

Progeria and Aging—Omics Based Comparative Analysis

 ,
,  and
and
Abstract
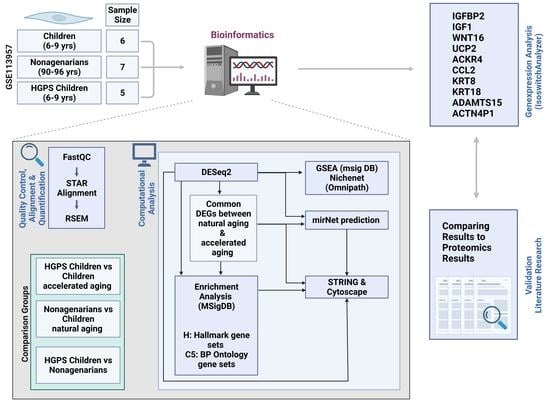
:
1. Introduction
2. Materials and Methods
2.1. Hardware and Software
2.2. RNA-Seq Data
2.3. Data Preprocessing
2.4. Identification of DEGs
2.5. Data Visualization
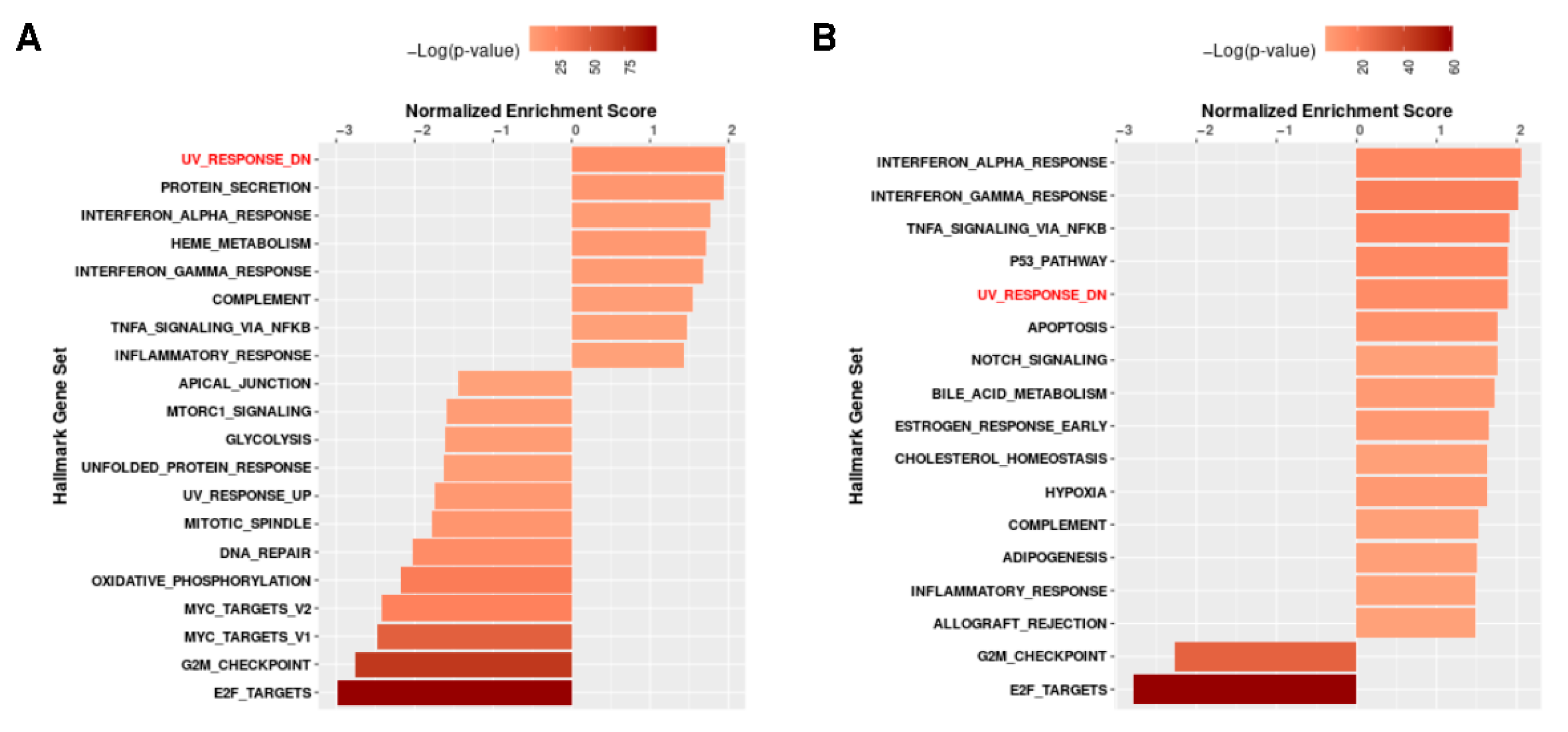
2.6. Pathway Enrichment Analysis
2.7. Protein–Protein Interactions
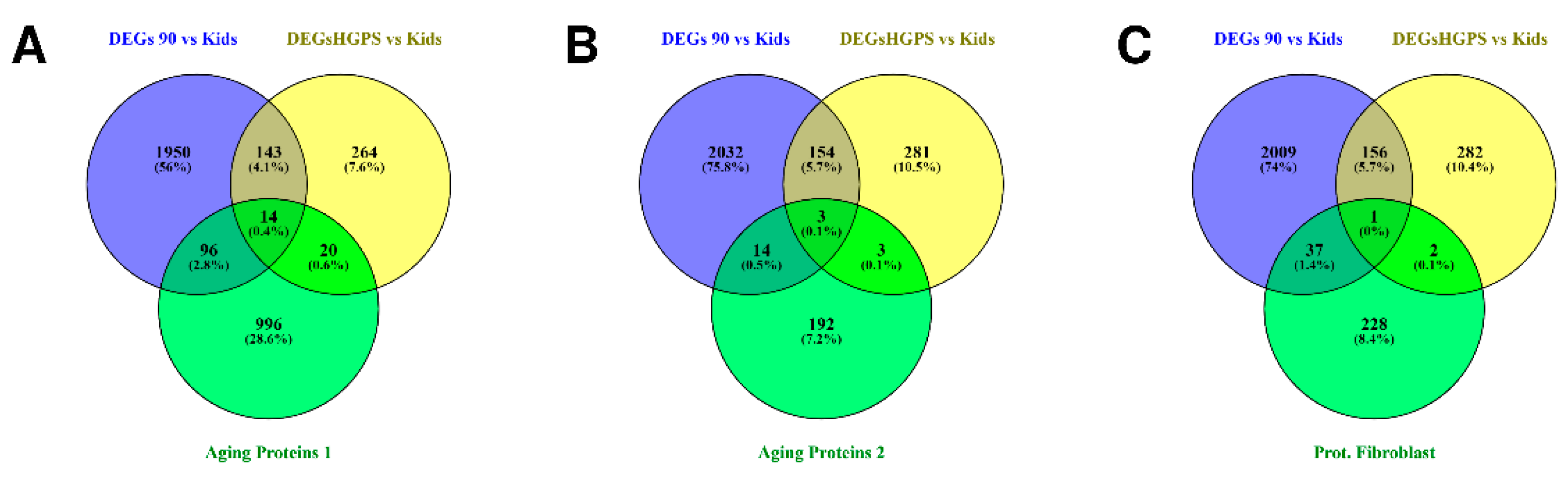
2.8. Venn Diagrams
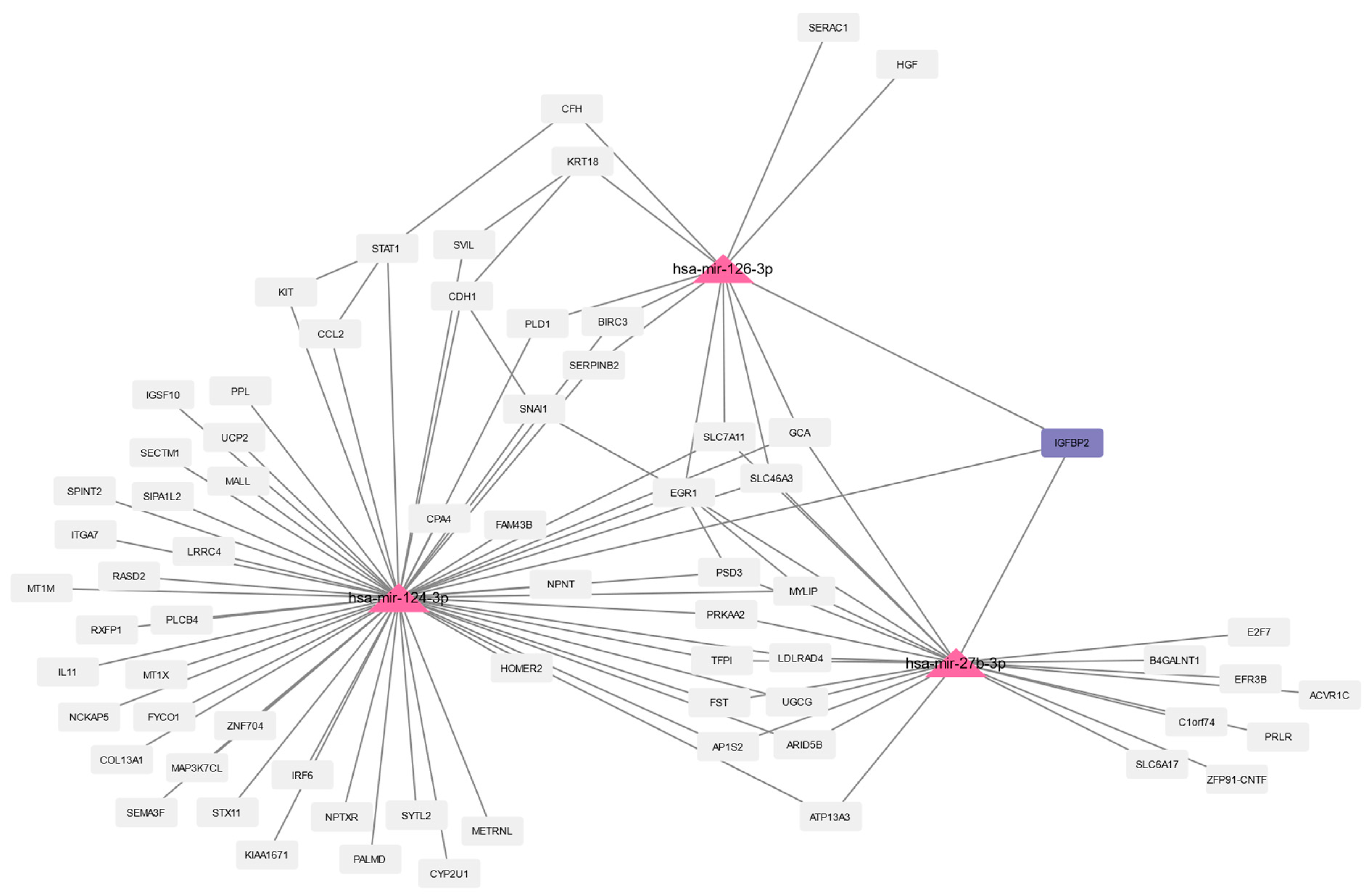
2.9. miRNA Prediction
2.10. NicheNet: Finding Ligand–Receptor Interactions Based on Prior Knowledge
2.11. Figures and Additional Packages
3. Results
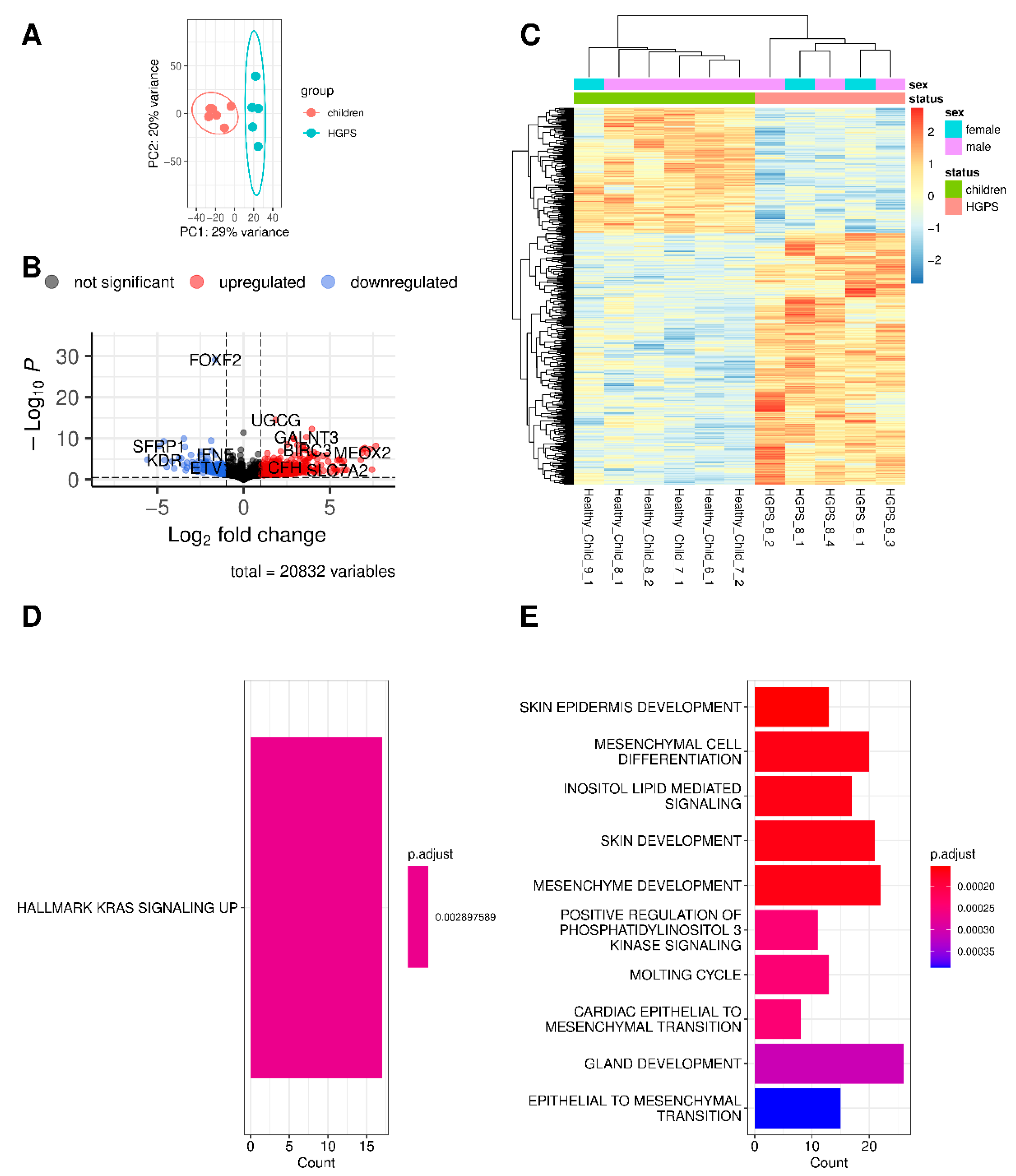
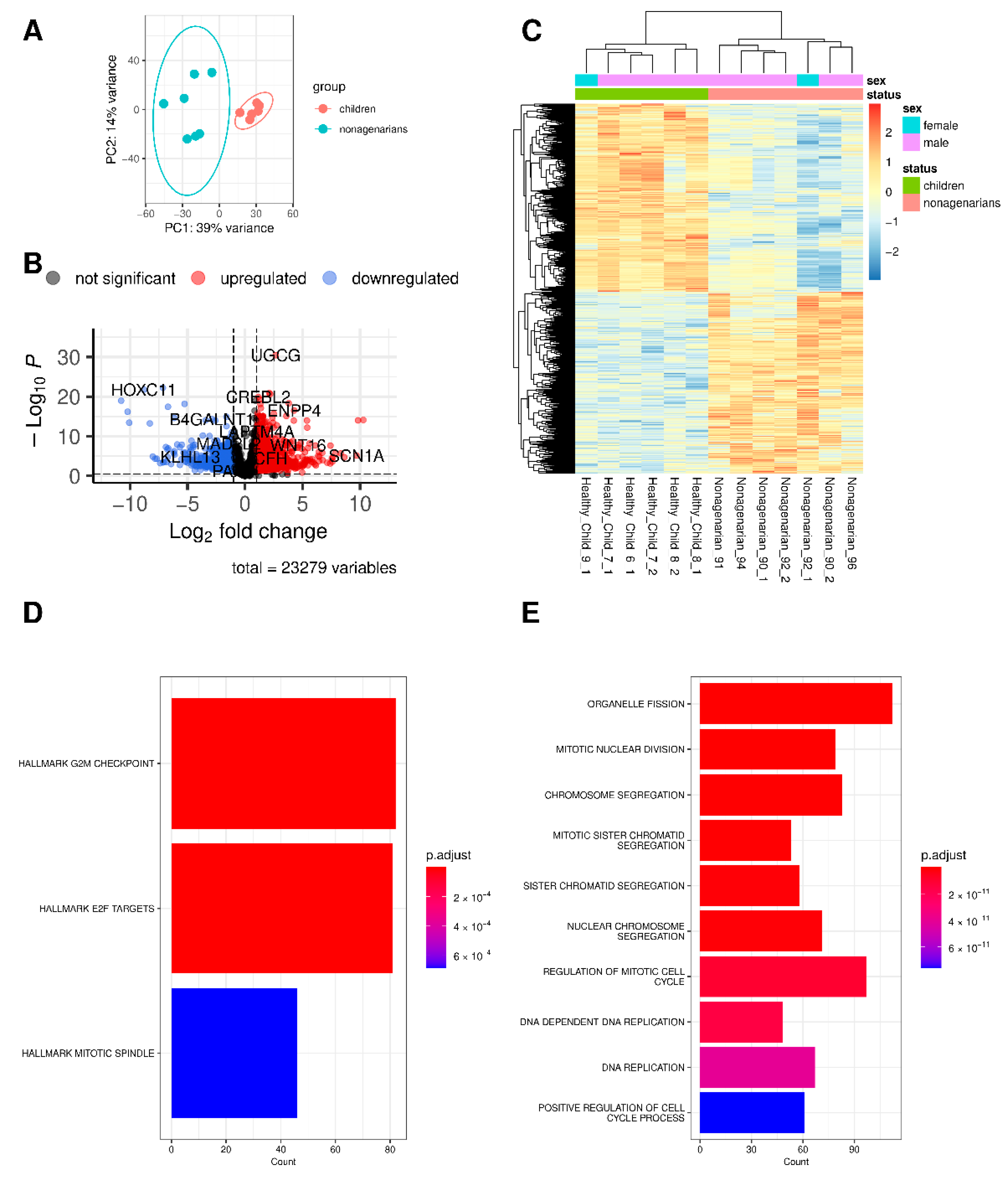
3.1. Differences and Similarities between Old Age and HGPS
3.2. Changes in Gene Expression in Progeria and Normal Aging
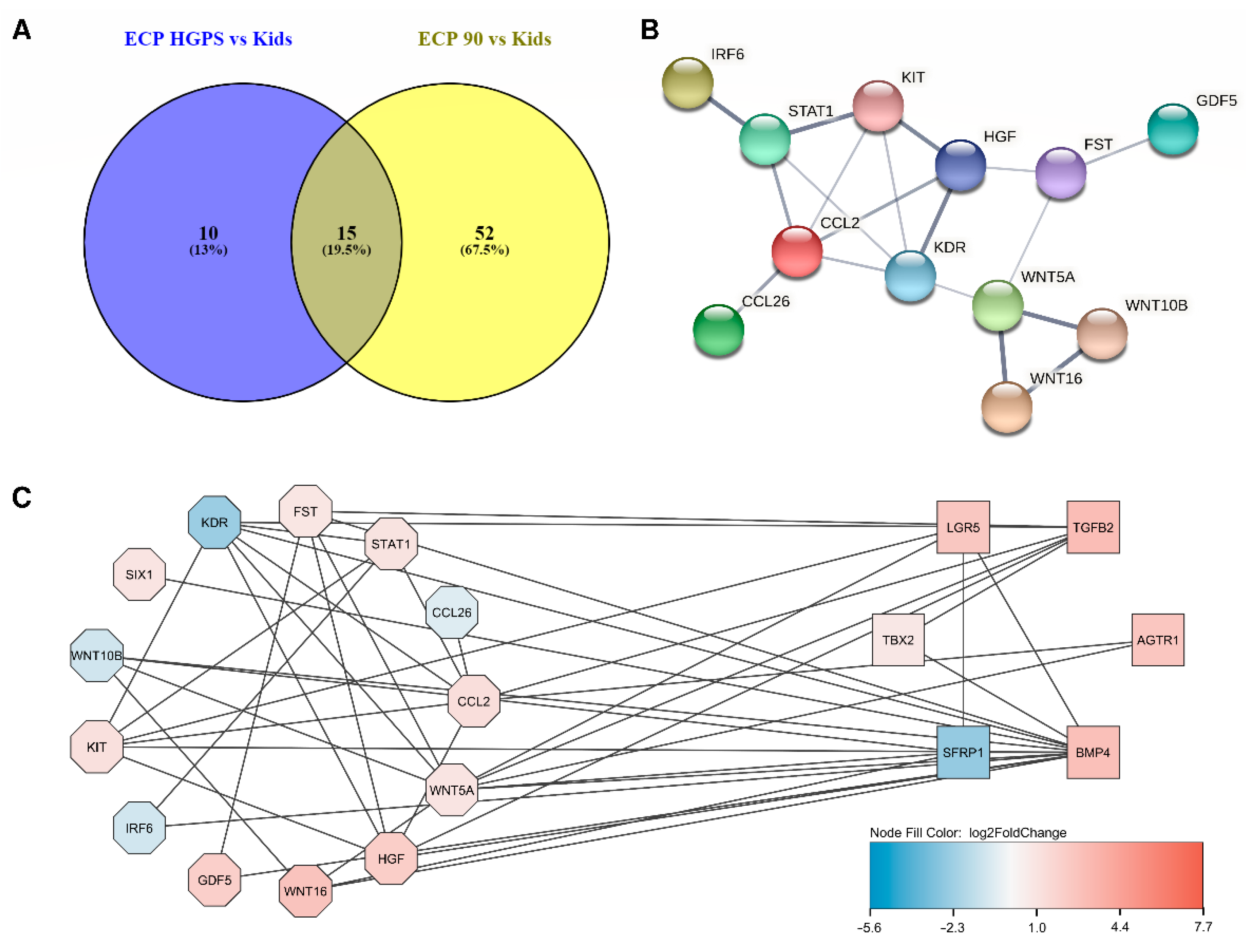
3.3. The Different Pathways Involved in Progeria, Aging, and Both Conditions
3.4. Prediction of microRNAs and Visual Exploration of Interaction Partners of WNT16, IGFBP2, and UCP2
3.5. Predicting Interactions Using NicheNet and Omnipath
3.6. Proteomics
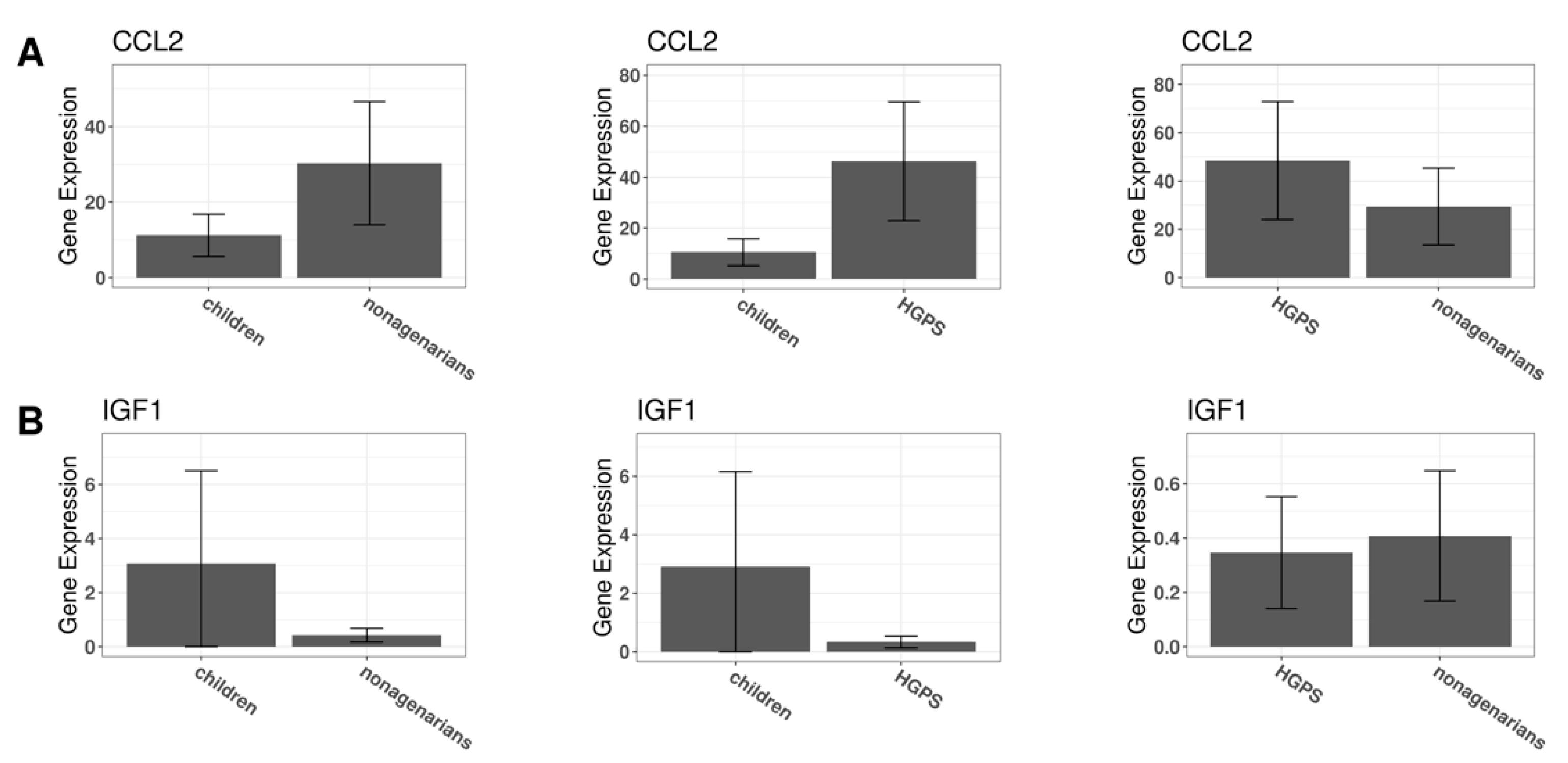
3.7. Validation Using a Different RNA-Seq Dataset
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbrevations
| CC | Cellular component |
| CD4+ T cells | T helper cells also known as CD4-positive cells (CD4 = cluster of differentiation 4) |
| DEGs | differentially expressed genes |
| E2F | group of genes encoding transcription factors in higher eukaryotes |
| ECP | Epithelial Cell Proliferation |
| FDA | U.S. Food and Drug Administration |
| FTIs | farnesyltransferase inhibitors |
| G2M checkpoint | G2/M checkpoint |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| GO BPs | Gene Ontology enriched biological processes |
| GSEA | Gene Set Enrichment Analysis |
| HGPS | Hutchinson-Gilford progeria syndrome |
| hsa-mir- | human microRNA |
| iTRAQ | isobaric tag for relative and absolute quantification |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MF | molecular function |
| miR- | microRNA |
| miRNAs | microRNAs |
| miRs | microRNAs |
| MSigDB | Molecular Signatures Database |
