
Inflammasome-Mediated Cytokines: A Key Connection between Obesity-Associated NASH and Liver Cancer Progression

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammasomes as a Mediator of Pro-Inflammatory Cytokines
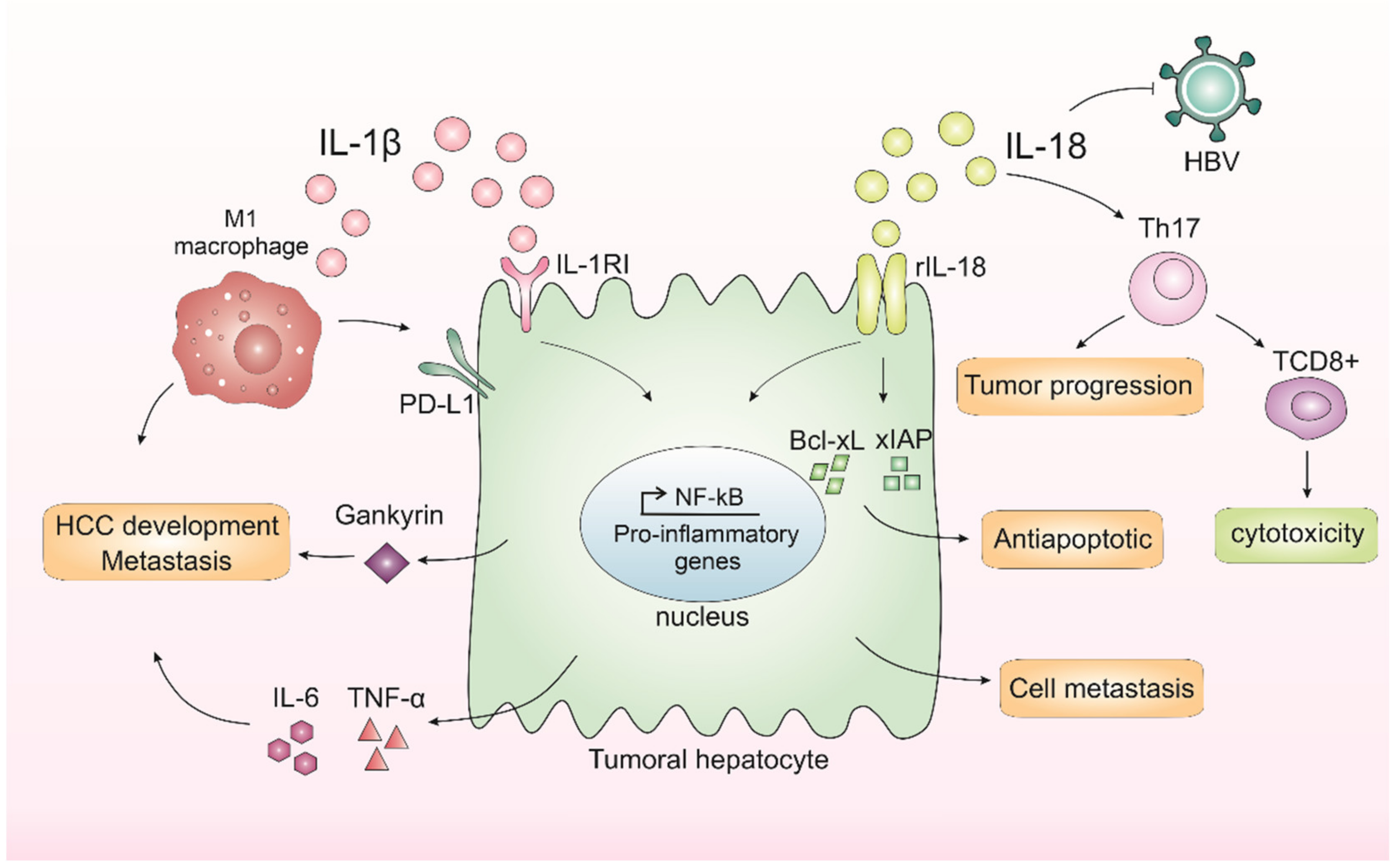
3. IL-1β and IL-18 in Liver Cancer Progression
4. Obesity Chronic Inflammation as a Key in Liver Disorder
5. Obesity-Associated NASH IL-1β and IL-18 in Liver Cancer Progression
6. Modulating IL-1β and IL-18 as a Therapeutic Target in HCC
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; López, S.A.; Fajes, J.L.H.; Martín, L.C. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Lin, Y.; Yuan, X.; Shen, L.; Chen, J.; Chen, L.; Qin, L.; Shen, B. Biomarker MicroRNAs for Diagnosis, Prognosis and Treatment of Hepatocellular Carcinoma: A Functional Survey and Comparison. Sci. Rep. 2016, 6, 38311. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M. Epidemiology and Clinical Presentation of Hepatocellular Carcinoma. J. Vasc. Interv. Radiol. 2002, 13, 169–171. [Google Scholar] [CrossRef]
- Balogh, J.; Balogh, J.; Victor, D., III; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Cajal, S.R.Y.; Capdevila, C.; Hernandez-Losa, J.; De Mattos-Arruda, L.; Ghosh, A.; Lorent, J.; Larsson, O.; Aasen, T.; Postovit, L.-M.; Topisirovic, I. Cancer as an ecomolecular disease and a neoplastic consortium. Biochim. Biophys. Acta-Rev. Cancer 2017, 1868, 484–499. [Google Scholar] [CrossRef]
- Giraud, J.; Chalopin, D.; Blanc, J.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef]
- Brunt, E.M. Non Alcoholic Steathohepatitis (NASH). Semin. Liver Dis. 2004, 24, 3–20. [Google Scholar]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two ‘Hits’? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Hashimoto, E.; Taniai, M.; Tokushige, K. Characteristics and diagnosis of NAFLD/NASH. J. Gastroenterol. Hepatol. 2013, 28, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Saitta, C.; Pollicino, T.; Raimondo, G. Obesity and liver cancer. Ann. Hepatol. 2019, 18, 810–815. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Knight, R.; Leibel, R.L. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol. Metab. 2015, 26, 493–501. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Immune regulation of metabolic homeostasis in health and disease. Cell 2015, 161, 146–160. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 27, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR Gene Family: A Standard Nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef]
- Dagenais, M.; Skeldon, A.; Saleh, M. The inflammasome: In memory of Dr. Jurg Tschopp. Cell Death Differ. 2012, 19, 5–12. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice Deficient in IL-l beta-Converting Enzyme Are Defective in Production of Mature IL-lp and Resistant to Endotoxic Shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef] [Green Version]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Andersen, K.; Eltrich, N.; Lichtnekert, J.; Anders, H.J.; Vielhauer, V. The NLRP3/ASC inflammasome promotes T-cell-dependent immune complex glomerulonephritis by canonical and noncanonical mechanisms. Kidney Int. 2014, 86, 965–978. [Google Scholar] [CrossRef]
