 8, e42693. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Kato, T.; Tsuboi, Y.; Miyauchi, E.; Suda, W.; Sato, K.; Nakajima, M.; Yokoji-Takeuchi, M.; Yamada-Hara, M.; Tsuzuno, T.; et al. Oral Pathobiont-Induced Changes in Gut Microbiota Aggravate the Pathology of Nonalcoholic Fatty Liver Disease in Mice. Front. Immunol. 2021, 12, 766170. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, A.; Takeuchi, Y.; Katagiri, S.; Suda, W.; Maekawa, S.; Shiba, T.; Komazaki, R.; Udagawa, S.; Sasaki, N.; Hattori, M.; et al. Influence of Porphyromonas Gingivalis in Gut Microbiota of Streptozotocin-Induced Diabetic Mice. Oral. Dis. 2019, 25, 868–880. [Google Scholar] [CrossRef]
- Kato, T.; Yamazaki, K.; Nakajima, M.; Date, Y.; Kikuchi, J.; Hase, K.; Ohno, H.; Yamazaki, K. Oral Administration of Porphyromonas Gingivalis Alters the Gut Microbiome and Serum Metabolome. mSphere 2018, 3, e00460-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, D.; Shapira, L. An Update on the Evidence for Pathogenic Mechanisms That May Link Periodontitis and Diabetes. J. Clin. Periodontol. 2018, 45, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Ceriello, A.; Buysschaert, M.; Chapple, I.; Demmer, R.T.; Graziani, F.; Herrera, D.; Jepsen, S.; Lione, L.; Madianos, P.; et al. Scientific Evidence on the Links between Periodontal Diseases and Diabetes: Consensus Report and Guidelines of the JoInt. Workshop on Periodontal Diseases and Diabetes by the International Diabetes Federation and the European Federation of Periodontology. J. Clin. Periodontol. 2018, 45, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Lux, R.; Klokkevold, P.; Chang, M.; Barnard, E.; Haake, S.; Li, H. The Subgingival Microbiome Associated with Periodontitis in Type 2 Diabetes Mellitus. ISME J. 2020, 14, 519–530. [Google Scholar] [CrossRef]
- Śmiga, M.; Smalley, J.W.; Ślęzak, P.; Brown, J.L.; Siemińska, K.; Jenkins, R.E.; Yates, E.A.; Olczak, T. Glycation of Host Proteins Increases Pathogenic Potential of Porphyromonas Gingivalis. Int. J. Mol. Sci. 2021, 22, 12084. [Google Scholar] [CrossRef]
- Xiao, E.; Mattos, M.; Vieira, G.H.A.; Chen, S.; Corrêa, J.D.; Wu, Y.; Albiero, M.L.; Bittinger, K.; Graves, D.T. Diabetes Enhances IL-17 Expression and Alters the Oral Microbiome to Increase Its Pathogenicity. Cell Host Microbe 2017, 22, 120–128.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.J.; Preshaw, P.M.; Lalla, E. A Review of the Evidence for Pathogenic Mechanisms That May Link Periodontitis and Diabetes. J. Periodontol. 2013, 84, S113–S134. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; de Brito Bezerra, B.; Pacios, S.; Andriankaja, O.; Li, Y.; Tsiagbe, V.; Schreiner, H.; Fine, D.H.; Graves, D.T. Aggregatibacter Actinomycetemcomitans Infection Enhances Apoptosis in Vivo through a Caspase-3-DepenDent. Mechanism in Experimental Periodontitis. Infect. Immun. 2012, 80, 2247–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andriankaja, O.M.; Galicia, J.; Dong, G.; Xiao, W.; Alawi, F.; Graves, D.T. Gene Expression Dynamics during Diabetic Periodontitis. J. Dent. Res. 2012, 91, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Bal, H.S.; Desta, T.; Krothapalli, N.; Alyassi, M.; Luan, Q.; Graves, D.T. Diabetes Enhances Periodontal Bone Loss through Enhanced Resorption and Diminished Bone Formation. J. Dent. Res. 2006, 85, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Naguib, G.; Lu, H.; Leone, C.; Hsue, H.; Krall, E. Inflammation Is More Persistent in Type 1 Diabetic Mice. J. Dent. Res. 2005, 84, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Naguib, G.; Al-Mashat, H.; Desta, T.; Graves, D.T. Diabetes Prolongs the Inflammatory Response to a Bacterial Stimulus through Cytokine Dysregulation. J. Investig. Dermatol. 