| NES | normalized enrichment score |
| PCA | Principal component analysis |
| PRF | Progeria Research Foundation |
| RNA | ribonucleic acid |
| RNA-Seq | RNA sequencing |
| STRING | Search Tool for Retrieval of Interacting Genes/Proteins |
| UV | ultraviolet |
| UVB | type B ultraviolet |
| Genes | |
| ACE2 | Angiotensin-converting enzyme 2 |
| ACKR2 | Atypical chemokine receptor 2 |
| ACKR4 | Atypical chemokine receptor 4 |
| ACTN4P1 | Actinin alpha 4 pseudogene 1 |
| ADAM9 | ADAM Metallopeptidase Domain 9 |
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin motifs |
| ADAMTS15 | ADAM metallopeptidase with thrombospondin type 1 motif 15 |
| ANLN | Anillin, Actin Binding Protein |
| APBA2 | Amyloid Beta Precursor Protein Binding Family A Member 2 |
| ASNS | Asparagine Synthetase (Glutamine-Hydrolyzing) |
| ASPA | Aspartoacylase |
| CCL2 | C-C Motif Chemokine Ligand 2 |
| CCL26 | C-C Motif Chemokine Ligand 26 |
| CCR10 | C-C chemokine receptor type 10 |
| CCR11 | Abbreviation for Atypical chemokine receptor 4 (ACKR4) |
| CDH1 | Cadherin 1 |
| CDK1 | Cyclin Dependent Kinase 1 |
| CDK4 | Cyclin Dependent Kinase 4 |
| CDKN2B | Cyclin Dependent Kinase Inhibitor 2B |
| CFH | Complement Factor H |
| CGAS | Cyclic GMP-AMP Synthase |
| CITED2 | Cbp/P300 Interacting Transactivator With Glu/Asp Rich Carboxy-Terminal Domain 2 |
| cKRT18 | caspase-cleaved fragment of keratin 18 (KRT18) |
| CLIP4 | CAP-Gly Domain Containing Linker Protein Family Member 4 |
| CPNE1 | Copine 1 |
| DLGAP5 | DLG Associated Protein 5 |
| DTYMK | Deoxythymidylate Kinase |
| ECM2 | Extracellular Matrix Protein 2 |
| EDIL3 | EGF Like Repeats And Discoidin Domains 3 |
| EFEMP1 | EGF containing fibulin extracellular matrix protein 1 |
| EGR1 | Early Growth Response 1 |
| FAM8A1 | Family With Sequence Similarity 8 Member A1 |
| FBN2 | Fibrillin 2 |
| FBLN5 | Fibulin 5 |
| FST | Follistatin |
| GDF5 | Growth Differentiation Factor 5 |
| HGF | Hepatocyte Growth Factor |
| HS3ST3A1 | Heparan Sulfate-Glucosamine 3-Sulfotransferase 3A1 |
| IGF1 | insulin-like growth factor 1 |
| IGFBP1 | Insulin Like Growth Factor Binding Protein 1 |
| IGFBP2 | Insulin Like Growth Factor Binding Protein 2 |