- Mariathasan, S.; Monack, D.M. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 2007, 7, 31–40. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.; Ting, J.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, X.; Na Jiang, N.; Mou, S.; Gu, L.; Li, S.; Lin, Q.; He, Y.; Zhang, M.; Zhou, W.; et al. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis. 2018, 9, 983. [Google Scholar] [CrossRef]
- Liu, X.; Yin, L.; Shen, S.; Hou, Y. Inflammation and cancer: Paradoxical roles in tumorigenesis and implications in immunotherapies. Genes Dis. 2021, 1–14. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [PubMed]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; DE Amici, M.; Quaglini, S.; Paglino, C.; Tagliani, F.; Boncimino, A.; Moratti, R.; Corazza, G.R. Circulating interleukin-6 as a tumor marker for hepatocellular carcinoma. Ann. Oncol. 2008, 19, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001, 12, 53–72. [Google Scholar] [CrossRef]
- Lin, A.; Karin, M. NF-kappaB in cancer: A marked target. Semin. Cancer Biol. 2003, 13, 107–114. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar]
- Su, B.; Luo, T.; Zhu, J.; Fu, J.; Zhao, X.; Chen, L.; Zhang, H.; Ren, Y.; Yu, L.; Yang, X.; et al. Interleukin-1β/Iinterleukin-1 receptor-associated kinase 1 inflammatory signaling contributes to persistent Gankyrin activation during hepatocarcinogenesis. Hepatology 2015, 61, 585–597. [Google Scholar] [CrossRef]
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 macrophages induce PD-L1 expression in hepatocellular carcinoma cells through IL-1β signaling. Front. Immunol. 2019, 10, 1643. [Google Scholar] [CrossRef]
- Numata, Y.; Akutsu, N.; Ishigami, K.; Koide, H.; Wagatsuma, K.; Motoya, M.; Sasaki, S.; Nakase, H. Synergistic effect of IFN-γ and IL-1β on PD-L1 expression in hepatocellular carcinoma. Biochem. Biophys. Rep. 2022, 30, 2405–5808. [Google Scholar] [CrossRef] [PubMed]
- Tak, K.H.; Yu, G.I.; Lee, M.Y.; Shin, D.H. Association between polymorphisms of interleukin 1 family genes and hepatocellular carcinoma. Med. Sci. Monit. 2018, 24, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Kono, H.; Amemiya, H.; Matsuda, M.; Suzuki, T.; Maki, A.; Fujii, H. Role of interleukin-18 and its receptor in hepatocellular carcinoma associated with hepatitis C virus infection. Int. J. Cancer 2006, 118, 564–570. [Google Scholar] [CrossRef]
- Eldesoky, A.A.; Ahmed, N.A.F.; Zaghloul, H.E.; Aziz, A.A.A. Interleukin-18 polymorphism as a diagnostic tumor marker for hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Egypt Liver J. 2020, 10, 51. [Google Scholar] [CrossRef]
- Sharafelldin, H.; Mors, A.; Elghobary, H.; Osman, E.; Rady, N. Association between TNF-α, Interleukin-18 Polymorphisms and Risk of Hepatocellular Carcinoma in Egyptian patients. Asian Pac. J. Cancer Prev. 2021, 22, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Zhao, M.; Gao, G.; Yang, J.; Wang, Z.; Liu, Y. Prognostic Role of IL-18 in Various Human Cancers and Radiation Injuries: A Meta-Analysis. Dose-Response 2020, 18, 1–7. [Google Scholar] [CrossRef]