2004, 123, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, H.-C.; Fu, M.M.-J.; Yang, T.-S.; Fu, E.; Chiang, C.-Y.; Tu, H.-P.; Chin, Y.-T.; Lin, F.-G.; Shih, K.-C. Effect of High Glucose, Porphyromonas Gingivalis Lipopolysaccharide and Advanced Glycation End-Products on Production of Interleukin-6/-8 by Gingival Fibroblasts. J. Periodontal Res. 2017, 52, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Bender, O.; Weinberg, E.; Moses, O.; Nemcovsky, C.E.; Weinreb, M. Porphyromonas Gingivalis Lipopolysaccharide and Glycated Serum Albumin Increase the Production of Several Pro-Inflammatory Molecules in Human Gingival Fibroblasts via NFκB. Arch. Oral. Biol. 2020, 116, 104766. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Sakamoto, E.; Yoshida, K.; Abe, K.; Naruishi, K.; Yamamoto, T.; Shinohara, Y.; Kido, J.-I.; Geczy, C.L. Advanced Glycation End-Products and Porphyromonas Gingivalis Lipopolysaccharide Increase Calprotectin Expression in Human Gingival Epithelial Cells. J. Cell BioChem. 2018, 119, 1591–1603. [Google Scholar] [CrossRef]
- Amir, J.; Waite, M.; Tobler, J.; Catalfamo, D.L.; Koutouzis, T.; Katz, J.; Wallet, S.M. The Role of Hyperglycemia in Mechanisms of Exacerbated Inflammatory Responses within the Oral Cavity. Cell Immunol. 2011, 272, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Koh, T.J.; DiPietro, L.A. Inflammation and Wound Healing: The Role of the Macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Chen, F.; Wang, J.; Zeng, Z.; Yang, Q.; Shao, S. Th17 and Treg Lymphocytes in Obesity and Type 2 Diabetic Patients. Clin. Immunol. 2018, 197, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.E. Pro-Resolving Mediators in the Regulation of Periodontal Disease. Mol. Aspects Med. 2017, 58, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in Inflammation: Emergence of the pro-Resolving Superfamily of Mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Hasturk, H.; Kantarci, A.; Goguet-Surmenian, E.; Blackwood, A.; Andry, C.; Serhan, C.N.; Van Dyke, T.E. Resolvin E1 Regulates Inflammation at the Cellular and Tissue Level and RestoRes. Tissue Homeostasis in Vivo. J. Immunol. 2007, 179, 7021–7029. [Google Scholar] [CrossRef] [Green Version]
- Sima, C.; Rhourida, K.; Van Dyke, T.E.; Gyurko, R. Type 1 Diabetes Predisposes to Enhanced Gingival Leukocyte Margination and Macromolecule Extravasation in Vivo. J. Periodontal Res. 2010, 45, 748–756. [Google Scholar] [CrossRef]
- Domingueti, C.P.; Dusse, L.M.S.; das Graças Carvalho, M.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes Mellitus: The Linkage between Oxidative Stress, Inflammation, Hypercoagulability and Vascular Complications. J. Diabetes Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef]
- Sun, X.; Mao, Y.; Dai, P.; Li, X.; Gu, W.; Wang, H.; Wu, G.; Ma, J.; Huang, S. Mitochondrial Dysfunction Is Involved in the Aggravation of Periodontitis by Diabetes. J. Clin. Periodontol. 2017, 44, 463–471. [Google Scholar] [CrossRef]
- Kajiura, Y.; Bando, M.; Inagaki, Y.; Nagata, T.; Kido, J. Glycated Albumin and Calprotectin Levels in Gingival Crevicular Fluid from Patients with Periodontitis and Type 2 Diabetes. J. Periodontol. 2014, 85, 1667–1675. [Google Scholar] [CrossRef] [Green Version]