| IGFBP7 | Insulin Like Growth Factor Binding Protein 7 |
| IL11 | Interleukin 11 |
| IL13RA2 | Interleukin 13 Receptor Subunit Alpha 2 |
| IRF6 | Interferon Regulatory Factor 6 |
| KDR | Kinase Insert Domain Receptor |
| KIFC1 | Kinesin Family Member C1 |
| KIT | KIT Proto-Oncogene, Receptor Tyrosine Kinase |
| KLHL24 | Kelch Like Family Member 24 |
| KRAS | Kristen rat sarcoma virus |
| KRT18 | Keratin 18 |
| KRT8 | Keratin 8 |
| LEPR | Leptin Receptor |
| LMNA | Lamin A/C |
| LMNB1 | Lamin B1 |
| LMNB2 | Lamin B2 |
| MAF | MAF BZIP Transcription Factor |
| MCP-1 | monocyte chemoattractant protein-1 |
| MKI67 | Marker Of Proliferation Ki-67 |
| MMP10 | Matrix Metallopeptidase 10 |
| MSR1 | Macrophage Scavenger Receptor 1 |
| MYL9 | Myosin Light Chain 9 |
| NEIL1 | Nei Like DNA Glycosylase 1 |
| NTN4 | Netrin 4 |
| NKX3-1 | NK3 Homeobox 1 |
| NOD1 | Nucleotide Binding Oligomerization Domain Containing 1 |
| PLSCR4 | Phospholipid Scramblase 4 |
| POLR2F | RNA Polymerase II, I And III Subunit F |
| POSTN | Periostin |
| PRPS1 | Phosphoribosyl Pyrophosphate Synthetase 1 |
| PTPRN | Protein Tyrosine Phosphatase Receptor Type N |
| SECTM1 | Secreted and Transmembrane 1 |
| SEMA3D | Semaphorin 3D |
| SEMA5B | Semaphorin 5B |
| SIX1 | SIX Homeobox 1/Sine Oculis Homeobox Homolog 1 |
| SNAI1 | Snail Family Transcriptional Repressor 1 |
| SNAP23 | Synaptosome Associated Protein 23 |
| SPINT2 | Serine Peptidase Inhibitor, Kunitz Type 2 |
| SPTB | Spectrin Beta, Erythrocytic |
| STAT1 | Signal Transducer And Activator Of Transcription 1 |
| STAT4 | Signal Transducer And Activator Of Transcription 4 |
| STRA6 | Signaling Receptor And Transporter Of Retinol STRA6 |
| SVEP1 | Sushi, Von Willebrand Factor Type A, EGF And Pentraxin Domain Containing 1 |
| TACC3 | Transforming Acidic Coiled-Coil Containing Protein 3 |
| TLR3 | Toll Like Receptor 3 |
| TLR4 | Toll Like Receptor 4 |
| TNXB | Tenascin XB |
| TOR1AIP1 | Torsin 1A Interacting Protein 1 (also known as LAP1B) |
| U2AF1 | U2 Small Nuclear RNA Auxiliary Factor 1 |
| UBE2D1 | Ubiquitin Conjugating Enzyme E2 D1 |
| UCP2 | Uncoupling Protein 2 |