- Markowitz, G.J.; Yang, P.; Fu, J.; Michelotti, G.A.; Chen, R.; Sui, J.; Yang, B.; Qin, W.-H.; Zhang, Z.; Wang, F.-S.; et al. Inflammation-dependent IL18 signaling restricts hepatocellular carcinoma growth by enhancing the accumulation and activity of tumor- infiltrating lymphocytes. Cancer Res. 2016, 76, 2394–2405. [Google Scholar] [CrossRef]
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.-X.; He, H.-W.; Cai, X.-Y.; Zhou, J.; Cheng, Y.-F.; Fan, J.; et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Ma, Y.; Liu, S.; She, Y.; Zhao, P.; Jing, M.; Han, T.; Yan, C.; Wu, Z.; et al. Dual effects of interleukin-18: Inhibiting hepatitis B virus replication in Hepg2.2.15 cells and promoting hepatoma cells metastasis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, 565–573. [Google Scholar] [CrossRef]
- Gu, F.M.; Li, Q.L.; Gao, Q.; Jiang, J.H.; Zhu, K.; Huang, X.Y.; Pan, J.F.; Yan, J.; Hu, J.H.; Wang, Z.; et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol. Cancer 2011, 10, 150. [Google Scholar] [CrossRef]
- Li, P.-L.; Boini, K.M.; Xia, M.; Koka, S.; Gehr, T.W. Sphingolipids in obesity and related complications. Front. Biosci. (Landmark Ed.) 2017, 22, 96–116. [Google Scholar] [CrossRef]
- Kellen, C.; Rodrigues, C.; Pereira, R.M.; De Campos, T.D.P. The Role of Physical Exercise to Improve the Browning of White Adipose Tissue via POMC Neurons. Front. Cell Neurosci. 2018, 12, 88. [Google Scholar]
- Brandão, B.B.; Guerra, B.A.; Mori, M.A. Shortcuts to a functional adipose tissue: The role of small non-coding RNAs. Redox Biol. 2017, 12, 82–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yan, Z.; Chen, Y.; Liu, F. A social contagious model of the obesity epidemic. Sci. Rep. 2016, 6, 37961. [Google Scholar] [CrossRef] [Green Version]
- Leroux, J.S.; Moore, S.; Dubé, L. Beyond the “I” in the Obesity Epidemic: A Review of Social Relational and Network Interventions on Obesity. J. Obes. 2013, 2013, 348249. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Oikawa, T.; Nakano, M.; Saeki, C.; Torisu, Y.; Kajihara, M.; Saruta, M. Recent Insights Into the Multiple Pathways Driving Non-alcoholic Steatohepatitis-Derived Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 762. [Google Scholar] [CrossRef] [PubMed]
- Walle, P.; Takkunen, M.; Männistö, V.; Vaittinen, M.; Lankinen, M.; Kärjä, V.; Käkelä, P.; Ågren, J.; Tiainen, M.; Schwab, U.; et al. Fatty acid metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism 2016, 65, 655–666. [Google Scholar] [CrossRef]
- Kanwar, P.; Kowdley, K.V. The Metabolic Syndrome and Its Influence on Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2016, 20, 225–243. [Google Scholar] [CrossRef]
- Aller, R.; Izaola, O.; Ruiz-Rebollo, L.; Pacheco, D.; de Luis, D.A. Predictive factors of non-alcoholic steatohepatitis: Relationship with metabolic syndrome. Nutr. Hosp. 2015, 31, 2496–2502. [Google Scholar]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef]
- Wani, K.; Alharthi, H.; Alghamdi, A.; Sabico, S.; Al-Daghri, N.M. Role of NLRP3 inflammasome activation in obesity-mediated metabolic disorders. Int. J. Environ. Res. Public Health 2021, 18, 511. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Flavell, R.A. Inflammasomes and Metabolic Disease. Annu. Rev. Physiol. 2014, 76, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hong, W.; Lu, S.; Li, Y.; Guan, Y.; Weng, X.; Feng, Z. The NLRP3 Inflammasome in Non-Alcoholic Fatty Liver Disease and Steatohepatitis: Therapeutic Targets and Treatment. Front. Pharmacol. 2022, 13, 682. [Google Scholar] [CrossRef]