- Akram, Z.; Alqahtani, F.; Alqahtani, M.; Al-Kheraif, A.A.; Javed, F. Levels of Advanced Glycation End Products in Gingival Crevicular Fluid of Chronic Periodontitis Patients with and without Type-2 Diabetes Mellitus. J. Periodontol. 2020, 91, 396–402. [Google Scholar] [CrossRef]
- Zizzi, A.; Tirabassi, G.; Aspriello, S.D.; Piemontese, M.; Rubini, C.; Lucarini, G. Gingival Advanced Glycation End-Products in Diabetes Mellitus-Associated Chronic Periodontitis: An Immunohistochemical Study. J. Periodontal Res. 2013, 48, 293–301. [Google Scholar] [CrossRef]
- Chang, P.-C.; Chien, L.-Y.; Yeo, J.F.; Wang, Y.-P.; Chung, M.-C.; Chong, L.Y.; Kuo, M.Y.-P.; Chen, C.-H.; Chiang, H.-C.; Ng, B.N.; et al. Progression of Periodontal Destruction and the Roles of Advanced Glycation End Products in Experimental Diabetes. J. Periodontol. 2013, 84, 379–388. [Google Scholar] [CrossRef]
- Yi, X.; Zhang, L.; Lu, W.; Tan, X.; Yue, J.; Wang, P.; Xu, W.; Ye, L.; Huang, D. The Effect of NLRP Inflammasome on the Regulation of AGEs-Induced Inflammatory Response in Human Periodontal Ligament Cells. J. Periodontal Res. 2019, 54, 681–689. [Google Scholar] [CrossRef]
- Nonaka, K.; Kajiura, Y.; Bando, M.; Sakamoto, E.; Inagaki, Y.; Lew, J.H.; Naruishi, K.; Ikuta, T.; Yoshida, K.; Kobayashi, T.; et al. Advanced Glycation End-Products Increase IL-6 and ICAM-1 Expression via RAGE, MAPK and NF-ΚB Pathways in Human Gingival Fibroblasts. J. Periodontal Res. 2018, 53, 334–344. [Google Scholar] [CrossRef]
- Lalla, E.; Lamster, I.B.; Feit, M.; Huang, L.; Spessot, A.; Qu, W.; Kislinger, T.; Lu, Y.; Stern, D.M.; Schmidt, A.M. Blockade of RAGE Suppresses Periodontitis-Associated Bone Loss in Diabetic Mice. J. Clin. Investig. 2000, 105, 1117–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tella, E.; Aldahlawi, S.; Eldeeb, A.; El Gazaerly, H. The Effect of Systemic Delivery of Aminoguanidine versus Doxycycline on the Resorptive Phase of Alveolar Bone Following Modified Widman Flap in Diabetic Rats: A Histopathological and Scanning Electron Microscope (SEM) Study. Int. J. Health Sci. 2014, 8, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, Z.; Xie, X.; Zhang, Y.; Liu, H.; Zhou, Z.; Zhao, J.; Lee, R.S.; Xiao, Y.; Ivanoviski, S.; et al. Epigenetic Changes Caused by Diabetes and Their Potential Role in the Development of Periodontitis. J. Diabetes Investig. 2021, 12, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Kajikawa, T.; Hajishengallis, E.; Maekawa, T.; Reis, E.S.; Mastellos, D.C.; Yancopoulou, D.; Hasturk, H.; Lambris, J.D. Complement-DepenDent. Mechanisms and Interventions in Periodontal Disease. Front. Immunol. 2019, 10, 406. [Google Scholar] [CrossRef] [Green Version]
- Gursoy, U.K.; Marakoglu, I.; Oztop, A.Y. Relationship between Neutrophil Functions and Severity of Periodontitis in Obese and/or Type 2 Diabetic Chronic Periodontitis Patients. Quintessence Int. 2008, 39, 485–489. [Google Scholar] [PubMed]
- Manosudprasit, A.; Kantarci, A.; Hasturk, H.; Stephens, D.; Van Dyke, T.E. Spontaneous PMN Apoptosis in Type 2 Diabetes and the Impact of Periodontitis. J. Leukoc. Biol. 2017, 102, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Marhoffer, W.; Stein, M.; Schleinkofer, L.; Federlin, K. Evidence of Ex Vivo and in Vitro Impaired Neutrophil Oxidative Burst and Phagocytic Capacity in Type 1 Diabetes Mellitus. Diabetes Res. Clin. Pract. 1993, 19, 183–188. [Google Scholar] [CrossRef]