| WISP2 | WNT1-Inducible-Signaling Pathway Protein 2also known as CCN5 (Cellular Communication Network Factor 5) |
| Wnt | Wingless/Integrated |
| WNT10B | Wnt Family Member 10B |
| WNT16 | Wnt Family Member 16 |
| WNT5A | Wnt Family Member 5A |
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; de Magalhães, J.P. The hoverfly and the wasp: A critique of the hallmarks of aging as a paradigm. Ageing Res. Rev. 2021, 70, 101407. [Google Scholar] [CrossRef] [PubMed]
- Ubaida-Mohien, C.; Moaddel, R.; Moore, A.Z.; Kuo, P.-L.; Faghri, F.; Tharakan, R.; Tanaka, T.; Nalls, M.A.; Ferrucci, L. Proteomics and Epidemiological Models of Human Aging. Front. Physiol. 2021, 12, 674013. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Hutchinson, J. Congenital Absence of Hair and Mammary Glands with Atrophic Condition of the Skin and its Appendages, in a Boy whose Mother had been almost wholly Bald from Alopecia Areata from the age of Six. Lancet 1886, 69, 473–477. [Google Scholar] [CrossRef]
- Keith, A. Progeria and Ateleiosis. Lancet 1913, 181, 305–313. [Google Scholar] [CrossRef]
- Gilford, H. Progeria and Ateleiosis. Lancet 1913, 181, 412–413. [Google Scholar] [CrossRef]
- Hegele, R.A. Drawing the line in progeria syndromes. Lancet 2003, 362, 416–417. [Google Scholar] [CrossRef]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.M.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and Course of Hutchinson–Gilford Progeria Syndrome. New Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Hennekam, R.C.M. Hutchinson–Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. Part A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart Colin, L.; Munnich, A.; Le Merrer, M.; et al. Lamin A Truncation in Hutchinson-Gilford Progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Lonafarnib: First Approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef]
- The Progeria Research Foundation. News-European Medicines Agency recommends Zokinvy, the First and Only Therapy to Treat Ultra-rare, Rapid-Ageing Disease Progeria, for Approval in Europe. Available online: https://www.progeriaresearch.org/wp-content/uploads/2022/05/Zokinvy-CHMP-lay_health-joint-PR-FINAL-no-references.pdf (accessed on 23 May 2022).