- Nov, O.; Kohl, A.; Lewis, E.C.; Bashan, N.; Dvir, I.; Ben-Shlomo, S.; Fishman, S.; Wueest, S.; Konrad, D.; Rudich, A. Interleukin-1β may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology 2010, 151, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- Nov, O.; Shapiro, H.; Ovadia, H.; Tarnovscki, T.; Dvir, I.; Shemesh, E.; Kovsan, J.; Shelef, I.; Carmi, Y.; Voronov, E.; et al. Interleukin-1β Regulates Fat-Liver Crosstalk in Obesity by Auto-Paracrine Modulation of Adipose Tissue Inflammation and Expandability. PLoS ONE 2013, 8, e53626. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.; Esteve, E.; Casamitjana, R. Potential Role of Interleukin-18 in Liver Disease Associated with Insulin Resistance. Obes. Res. 2005, 13, 1925–1931. [Google Scholar]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef]
- Bijnen, M.; Josefs, T.; Cuijpers, I.; Maalsen, C.J.; Van De Gaar, J.; Vroomen, M.; Wijnands, E.; Rensen, S.S.; Greve, J.W.M.; Hofker, M.H.; et al. Adipose tissue macrophages induce hepatic neutrophil recruitment and macrophage accumulation in mice. Gut 2018, 67, 1317–1327. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef]
- Yilmaz, Y.; Younossi, Z.M. Obesity-Associated Nonalcoholic Fatty Liver Disease. Clin. Liver Dis. 2014, 18, 19–31. [Google Scholar] [CrossRef]
- Charrez, B.; Qiao, L.; Hebbard, L. Hepatocellular carcinoma and non-alcoholic steatohepatitis: The state of play. World J. Gastroenterol. 2016, 22, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed. Res. Int. 2016, 2016, 5170402. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. TGF-β Signaling in Hepatocytes Participates in Steatohepatitis Through Regulation of Cell Death and Lipid Metabolism. Hepatology 2013, 59, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, S.A.; Muhsin, N.I.A.; Jamal, R. Regulatory non-coding RNAs network in non-alcoholic fatty liver disease. Front. Physiol. 2019, 10, 279. [Google Scholar] [CrossRef]
- Diehl, A.M.; Li, Z.P.; Lin, H.Z.; Yang, S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 2005, 54, 303–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef]
- Niederreiter, L.; Tilg, H. Cytokines and fatty liver diseases. Liver Res. 2018, 2, 14–20. [Google Scholar] [CrossRef]
- Kugelmas, M.; Hill, D.B.; Vivian, B.; Marsano, L.; McClain, C.J. Cytokines and NASH: A pilot study of the effects of lifestyle modification and vitamin E. Hepatology 2003, 38, 413–419. [Google Scholar] [CrossRef]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D.A. Mechanisms of liver fibrosis and its role in liver cancer. Exp. Biol. Med. 2020, 2, 96–108. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Gieling, R.G.; Wallace, K.; Han, Y.P. Interleukin-1 participates in the progression from liver injury to fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Liang, T. HIF-1α/IL-1β signaling enhances hepatoma epithelial-mesenchymal transition via macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889. [Google Scholar] [CrossRef] [Green Version]