- Wong, S.L.; Wagner, D.D. Peptidylarginine Deiminase 4: A Nuclear Button Triggering Neutrophil Extracellular Traps in Inflammatory Diseases and Aging. FASEB J. 2018, 32, fj201800691R. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Ren, B.; Zou, L.; He, B.; Li, M. The Role of Neutrophil Extracellular Traps in Periodontitis. Front. Cell Infect. MicroBiol. 2021, 11, 639144. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; He, L.; Qi, X.; Lin, X. Injecting Immunosuppressive M2 Macrophages Alleviates the Symptoms of Periodontitis in Mice. Front. Mol. BioSci. 2020, 7, 603817. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Netea-Maier, R.T.; Riksen, N.P.; Joosten, L.A.B.; Netea, M.G. Specific and Complex Reprogramming of Cellular Metabolism in Myeloid Cells during Innate Immune Responses. Cell Metab. 2017, 26, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.C.; Kraakman, M.J.; Tikellis, C.; Lee, M.K.S.; Hanssen, N.M.J.; Kammoun, H.L.; Pickering, R.J.; Dragoljevic, D.; Al-Sharea, A.; Barrett, T.J.; et al. Transient Intermittent Hyperglycemia Accelerates Atherosclerosis by Promoting Myelopoiesis. Circ. Res. 2020, 127, 877–892. [Google Scholar] [CrossRef]
- Huang, N.; Dong, H.; Luo, Y.; Shao, B. Th17 Cells in Periodontitis and Its Regulation by A20. Front. Immunol. 2021, 12, 742925. [Google Scholar] [CrossRef]
- Ramamurthy, N.S.; Golub, L.M. Diabetes Increases Collagenase Activity in Extracts of Rat Gingiva and Skin. J. Periodontal Res. 1983, 18, 23–30. [Google Scholar] [CrossRef]
- Sasaki, T.; Ramamurthy, N.S.; Yu, Z.; Golub, L.M. Tetracycline Administration Increases Protein (Presumably Procollagen) Synthesis and Secretion in Periodontal Ligament Fibroblasts of Streptozotocin-Induced Diabetic Rats. J. Periodontal Res. 1992, 27, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Balci Yuce, H.; Karatas, Ö.; Tulu, F.; Altan, A.; Gevrek, F. Effect of Diabetes on Collagen Metabolism and Hypoxia in Human Gingival Tissue: A Stereological, Histopathological, and Immunohistochemical Study. Biotech. HistoChem. 2019, 94, 65–73. [Google Scholar] [CrossRef]
- Ren, L.; Fu, Y.; Deng, Y.; Qi, L.; Jin, L. Advanced Glycation End Products Inhibit the Expression of Collagens Type I and III by Human Gingival Fibroblasts. J. Periodontol. 2009, 80, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhan, D.; Guo, L.; Zheng, L. Inhibition of the Receptor for Advanced Glycation Promotes Proliferation and Repair of Human Periodontal Ligament Fibroblasts in Response to High Glucose via the NF-ΚB Signaling Pathway. Arch. Oral. Biol. 2018, 87, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Checchi, V.; Maravic, T.; Bellini, P.; Generali, L.; Consolo, U.; Breschi, L.; Mazzoni, A. The Role of Matrix Metalloproteinases in Periodontal Disease. Int. J. Environ. Res. Public Health 2020, 17, 4923. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Vamsi, G.; Sripriya, R.; Sehgal, P.K. Expression of Matrix Metalloproteinases (MMP-8 and -9) in Chronic Periodontitis Patients with and without Diabetes Mellitus. J. Periodontol. 2006, 77, 1803–1808. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.R.; Lima, J.A.; Gonçalves, T.E.D.; Bastos, M.F.; Figueiredo, L.C.; Shibli, J.A.; Duarte, P.M. Receptor Activator of Nuclear Factor-Kappa B Ligand/Osteoprotegerin Ratio in Sites of Chronic Periodontitis of Subjects with Poorly and Well-Controlled Type 2 Diabetes. J. Periodontol. 