- Fleischer, J.G.; Schulte, R.; Tsai, H.H.; Tyagi, S.; Ibarra, A.; Shokhirev, M.N.; Huang, L.; Hetzer, M.W.; Navlakha, S. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 2018, 19, 221. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Köhler, F.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Corless, S.; Musch, T.; Lonsdorf, A.S.; Erhardt, S.; Lyko, F.; Rodríguez-Paredes, M. Epigenetic deregulation of lamina-associated domains in Hutchinson-Gilford progeria syndrome. Genome Med. 2020, 12, 46. [Google Scholar] [CrossRef]
- Ikegami, K.; Secchia, S.; Almakki, O.; Lieb, J.D.; Moskowitz, I.P. Phosphorylated Lamin A/C in the Nuclear Interior Binds Active Enhancers Associated with Abnormal Transcription in Progeria. Dev. Cell 2020, 52, 699–713.e611. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Ding, D.; Li, X.; Shen, T.; Fu, H.; Zhong, H.; Wei, G.; Ni, T. Prevalent intron retention fine-tunes gene expression and contributes to cellular senescence. Aging Cell 2020, 19, e13276. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shivashankar, G.V. Analysis of transcriptional modules during human fibroblast ageing. Sci. Rep. 2020, 10, 19086. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Cavalier, A.N.; Wahl, D. Repetitive elements as a transcriptomic marker of aging: Evidence in multiple datasets and models. Aging Cell 2020, 19, e13167. [Google Scholar] [CrossRef] [PubMed]
- McCauley, B.S.; Sun, L.; Yu, R.; Lee, M.; Liu, H.; Leeman, D.S.; Huang, Y.; Webb, A.E.; Dang, W. Altered chromatin states drive cryptic transcription in aging mammalian stem cells. Nat. Aging 2021, 1, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Bickler, S.W.; Cauvi, D.M.; Fisch, K.M.; Prieto, J.M.; Sykes, A.G.; Thangarajah, H.; Lazar, D.A.; Ignacio, R.C.; Gerstmann, D.R.; Ryan, A.F.; et al. Extremes of age are associated with differences in the expression of selected pattern recognition receptor genes and ACE2, the receptor for SARS-CoV-2: Implications for the epidemiology of COVID-19 disease. BMC Med. Genom. 2021, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Parry, A.; Santos, F.; Okkenhaug, H.; Todd, C.D.; Hernando-Herraez, I.; Stubbs, T.M.; Milagre, I.; Reik, W. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. eLife 2022, 11, e71624. [Google Scholar] [CrossRef]
- Meyer, D.H.; Schumacher, B. BiT age: A transcriptome-based aging clock near the theoretical limit of accuracy. Aging Cell 2021, 20, e13320. [Google Scholar] [CrossRef]
- Roth, S.H.; Levanon, E.Y.; Eisenberg, E. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat. Methods 2019, 16, 1131–1138. [Google Scholar] [CrossRef]
- Yim, A.; Koti, P.; Bonnard, A.; Marchiano, F.; Dürrbaum, M.; Garcia-Perez, C.; Villaveces, J.; Gamal, S.; Cardone, G.; Perocchi, F.; et al. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Nucleic Acids Res. 2020, 48, 605–632. [Google Scholar] [CrossRef] [Green Version]
- Kramme, C.; Plesa, A.M.; Wang, H.H.; Wolf, B.; Smela, M.P.; Guo, X.; Kohman, R.E.; Chatterjee, P.; Church, G.M. An integrated pipeline for mammalian genetic screening. Cell Rep. Methods 2021, 1, 100082. [Google Scholar] [CrossRef] [PubMed]
- Gabay, O.; Shoshan, Y.; Kopel, E.; Ben-Zvi, U.; Mann, T.D.; Bressler, N.; Cohen-Fultheim, R.; Schaffer, A.A.; Roth, S.H.; Tzur, Z.; et al. Landscape of adenosine-to-inosine RNA recoding across human tissues. Nat. Commun. 2022, 13, 1184. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/pdf (accessed on 29 June 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.; Robinson, M. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. R Package. 2022. Available online: https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html (accessed on 29 June 2022).
- Kolde, R. pheatmap: Pretty Heatmaps. R Package. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 29 June 2022).
- Vitting-Seerup, K.; Sandelin, A. The Landscape of Isoform Switches in Human Cancers. Mol. Cancer Res. 2017, 15, 1206–1220. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha Vamsi, K.; Mukherjee, S.; Ebert Benjamin, L.; Gillette Michael, A.; Paulovich, A.; Pomeroy Scott, L.; Golub Todd, R.; Lander Eric, S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Bao, L.; Zhang, X. Investigating potential molecular mechanisms of serum exosomal miRNAs in colorectal cancer based on bioinformatics analysis. Medicine 2020, 99, e22199. [Google Scholar] [CrossRef]
- Chagoyen, M.; Pazos, F. Quantifying the biological significance of gene ontology biological processes—Implications for the analysis of systems-wide data. Bioinformatics 2010, 26, 378–384. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.16.2. Available online: https://yulab-smu.top/biomedical-knowledge-mining-book/ (accessed on 29 June 2022).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Venn, J.I. On the diagrammatic and mechanical representation of propositions and reasonings. Philos. Mag. J. Sci. 1880, 10, 1–18. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 29 June 2022).
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef]
- Türei, D.; Valdeolivas, A.; Gul, L.; Palacio-Escat, N.; Klein, M.; Ivanova, O.; Ölbei, M.; Gábor, A.; Theis, F.; Módos, D.; et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 2021, 17, e9923. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wilke, C.O. cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’, R Package. 2020. Available online: https://wilkelab.org/cowplot/ (accessed on 29 June 2022).
- Yu, G. ggplotify: Convert Plot to ‘grob’ or ‘ggplot’ Object. R Package. Available online: https://cran.r-project.org/web/packages/ggplotify/index.html (accessed on 29 June 2021).
- Ooms, J. magick: Advanced Graphics and Image-Processing in R, R Package. 2021. Available online: https://cran.r-project.org/web/packages/magick/vignettes/intro.html (accessed on 29 June 2022).
- Ligges, U.; Maechler, M. scatterplot3d-An R Package for Visualizing Multivariate Data. J. Stat. Softw. 2003, 8, 1–20. [Google Scholar] [CrossRef]
- Wickham, H.; Seidel, D. scales: Scale Functions for Visualization, R package. 2022. Available online: https://cran.r-project.org/web/packages/scales/index.html (accessed on 29 June 2022).
- Garnier, S.; Ross, N.; Rudis, R.; Camargo, A.P.; Sciaini, M.; Scherer, C. Rvision-Colorblind-Friendly Color Maps for R, R package. 2021. Available online: https://rdrr.io/cran/viridis/ (accessed on 29 June 2022).
- Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny; Chapman and Hall/CRC: London, UK, 2020. [Google Scholar]
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes, R package. 2022. Available online: https://rdrr.io/cran/RColorBrewer/ (accessed on 29 June 2022).
- Ahlmann-Eltze, C. ggupset: Combination Matrix Axis for ‘ggplot2’ to Create ‘UpSet’ Plots, R package. 2020. Available online: https://rdrr.io/cran/ggupset/ (accessed on 29 June 2022).
- Campitelli, E. ggnewscale: Multiple Fill and Colour Scales in ‘ggplot2’, R package. 2022. Available online: https://cran.r-project.org/web/packages/ggnewscale/index.html (accessed on 29 June 2022).
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Wilke, C.O. ggridges: Ridgeline Plots in ‘ggplot2’. R Package. 2021. Available online: https://github.com/wilkelab/ggridges (accessed on 29 June 2022).
- Jahn, N. europepmc: R Interface to the Europe PubMed Central RESTful Web Service, R package. 2021. Available online: https://CRAN.R-project.org/package=europepmc (accessed on 29 June 2022).