- Yamanishi, K.; Maeda, S.; Kuwahara-Otani, S.; Watanabe, Y.; Yoshida, M.; Ikubo, K.; Okuzaki, D.; El-Darawish, Y.; Li, W.; Nakasho, K.; et al. Interleukin-18–deficient mice develop dyslipidemia resulting in nonalcoholic fatty liver disease and steatohepatitis. Transl. Res. 2016, 173, 101–114. [Google Scholar] [CrossRef]
- Cyr, B.; Keane, R.W.; de Rivero Vaccari, J.P. Asc, IL-18 and galectin-3 as biomarkers of non-alcoholic steatohepatitis: A proof of concept study. Int. J. Mol. Sci. 2020, 21, 8580. [Google Scholar] [CrossRef]
- Flisiak-Jackiewicz, M.; Bobrus-Chociej, A.; Tarasów, E.; Wojtkowska, M.; Białokoz-Kalinowska, I.; Lebensztejn, D.M. Predictive Role of Interleukin-18 in Liver Steatosis in Obese Children. Can. J. Gastroenterol. Hepatol. 2018, 2018, 3870454. [Google Scholar] [CrossRef]
- Tapan, S.; Dogru, T.; Kara, M.; Ercin, C.N.; Kilciler, G.; Genc, H.; Sertoglu, E.; Acikel, C.; Kilic, S.; Karslioglu, Y.; et al. Circulating levels of interleukin-18 in patients with non-alcoholic fatty liver disease. Scand. J. Clin. Lab. Investig. 2010, 70, 399–403. [Google Scholar] [CrossRef]
- Brandl, K.; Schnabl, B. Intestinal microbiota and nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2017, 33, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Farhadi, A.; Gundlapalli, S.; Shaikh, M.; Frantzides, C.; Harrell, L.; Kwasny, M.M.; Keshavarzian, A. Susceptibility to gut leakiness: A possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008, 28, 1026–1033. [Google Scholar] [CrossRef]
- Augustyn, M.; Grys, I.; Kukla, M. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease. Clin. Exp. Hepatol. 2019, 5, 1–10. [Google Scholar] [CrossRef]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 300, 433–441. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver cancer: Connections with obesity, fatty liver, and cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Ye, S.L.; Takayama, T.; Geschwind, J.; Marrero, J.A.; Bronowicki, J.P. Current Approaches to the Treatment of Early Hepatocellular Carcinoma. The Oncologist 2010, 15, 34–41. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 2003, 37, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Brar, G.; Kesselman, A.; Malhotra, A.; Shah, M.A. Redefining Intermediate-Stage HCC Treatment in the Era of Immune Therapies. JCO Oncol. Pract. 2022, 18, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lurje, I.; Czigany, Z.; Bednarsch, J.; Roderburg, C.; Isfort, P.; Neumann, U.P.; Lurje, G. Treatment strategies for hepatocellular carcinoma—A multidisciplinary approach. Int. J. Mol. Sci. 2019, 20, 1465. [Google Scholar] [CrossRef] [PubMed]
- Montanari, N.R.; Anugwom, C.M.; Boonstra, A.; Debes, J.D. The Role of Cytokines in the Different Stages of Hepatocellular Carcinoma. Cancers 2021, 13, 4876. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, X.; Liang, J.; Liu, Y.; Hou, X.; Zhang, M.; Li, Y.; Jiang, X. Immunotherapy for Hepatocellular Carcinoma: Current Status and Future Prospects. Front. Immunol. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Zhang, W.E.I.; Song, T.Q.; Zhang, T.I.; Wu, Q. Adjuvant interferon for early or late recurrence of hepatocellular carcinoma and mortality from hepatocellular carcinoma following curative treatment: A meta—Analysis with comparison of different types of hepatitis. Mol. Clin. Oncol. 2014, 2, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yin, Z.; Cao, L.; Xu, X.; Yan, T.; Liu, C.; Li, D. Adjuvant pegylated interferon therapy improves the survival outcomes in patients with hepatitis-related hepatocellular carcinoma after curative treatment. Medicine 2018, 97, e11295. [Google Scholar] [CrossRef]