2010, 81, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Alshabab, A.; Albiero, M.L.; Mattos, M.; Corrêa, J.D.; Chen, S.; Yang, Y. Osteocytes Play an Important Role in Experimental Periodontitis in Healthy and Diabetic Mice through Expression of RANKL. J. Clin. Periodontol. 2018, 45, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Pacios, S.; Xiao, W.; Mattos, M.; Lim, J.; Tarapore, R.S.; Alsadun, S.; Yu, B.; Wang, C.-Y.; Graves, D.T. Osteoblast Lineage Cells Play an Essential Role in Periodontal Bone Loss Through Activation of Nuclear Factor-Kappa B. Sci. Rep. 2015, 5, 16694. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Flegler, A.; Kozlov, A.; Stern, P.H. Direct Inhibitory and Indirect Stimulatory Effects of RAGE Ligand S100 on SRANKL-Induced Osteoclastogenesis. J. Cell BioChem. 2009, 107, 917–925. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, A.R.; Choi, Y.H.; Jang, S.; Woo, G.-H.; Cha, J.-H.; Bak, E.-J.; Yoo, Y.-J. Tumor Necrosis Factor-α Antagonist Diminishes Osteocytic RANKL and Sclerostin Expression in Diabetes Rats with Periodontitis. PLoS ONE 2017, 12, e0189702. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.-W.; He, H.-B. Apoptosis of Periodontium Cells in Streptozototocin- and Ligature-Induced Experimental Diabetic Periodontitis in Rats. Acta Odontol. Scand. 2013, 71, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Bal, H.S.; Desta, T.; Behl, Y.; Graves, D.T. Tumor Necrosis Factor-Alpha Mediates Diabetes-Enhanced Apoptosis of Matrix-Producing Cells and Impairs Diabetic Healing. Am. J. Pathol. 2006, 168, 757–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Y.-M.; Li, L.; Wang, X.-Q.; Zhang, M.; Zhu, L.-F.; Fu, Y.-W.; Xu, Y. AGEs Induces Apoptosis and Autophagy via Reactive Oxygen Species in Human Periodontal Ligament Cells. J. Cell BioChem. 2019, 121, 3764–3779. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Sun, Y.; Liu, H.; Wang, W.; Wang, J.; Zhang, F. Selective Adipogenic Differentiation of Human Periodontal Ligament Stem Cells Stimulated with High Doses of Glucose. PLoS ONE 2018, 13, e0199603. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Taguchi, Y.; Tominaga, K.; Kimura, D.; Yamawaki, I.; Noguchi, M.; Yamauchi, N.; Tamura, I.; Tanaka, A.; Umeda, M. High Glucose Concentrations Suppress the Proliferation of Human Periodontal Ligament Stem Cells and Their Differentiation Into Osteoblasts. J. Periodontol. 2016, 87, e44–e51. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penkov, S.; Mitroulis, I.; Hajishengallis, G.; Chavakis, T. Immunometabolic Crosstalk: An Ancestral Principle of Trained Immunity? Trends Immunol. 2019, 40, 1–11. [Google Scholar] [CrossRef]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The Fate and Lifespan of Human Monocyte Subsets in Steady State and Systemic Inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef]
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic Progenitor Cells as Integrative Hubs for Adaptation to and Fine-Tuning of Inflammation. Nat. Immunol. 2019, 20, 802–811. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.-S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonça, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.-C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, D.R.; Bultman, S.J. Metaboloepigenetics: Interrelationships between Energy Metabolism and Epigenetic Control of Gene Expression. J. Cell Physiol. 2012, 227, 3169–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. MTOR- and HIF-1α-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.-C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic Programming of Monocyte-to-Macrophage Differentiation and Trained Innate Immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e9. [Google Scholar] [CrossRef] [Green Version]