- Morgan, M. BiocManager: Access the Bioconductor Project Package Repository, R package. 2022. Available online: https://CRAN.R-project.org/package=BiocManager (accessed on 29 June 2022).
- Carlson, M. org.Hs.eg.db: Genome wide Annotation for Human, R package. 2022. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html (accessed on 29 June 2022).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation, R package. 2022. Available online: https://dplyr.tidyverse.org and https://github.com/tidyverse/dplyr (accessed on 29 June 2022).
- Leslie, B.G.; Brown, W.T.; Collins, F.S. Hutchinson-Gilford Progeria Syndrome. 2003 Dec 12 [Updated 2019 Jan 17]. In GeneReviews® [Internet]; University of Washington, Seattle: Seattle, WA, USA, 2022. [Google Scholar]
- Dangwal, S.; Thum, T. microRNA Therapeutics in Cardiovascular Disease Models. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 185–203. [Google Scholar] [CrossRef]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef]
- Lesiak, A.; Bednarski, I.; Rogowski-Tylman, M.; Sobjanek, M.; Woźniacka, A.; Danilewicz, M.; Young, A.; Narbutt, J. One week of exposure to sunlight induces progerin expression in human skin. Adv. Dermatol. Allergol./Postępy Dermatol. I Alergol. 2017, 34, 629–631. [Google Scholar] [CrossRef]
- McClintock, D.; Gordon Leslie, B.; Djabali, K. Hutchinson–Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc. Natl. Acad. Sci. 2006, 103, 2154–2159. [Google Scholar] [CrossRef]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Q.; Zhang, B. Leveraging the complementary nature of RNA-Seq and shotgun proteomics data. Proteomics 2014, 14, 2676–2687. [Google Scholar] [CrossRef]
- Johnson, A.A.; Shokhirev, M.N.; Wyss-Coray, T.; Lehallier, B. Systematic review and analysis of human proteomics aging studies unveils a novel proteomic aging clock and identifies key processes that change with age. Ageing Res. Rev. 2020, 60, 101070. [Google Scholar] [CrossRef]
- Moaddel, R.; Ubaida-Mohien, C.; Tanaka, T.; Lyashkov, A.; Basisty, N.; Schilling, B.; Semba, R.D.; Franceschi, C.; Gorospe, M.; Ferrucci, L. Proteomics in aging research: A roadmap to clinical, translational research. Aging Cell 2021, 20, e13325. [Google Scholar] [CrossRef]
- Tsitsipatis, D.; Martindale, J.L.; Ubaida-Mohien, C.; Lyashkov, A.; Yanai, H.; Kashyap, A.; Shin, C.H.; Herman, A.B.; Ji, E.; Yang, J.-H.; et al. Proteomes of primary skin fibroblasts from healthy individuals reveal altered cell responses across the life span. Aging Cell 2022, 21, e13609. [Google Scholar] [CrossRef]
- Tin, A.; Yu, B.; Ma, J.; Masushita, K.; Daya, N.; Hoogeveen, R.C.; Ballantyne, C.M.; Couper, D.; Rebholz, C.M.; Grams, M.E.; et al. Reproducibility and Variability of Protein Analytes Measured Using a Multiplexed Modified Aptamer Assay. J. Appl. Lab. Med. 2019, 4, 30–39. [Google Scholar] [CrossRef]
- Tanaka, T.; Biancotto, A.; Moaddel, R.; Moore, A.Z.; Gonzalez-Freire, M.; Aon, M.A.; Candia, J.; Zhang, P.; Cheung, F.; Fantoni, G.; et al. Plasma proteomic signature of age in healthy humans. Aging Cell 2018, 17, e12799. [Google Scholar] [CrossRef]
- Hennrich, M.L.; Romanov, N.; Horn, P.; Jaeger, S.; Eckstein, V.; Steeples, V.; Ye, F.; Ding, X.; Poisa-Beiro, L.; Lai, M.C.; et al. Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nat. Commun. 2018, 9, 4004. [Google Scholar] [CrossRef]
- Lehallier, B.; Gate, D.; Schaum, N.; Nanasi, T.; Lee, S.E.; Yousef, H.; Moran Losada, P.; Berdnik, D.; Keller, A.; Verghese, J.; et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 2019, 25, 1843–1850. [Google Scholar] [CrossRef]
- Heinze, I.; Bens, M.; Calzia, E.; Holtze, S.; Dakhovnik, O.; Sahm, A.; Kirkpatrick, J.M.; Szafranski, K.; Romanov, N.; Sama, S.N.; et al. Species comparison of liver proteomes reveals links to naked mole-rat longevity and human aging. BMC Biol. 2018, 16, 82. [Google Scholar] [CrossRef]
- Lu, J.; Huang, Y.; Wang, Y.; Li, Y.; Zhang, Y.; Wu, J.; Zhao, F.; Meng, S.; Yu, X.; Ma, Q.; et al. Profiling Plasma Peptides for the Identification of Potential Ageing Biomarkers in Chinese Han Adults. PLoS ONE 2012, 7, e39726. [Google Scholar] [CrossRef]
- Lind, L.; Sundström, J.; Larsson, A.; Lampa, E.; Ärnlöv, J.; Ingelsson, E. Longitudinal effects of aging on plasma proteins levels in older adults–associations with kidney function and hemoglobin levels. PLoS ONE 2019, 14, e0212060. [Google Scholar] [CrossRef]
- Enroth, S.; Johansson, Å.; Enroth, S.B.; Gyllensten, U. Strong effects of genetic and lifestyle factors on biomarker variation and use of personalized cutoffs. Nat. Commun. 2014, 5, 4684. [Google Scholar] [CrossRef]
- Di Narzo, A.F.; Telesco, S.E.; Brodmerkel, C.; Argmann, C.; Peters, L.A.; Li, K.; Kidd, B.; Dudley, J.; Cho, J.; Schadt, E.E.; et al. High-Throughput Characterization of Blood Serum Proteomics of IBD Patients with Respect to Aging and Genetic Factors. PLOS Genet. 2017, 13, e1006565. [Google Scholar] [CrossRef]
- Baird, G.S.; Nelson, S.K.; Keeney, T.R.; Stewart, A.; Williams, S.; Kraemer, S.; Peskind, E.R.; Montine, T.J. Age-Dependent Changes in the Cerebrospinal Fluid Proteome by Slow Off-Rate Modified Aptamer Array. Am. J. Pathol. 2012, 180, 446–456. [Google Scholar] [CrossRef]
- Ignjatovic, V.; Lai, C.; Summerhayes, R.; Mathesius, U.; Tawfilis, S.; Perugini, M.A.; Monagle, P. Age-Related Differences in Plasma Proteins: How Plasma Proteins Change from Neonates to Adults. PLoS ONE 2011, 6, e17213. [Google Scholar] [CrossRef] [Green Version]
- Bakun, M.; Senatorski, G.; Rubel, T.; Lukasik, A.; Zielenkiewicz, P.; Dadlez, M.; Paczek, L. Urine proteomes of healthy aging humans reveal extracellular matrix (ECM) alterations and immune system dysfunction. Age 2014, 36, 299–311. [Google Scholar] [CrossRef]
- Cominetti, O.; Núñez Galindo, A.; Corthésy, J.; Valsesia, A.; Irincheeva, I.; Kussmann, M.; Saris, W.H.M.; Astrup, A.; McPherson, R.; Harper, M.-E.; et al. Obesity shows preserved plasma proteome in large independent clinical cohorts. Sci. Rep. 2018, 8, 16981. [Google Scholar] [CrossRef]
- Zhang, J.; Goodlett, D.R.; Peskind, E.R.; Quinn, J.F.; Zhou, Y.; Wang, Q.; Pan, C.; Yi, E.; Eng, J.; Aebersold, R.H.; et al. Quantitative proteomic analysis of age-related changes in human cerebrospinal fluid. Neurobiol. Aging 2005, 26, 207–227. [Google Scholar] [CrossRef]
- Mateos, J.; Fafián-Labora, J.; Morente-López, M.; Lesende-Rodriguez, I.; Monserrat, L.; Ódena, M.A.; Oliveira, E.d.; de Toro, J.; Arufe, M.C. Next-Generation Sequencing and Quantitative Proteomics of Hutchinson-Gilford progeria syndrome-derived cells point to a role of nucleotide metabolism in premature aging. PLoS ONE 2018, 13, e0205878. [Google Scholar] [CrossRef]
- Jones, A.M.; Nühse, T.S. Phosphoproteomics using iTRAQ. Methods Mol. Biol. 2011, 779, 287–302. [Google Scholar] [CrossRef]
- Rowe John, W.; Kahn Robert, L. Human Aging: Usual and Successful. Science 1987, 237, 143–149. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Khalil, A.; Cohen, A.A.; Witkowski, J.M. Are We Ill Because We Age? Front. Physiol. 2019, 10, 1508. [Google Scholar] [CrossRef]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B., Sr.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Li, Y.; Li, L.-L.; Fan, X.-X.; Wang, Y.-W.; Wei, C.-L.; Liu, L.; Leung, E.L.-H.; Yao, X.-J. Identification of a New Potent Inhibitor Targeting KRAS in Non-small Cell Lung Cancer Cells. Front. Pharm. 2017, 8, 823. [Google Scholar] [CrossRef] [PubMed]