- Sakisaka, M.; Haruta, M.; Komohara, Y.; Umemoto, S.; Matsumura, K.; Ikeda, T.; Takeya, M.; Inomata, Y.; Nishimura, Y.; Senju, S. Therapy of primary and metastatic liver cancer by human iPS cell-derived myeloid cells producing interferon-β. J. Hepatobiliary Pancreat Sci. 2017, 24, 109–119. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, H.; Chen, G.; Huang, X. Genetically engineered recombinant adenovirus expressing interleukin-2 for hepatocellular carcinoma therapy. Mol. Med. Rep. 2018, 17, 300–306. [Google Scholar] [CrossRef]
- Xu, J.; Lin, H.; Wu, G.; Zhu, M.; Li, M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 760971. [Google Scholar] [CrossRef]
- Teng, D.; Ding, L.; Cai, B.; Luo, Q.; Wang, H. Cytokine Interleukin-7 enhances anti-tumor activity of CD8 + T cells in patients with hepatocellular carcinoma. Cytokine 2018, 118, 115–123. [Google Scholar] [CrossRef]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF- α is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, E.; Vaquero, J.; Férnandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF- β Pathway: A Pharmacological Target in Hepatocellular Carcinoma ? Cancers 2021, 13, 3248. [Google Scholar]
- Masuzaki, R.; Kanda, T.; Sasaki, R.; Matsumoto, N.; Nirei, K.; Ogawa, M.; Karp, S.J.; Moriyama, M.; Kogure, H. Suppressors of Cytokine Signaling and Hepatocellular Carcinoma. Cancers 2022, 14, 2549. [Google Scholar] [CrossRef] [PubMed]
- García-Pras, E.; Fernández-Iglesias, A.; Gracia-Sancho, J.; Pérez-Del-Pulgar, S. Cell Death in Hepatocellular Carcinoma: Pathogenesis and Therapeutic Opportunities. Cancers 2022, 14, 48. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Cruz, N.S.; Pasquarelli-do-Nascimento, G.; e Oliveira, A.C.P.; Magalhães, K.G. Inflammasome-Mediated Cytokines: A Key Connection between Obesity-Associated NASH and Liver Cancer Progression. Biomedicines 2022, 10, 2344. https://doi.org/10.3390/biomedicines10102344
da Cruz NS, Pasquarelli-do-Nascimento G, e Oliveira ACP, Magalhães KG. Inflammasome-Mediated Cytokines: A Key Connection between Obesity-Associated NASH and Liver Cancer Progression. Biomedicines. 2022; 10(10):2344. https://doi.org/10.3390/biomedicines10102344
Chicago/Turabian Styleda Cruz, Nathalia Soares, Gabriel Pasquarelli-do-Nascimento, Augusto Cézar Polveiro e Oliveira, and Kelly Grace Magalhães. 2022. "Inflammasome-Mediated Cytokines: A Key Connection between Obesity-Associated NASH and Liver Cancer Progression" Biomedicines 10, no. 10: 2344. https://doi.org/10.3390/biomedicines10102344
APA Styleda Cruz, N. S., Pasquarelli-do-Nascimento, G., e Oliveira, A. C. P., & Magalhães, K. G. (2022). Inflammasome-Mediated Cytokines: A Key Connection between Obesity-Associated NASH and Liver Cancer Progression. Biomedicines, 10(10), 2344. https://doi.org/10.3390/biomedicines10102344