- Ishai, A.; Osborne, M.T.; El Kholy, K.; Takx, R.A.P.; Ali, A.; Yuan, N.; Hsue, P.; Van Dyke, T.E.; Tawakol, A. Periodontal Disease Associates With Arterial Inflammation Via Potentiation of a Hematopoietic-Arterial Axis. JACC Cardiovasc. Imaging 2019, 12, 2271–2273. [Google Scholar] [CrossRef]
- Wright, H.J.; Matthews, J.B.; Chapple, I.L.C.; Ling-Mountford, N.; Cooper, P.R. Periodontitis Associates with a Type 1 IFN Signature in Peripheral Blood Neutrophils. J. Immunol. 2008, 181, 5775–5784. [Google Scholar] [CrossRef] [Green Version]
- Ling, M.R.; Chapple, I.L.C.; Matthews, J.B. Peripheral Blood Neutrophil Cytokine Hyper-Reactivity in Chronic Periodontitis. Innate Immun. 2015, 21, 714–725. [Google Scholar] [CrossRef] [Green Version]
- Arts, R.J.W.; Joosten, L.A.B.; Netea, M.G. Immunometabolic Circuits in Trained Immunity. Semin. Immunol. 2016, 28, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-DepenDent. Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [Green Version]
- Alrdahe, S.; Al Sadoun, H.; Torbica, T.; McKenzie, E.A.; Bowling, F.L.; Boulton, A.J.M.; Mace, K.A. Dysregulation of Macrophage Development and Phenotype in Diabetic Human Macrophages Can Be Rescued by Hoxa3 Protein Transduction. PLoS ONE 2019, 14, e0223980. [Google Scholar] [CrossRef] [PubMed]
- Edgar, L.; Akbar, N.; Braithwaite, A.T.; Krausgruber, T.; Gallart-Ayala, H.; Bailey, J.; Corbin, A.L.; Khoyratty, T.E.; Chai, J.T.; Alkhalil, M.; et al. Hyperglycemia Induces Trained Immunity in Macrophages and Their Precursors and Promotes Atherosclerosis. Circulation 2021, 144, 961–982. [Google Scholar] [CrossRef] [PubMed]
- Ayala, T.S.; Tessaro, F.H.G.; Jannuzzi, G.P.; Bella, L.M.; Ferreira, K.S.; Martins, J.O. High Glucose Environments Interfere with Bone Marrow-Derived Macrophage Inflammatory Mediator Release, the TLR4 Pathway and Glucose Metabolism. Sci. Rep. 2019, 9, 11447. [Google Scholar] [CrossRef]
- Choudhury, R.P.; Edgar, L.; Rydén, M.; Fisher, E.A. Diabetes and Metabolic Drivers of Trained Immunity: New Therapeutic Targets Beyond Glucose. Arterioscler Thromb Vasc Biol. 2021, 41, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.A.; Pereira, A.D.; Medeiros, C.A.; Brito, G.A.; Leitão, R.F.; Araújo, L.D.; Guedes, P.M.; Hiyari, S.; Pirih, F.Q.; Araújo Júnior, R.F. Effects of Metformin on Inflammation, Oxidative Stress, and Bone Loss in a Rat Model of Periodontitis. PLoS ONE 2017, 12, e0183506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantzis, T.G.; Reeve, C.M.; Brown, A.L. The Ultrastructure of Capillary Basement Membranes in the Attached Gingiva of Diabetic and Nondiabetic Patients with Periodontal Disease. J. Periodontol. 1971, 42, 406–411. [Google Scholar] [CrossRef]
- Scardina, G.A.; Cacioppo, A.; Messina, P. Periodontal Microcirculation in Diabetics: An in Vivo Non-Invasive Analysis by Means of Videocapillaroscopy. Med. Sci. Monit. 2012, 18, CR58–CR64. [Google Scholar] [CrossRef] [PubMed]