- Macicior, J.; Marcos-Ramiro, B.; Ortega-Gutiérrez, S. Small-Molecule Therapeutic Perspectives for the Treatment of Progeria. Int. J. Mol. Sci. 2021, 22, 7190. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851, Erratum in RNA 2016, 2022, 1641. [Google Scholar] [CrossRef]
- Miao, Q.; Xu, Y.; Yin, H.; Zhang, H.; Ye, J. KRT8 phosphorylation regulates the epithelial-mesenchymal transition in retinal pigment epithelial cells through autophagy modulation. J. Cell. Mol. Med. 2020, 24, 3217–3228. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Fernandes, A.; Aguilar-Pimentel, J.A.; de Angelis, M.H.; Guedes, J.R.; Brito, M.A.; Ortolano, S.; Pani, G.; Athanasopoulou, S.; Gonos, E.S.; et al. Towards frailty biomarkers: Candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res. Rev. 2018, 47, 214–277. [Google Scholar] [CrossRef]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef]
- Talpin, A.; Costantino, F.; Bonilla, N.; Leboime, A.; Letourneur, F.; Jacques, S.; Dumont, F.; Amraoui, S.; Dutertre, C.-A.; Garchon, H.-J.; et al. Monocyte-derived dendritic cells from HLA-B27+ axial spondyloarthritis (SpA) patients display altered functional capacity and deregulated gene expression. Arthritis Res. Ther. 2014, 16, 417. [Google Scholar] [CrossRef]
- Cheetham, S.W.; Faulkner, G.J.; Dinger, M.E. Overcoming challenges and dogmas to understand the functions of pseudogenes. Nat. Rev. Genet. 2020, 21, 191–201. [Google Scholar] [CrossRef]
- Pink, R.C.; Wicks, K.; Caley, D.P.; Punch, E.K.; Jacobs, L.; Carter, D.R.F. Pseudogenes: Pseudo-functional or key regulators in health and disease? RNA 2011, 17, 792–798. [Google Scholar] [CrossRef]
- Kovalenko, T.F.; Patrushev, L.I. Pseudogenes as Functionally Significant Elements of the Genome. Biochem. 2018, 83, 1332–1349. [Google Scholar] [CrossRef]
- Marthandan, S.; Baumgart, M.; Priebe, S.; Groth, M.; Schaer, J.; Kaether, C.; Guthke, R.; Cellerino, A.; Platzer, M.; Diekmann, S.; et al. Conserved Senescence Associated Genes and Pathways in Primary Human Fibroblasts Detected by RNA-Seq. PLoS ONE 2016, 11, e0154531. [Google Scholar] [CrossRef] [PubMed]
- Todd, H.; Galea, G.L.; Meakin, L.B.; Delisser, P.J.; Lanyon, L.E.; Windahl, S.H.; Price, J.S. Wnt16 Is Associated with Age-Related Bone Loss and Estrogen Withdrawal in Murine Bone. PLoS ONE 2015, 10, e0140260. [Google Scholar] [CrossRef] [PubMed]
- Binet, R.; Ythier, D.; Robles, A.I.; Collado, M.; Larrieu, D.; Fonti, C.; Brambilla, E.; Brambilla, C.; Serrano, M.; Harris, C.C.; et al. WNT16B Is a New Marker of Cellular Senescence That Regulates p53 Activity and the Phosphoinositide 3-Kinase/AKT Pathway. Cancer Res. 2009, 69, 9183–9191. [Google Scholar] [CrossRef] [PubMed]
- Sola-Carvajal, A.; Revêchon, G.; Helgadottir, H.T.; Whisenant, D.; Hagblom, R.; Döhla, J.; Katajisto, P.; Brodin, D.; Fagerström-Billai, F.; Viceconte, N.; et al. Accumulation of Progerin Affects the Symmetry of Cell Division and Is Associated with Impaired Wnt Signaling and the Mislocalization of Nuclear Envelope Proteins. J. Investig. Dermatol. 2019, 139, 2272–2280.e2212. [Google Scholar] [CrossRef]
- Hirose, M.; Schilf, P.; Lange, F.; Mayer, J.; Reichart, G.; Maity, P.; Jöhren, O.; Schwaninger, M.; Scharffetter-Kochanek, K.; Sina, C.; et al. Uncoupling protein 2 protects mice from aging. Mitochondrion 2016, 30, 42–50. [Google Scholar] [CrossRef]
- Tian, X.Y.; Ma, S.; Tse, G.; Wong, W.T.; Huang, Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front. Physiol. 2018, 9, 1060. [Google Scholar] [CrossRef]
- Kukat, A.; Dogan, S.A.; Edgar, D.; Mourier, A.; Jacoby, C.; Maiti, P.; Mauer, J.; Becker, C.; Senft, K.; Wibom, R.; et al. Loss of UCP2 Attenuates Mitochondrial Dysfunction without Altering ROS Production and Uncoupling Activity. PLOS Genet. 2014, 10, e1004385. [Google Scholar] [CrossRef]
- Lopez-Mejia, I.C.; de Toledo, M.; Chavey, C.; Lapasset, L.; Cavelier, P.; Lopez-Herrera, C.; Chebli, K.; Fort, P.; Beranger, G.; Fajas, L.; et al. Antagonistic functions of LMNA isoforms in energy expenditure and lifespan. EMBO Rep. 2014, 15, 529–539. [Google Scholar] [CrossRef]
- van den Beld, A.; Carlson, O.; Doyle, M.E.; Rizopoulos, D.; Ferrucci, L.; Van der Lely, A.J.; Egan, J. IGFBP-2 And Aging; A 20 Year Longitudinal Study on IGFBP-2, IGF-I, BMI, Insulin Sensitivity and Mortality in an Aging Population. Eur. J. Endocrinol. 2018, EJE-18-0422. [Google Scholar] [CrossRef]
- Sisu, C. Pseudogenes as Biomarkers and Therapeutic Targets in Human Cancers. Methods Mol Biol 2021, 2324, 319–337. [Google Scholar] [CrossRef]
- Salmena, L. Pseudogenes: Four Decades of Discovery. Methods Mol Biol 2021, 2324, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Dangwal, S.; Schimmel, K.; Foinquinos, A.; Xiao, K.; Thum, T. Noncoding RNAs in Heart Failure. In Heart Failure; Bauersachs, J., Butler, J., Sandner, P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 423–445. [Google Scholar]
- Hinkel, R.; Batkai, S.; Bähr, A.; Bozoglu, T.; Straub, S.; Borchert, T.; Viereck, J.; Howe, A.; Hornaschewitz, N.; Oberberger, L.; et al. AntimiR-132 Attenuates Myocardial Hypertrophy in an Animal Model of Percutaneous Aortic Constriction. J. Am. Coll. Cardiol. 2021, 77, 2923–2935. [Google Scholar] [CrossRef] [PubMed]
- Gerasymchuk, M.; Cherkasova, V.; Kovalchuk, O.; Kovalchuk, I. The Role of microRNAs in Organismal and Skin Aging. Int. J. Mol. Sci. 2020, 21, 5281. [Google Scholar] [CrossRef] [PubMed]
- ElSharawy, A.; Keller, A.; Flachsbart, F.; Wendschlag, A.; Jacobs, G.; Kefer, N.; Brefort, T.; Leidinger, P.; Backes, C.; Meese, E.; et al. Genome-wide miRNA signatures of human longevity. Aging Cell 2012, 11, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Bonafè, M.; Spazzafumo, L.; Gobbi, M.; Prattichizzo, F.; Recchioni, R.; Marcheselli, F.; La Sala, L.; Galeazzi, R.; Rippo, M.R.; et al. Age- and glycemia-related miR-126-3p levels in plasma and endothelial cells. Aging 2014, 6, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Bi, Q.; Liu, J.; Wang, X.; Sun, F. Downregulation of miR-27b promotes skin wound healing in a rat model of scald burn by promoting fibroblast proliferation. Exp Med 2020, 20, 63. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Jinnin, M.; Wang, Z.; Hirano, A.; Tomizawa, Y.; Kira, T.; Igata, T.; Masuguchi, S.; Fukushima, S.; Ihn, H. The expression of miR-124 increases in aged skin to cause cell senescence and it decreases in squamous cell carcinoma. BioScience Trends 2016, 10, 454–459. [Google Scholar] [CrossRef]
- Yuan, F.; Chen, H.; Hu, P.; Su, P.; Guan, X. MiR-26a regulates the expression of serum IGF-1 in patients with osteoporosis and its effect on proliferation and apoptosis of mouse chondrocytes. J. Musculoskelet. Neuronal. Interact. 2021, 21, 298–307. [Google Scholar]
- Hu, D.; Pawlikowska, L.; Kanaya, A.; Hsueh, W.-C.; Colbert, L.; Newman, A.B.; Satterfield, S.; Rosen, C.; Cummings, S.R.; Harris, T.B.; et al. Serum Insulin-Like Growth Factor-1 Binding Proteins 1 and 2 and Mortality in Older Adults: The Health, Aging, and Body Composition Study. J. Am. Geriatr. Soc. 2009, 57, 1213–1218. [Google Scholar] [CrossRef]