- Develioglu, H.; Özdemir, H.; Bostanci, V. Comparative Analysis of the Blood Flow Values of Patients with Type 2 Diabetes Mellitus Presenting with Chronic Periodontitis, Patients with Chronic Periodontitis Only and Healthy Individuals. West Indian Med. J. 2014, 63, 359–363. [Google Scholar] [CrossRef] [Green Version]
- Scardina, G.; Citarrella, R.; Messina, P. Diabetic Microagiopathy of Oral Mucosa Depends on Disease Duration and Therapy. Med. Sci. Monit. 2017, 23, 5613–5619. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Uemura, M.; Suwa, F. Morphological Study of the Palatal Gingiva of the Maxillary First Molar in the Type 2 Diabetes Mellitus Model Rat. Okajimas Folia ANat. Jpn 2011, 88, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Suzuki, Y.; Sawada, N.; Kobayashi, Y.; Nakamura, N.; Miyabe, M.; Miyajima, S.-I.; Adachi, K.; Minato, T.; Mizutani, M.; et al. Therapeutic Potential for Insulin on Type 1 Diabetes-Associated Periodontitis: Analysis of Experimental Periodontitis in Streptozotocin-Induced Diabetic Rats. J. Diabetes Investig. 2020, 11, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Seppälä, B.; Sorsa, T.; Ainamo, J. Morphometric Analysis of Cellular and Vascular Changes in Gingival Connective Tissue in Long-Term Insulin-DepenDent. Diabetes. J. Periodontol. 1997, 68, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Nardi, G.M.; Ferrara, E.; Converti, I.; Cesarano, F.; Scacco, S.; Grassi, R.; Gnoni, A.; Grassi, F.R.; Rapone, B. Does Diabetes Induce the Vascular Endothelial Growth Factor (VEGF) Expression in Periodontal Tissues? A Systematic Review. Int. J. Environ. Res. Public Health 2020, 17, 2765. [Google Scholar] [CrossRef]
- Needleman, I.; Garcia, R.; Gkranias, N.; Kirkwood, K.L.; Kocher, T.; Iorio, A.D.; Moreno, F.; Petrie, A. Mean Annual Attachment, Bone Level, and Tooth Loss: A Systematic Review. J. Clin. Periodontol. 2018, 45 (Suppl. 20), S112–S129. [Google Scholar] [CrossRef]
- Dietrich, T.; Ower, P.; Tank, M.; West, N.X.; Walter, C.; Needleman, I.; Hughes, F.J.; Wadia, R.; Milward, M.R.; Hodge, P.J.; et al. Periodontal Diagnosis in the Context of the 2017 Classification System of Periodontal Diseases and Conditions—Implementation in Clinical Practice. Br. Dent. J. 2019, 226, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrich, C.G.; Araujo, M.W.B.; Lipman, R.D. Prediabetes and Diabetes Screening in Dental Care Settings: NHANES 2013 to 2016. JDR Clin. Trans. Res. 2019, 4, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Eke, P.I.; Dye, B. Assessment of Self-Report MeasuRes. for Predicting Population Prevalence of Periodontitis. J. Periodontol. 2009, 80, 1371–1379. [Google Scholar] [CrossRef]
- Boyko, E.J.; Fujimoto, W.Y.; Leonetti, D.L.; Newell-Morris, L. Visceral Adiposity and Risk of Type 2 Diabetes: A Prospective Study among Japanese Americans. Diabetes Care 2000, 23, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernicke, K.; Grischke, J.; Stiesch, M.; Zeissler, S.; Krüger, K.; Bauer, P.; Hillebrecht, A.; Eberhard, J. Influence of Physical Activity on Periodontal Health in Patients with Type 2 Diabetes Mellitus. A Blinded, Randomized, Controlled Trial. Clin. Oral. Investig. 2021, 25, 6101–6107. [Google Scholar] [CrossRef]
- Zhang, Y.; He, J.; He, B.; Huang, R.; Li, M. Effect of Tobacco on Periodontal Disease and Oral Cancer. Tob Induc Dis. 2019, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Bibars, A.R.M.; Obeidat, S.R.; Khader, Y.; Mahasneh, A.M.; Khabour, O.F. The Effect of Waterpipe Smoking on Periodontal Health. Oral. Health PRev. Dent. 2015, 13, 253–259. [Google Scholar] [CrossRef]
- Kulkarni, V.; Uttamani, J.R.; Bhatavadekar, N.B. Comparison of Clinical Periodontal Status among Habitual Smokeless-Tobacco Users and Cigarette Smokers. Int. Dent. J. 2016, 66, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, M.L.; Valdivia-Gandur, I.; Lozano de Luaces, V.; Varela Véliz, H.; Balasubbaiah, Y.; Chimenos-Küstner, E. Betel and Tobacco Chewing Habit and Its Relation to Risk Factors for Periodontal Disease. Oral. Dis. 2018, 24, 829–839. [Google Scholar] [CrossRef]
- Geisinger, M.L.; Geurs, N.C.; Ogdon, D.; Reddy, M.S. Commentary: Targeting Underlying Biologic Mechanisms in Selecting Adjunctive Therapies to Improve Periodontal Treatment in Smokers: A Commentary. J. Periodontol. 2017, 88, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Kotsakis, G.A.; Javed, F.; Hinrichs, J.E.; Karoussis, I.K.; Romanos, G.E. Impact of Cigarette Smoking on Clinical Outcomes of Periodontal Flap Surgical Procedures: A Systematic Review and Meta-Analysis. J. Periodontol. 2015, 86, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Bunaes, D.F.; Lie, S.A.; Enersen, M.; Aastrøm, A.N.; Mustafa, K.; Leknes, K.N. Site-Specific Treatment Outcome in Smokers Following Non-Surgical and Surgical Periodontal Therapy. J. Clin. Periodontol. 2015, 42, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Papantonopoulos, G.H. Smoking Influences Decision Making in Periodontal Therapy: A Retrospective Clinical Study. J. Periodontol. 1999, 70, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Silva, H. Tobacco Use and Periodontal Disease-The Role of Microvascular Dysfunction. Biology 2021, 10, 441. [Google Scholar] [CrossRef]
- Obradović, R.; Kesić, L.J.; Gasić, J.; Petrović, M.; Zivković, N. Role of Smoking in Periodontal Disease among Diabetic Patients. West Indian Med. J. 2012, 61, 98–101. [Google Scholar]
- Hajishengallis, G.; Chavakis, T.; Lambris, J.D. Current Understanding of Periodontal Disease Pathogenesis and Targets for Host-Modulation Therapy. Periodontology 2000 2020, 84, 14–34. [Google Scholar] [CrossRef]
- Hasturk, H.; Schulte, F.; Martins, M.; Sherzai, H.; Floros, C.; Cugini, M.; Chiu, C.-J.; Hardt, M.; Van Dyke, T. Safety and Preliminary Efficacy of a Novel Host-Modulatory Therapy for Reducing Gingival Inflammation. Front. Immunol. 2021, 12, 704163. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barutta, F.; Bellini, S.; Durazzo, M.; Gruden, G. Novel Insight into the Mechanisms of the Bidirectional Relationship between Diabetes and Periodontitis. Biomedicines 2022, 10, 178. https://doi.org/10.3390/biomedicines10010178
Barutta F, Bellini S, Durazzo M, Gruden G. Novel Insight into the Mechanisms of the Bidirectional Relationship between Diabetes and Periodontitis. Biomedicines. 2022; 10(1):178. https://doi.org/10.3390/biomedicines10010178
Chicago/Turabian StyleBarutta, Federica, Stefania Bellini, Marilena Durazzo, and Gabriella Gruden. 2022. "Novel Insight into the Mechanisms of the Bidirectional Relationship between Diabetes and Periodontitis" Biomedicines 10, no. 1: 178. https://doi.org/10.3390/biomedicines10010178
APA StyleBarutta, F., Bellini, S., Durazzo, M., & Gruden, G. (2022). Novel Insight into the Mechanisms of the Bidirectional Relationship between Diabetes and Periodontitis. Biomedicines, 10(1), 178. https://doi.org/10.3390/biomedicines10010178