- Kooijman, R. Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev. 2006, 17, 305–323. [Google Scholar] [CrossRef]
- Kemp, M.G.; Spandau, D.F.; Travers, J.B. Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin. Molecules 2017, 22, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.A.; Travers, J.B.; Machado, C.; Somani, A.-K.; Spandau, D.F. Reversing the aging stromal phenotype prevents carcinoma initiation. Aging 2011, 3, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Travers, J.B.; Somani, A.K.; Spandau, D.F. The IGF-1/IGF-1R signaling axis in the skin: A new role for the dermis in aging-associated skin cancer. Oncogene 2010, 29, 1475–1485. [Google Scholar] [CrossRef]
- Hruza, L.L.; Pentland, A.P. Mechanisms of UV-induced inflammation. J Invest Derm. 1993, 100, 35s–41s. [Google Scholar] [CrossRef] [PubMed]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [PubMed]
- Luciano-Mateo, F.; Cabré, N.; Baiges-Gaya, G.; Fernández-Arroyo, S.; Hernández-Aguilera, A.; Elisabet Rodríguez-Tomàs, E.; Arenas, M.; Camps, J.; Menéndez, J.A.; Joven, J. Systemic overexpression of C-C motif chemokine ligand 2 promotes metabolic dysregulation and premature death in mice with accelerated aging. Aging 2020, 12, 20001–20023. [Google Scholar] [CrossRef]
- Schweickart, V.L.; Epp, A.; Raport, C.J.; Gray, P.W. CCR11 Is a Functional Receptor for the Monocyte Chemoattractant Protein Family of Chemokines. J. Biol. Chem. 2000, 275, 9550–9556. [Google Scholar] [CrossRef]
- Jiao, S.; Meng, F.; Zhang, J.; Yang, X.; Zheng, X.; Wang, L. STAT1 mediates cellular senescence induced by angiotensin II and H2O2 in human glomerular mesangial cells. Mol. Cell. Biochem. 2012, 365, 9–17. [Google Scholar] [CrossRef]
- Kreienkamp, R.; Graziano, S.; Coll-Bonfill, N.; Bedia-Diaz, G.; Cybulla, E.; Vindigni, A.; Dorsett, D.; Kubben, N.; Batista, L.F.Z.; Gonzalo, S. A Cell-Intrinsic Interferon-like Response Links Replication Stress to Cellular Aging Caused by Progerin. Cell Rep. 2018, 22, 2006–2015. [Google Scholar] [CrossRef]
- Breitenbach, T.; Lorenz, K.; Dandekar, T. How to Steer and Control ERK and the ERK Signaling Cascade Exemplified by Looking at Cardiac Insufficiency. Int. J. Mol. Sci. 2019, 20, 2179. [Google Scholar] [CrossRef]
- Baker, M. Gene data to hit milestone. Nature 2012, 487, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Kodama, K.; Horikoshi, M.; Toda, K.; Yamada, S.; Hara, K.; Irie, J.; Sirota, M.; Morgan, A.A.; Chen, R.; Ohtsu, H.; et al. Expression-based genome-wide association study links the receptor CD44 in adipose tissue with type 2 diabetes. Proc. Natl. Acad. Sci. USA 2012, 109, 7049–7054. [Google Scholar] [CrossRef] [PubMed]
- Maleki, F.; Ovens, K.; McQuillan, I.; Kusalik, A.J. Size matters: How sample size affects the reproducibility and specificity of gene set analysis. Hum. Genom. 2019, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M.; on behalf of the International Nucleotide Sequence Database. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Vitting-Seerup, K.; Sandelin, A. IsoformSwitchAnalyzeR: Analysis of changes in genome-wide patterns of alternative splicing and its functional consequences. Bioinformatics 2019, 35, 4469–4471. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 29 June 2022).
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Title | Focus of the Study | Ref. |
|---|---|---|
| Progeria and Aging—Omics Based Comparative Analysis |
| [our study] |
| Epigenetic deregulation of lamina-associated domains in Hutchinson-Gilford progeria syndrome |
| [21] |
| Phosphorylated Lamin A/C in the Nuclear Interior Binds Active Enhancers Associated with Abnormal Transcription in Progeria |
| [22] |
| Prevalent intron retention fine-tunes gene expression and contributes to cellular senescence |
| [23] |
| Analysis of transcriptional modules during human fibroblast ageing |
| [24] |
| Repetitive elements as a tran-scriptomic marker of aging: Ev-idence in multiple datasets and models |
| [25] |
| Altered Chromatin States Drive Cryptic Transcription in Aging Mammalian Stem Cells |
| [26] |
| Extremes of age are associated with differences in the expression of selected pattern recognition receptor genes and ACE2, the receptor for SARS-CoV-2: implications for the epidemiology of COVID-19 disease |
| [27] |
| Multi-omic rejuvenation of human cells by maturation phase transient reprogramming |
| [28] |
| BiT age: A transcriptome-based aging clock near the theoretical limit of accuracy |
| [29] |
| Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity |
| [30] |
| mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations |
| [31] |
| An integrated pipeline for mammalian genetic screening |
| [32] |
| Landscape of adenosine-to-inosine RNA recoding across human tissues |
| [33] |
| Predicting age from the transcriptome of human dermal fibroblasts |
| [19] |
| Gene Name | Description | Reported Tissue Proteomics | Ref. | |
|---|---|---|---|---|
| Aging Proteomics 1 | IGFBP2 | insulin like growth factor binding protein 2 | Plasma, monocytes, macrophages and precursors | [90,93,94,95] |
| STAT1 | signal transducer and activator of transcription 1 | Plasma, liver | [90,93,96,97] | |
| TFPI | tissue factor pathway inhibitor | Plasma | [90,94,96] | |
| KRT18 | keratin 18 | Plasma, liver | [90,96,97,98] | |
| CCL2 | C-C motif chemokine ligand 2 | Plasma | [90,99,100,101] | |
| IGF1 | insulin like growth factor 1 | Plasma, cerebrospinal fluid | [90,96,102] | |
| HGF | hepatocyte growth factor | Plasma, cerebrospinal fluid | [90,93,99,100,102,103] | |
| MSR1 | macrophage scavenger receptor 1 | Plasma | [90,93,96] | |
| EFEMP1 | EGF containing fibulin extracellular matrix protein 1 | Plasma, urine | [90,93,94,104,105] | |
| GDF5 | growth differentiation factor 5 | Plasma, cerebrospinal fluid | [90,96,102] | |
| KDR | kinase insert domain receptor | Plasma | [90,93,96] | |
| FST | follistatin | Plasma | [90,93,99] | |
| SECTM1 | secreted and transmembrane 1 | Plasma | [90,94,96] | |
| HS3ST3A1 | heparan sulfate-glucosamine 3-sulfotransferase 3A1 | Plasma | [90,93,96] | |
| SPINT2 | serine peptidase inhibitor, Kunitz type 2 | Plasma, cerebrospinal fluid | [90,93,96,106] | |
| Aging Proteomics 2 | IGFBP2 | insulin like growth factor binding protein 2 | Plasma, monocytes, macrophages and precursors | [90,91,93,94,95] |
| STAT1 | signal transducer and activator of transcription 1 | Plasma, liver | [90,91,93,96,97] | |
| TFPI | tissue factor pathway inhibitor | Plasma | [90,91,94,96] | |
| Proteomics Fibroblasts | Wnt5A | Wnt family member 5A | Fibroblasts | [92] |
| Gene | Observed in … Proteomics Studies (Johnson et al.) | Tendency Observed in RNA-Seq Progeria (Mateos et al.) | Tendency Observed in iTRAQ Progeria (Mateos et al.) | Tendency Observed in Our Study (Aging) | Tendency Observed in Our Study (Progeria) |
|---|---|---|---|---|---|
| IGFBP2 | 3 | up | up | up | up |
| IGF1 | 2 | down | - | down | down |
| WNT16 | - | up | - | up | up |
| UCP2 | - | up | - | down | up |
| ACKR4 | - | - | - | up | down |
| CCL2 | 2 | up | - | up | up |
| KRT8 | - | up | - | down | up |
| KRT18 | 3 | up | - | down | up |
| ADAMTS15 | - | up | - | down | up |
| ACTN4P1 | - | - | - | down | up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caliskan, A.; Crouch, S.A.W.; Giddins, S.; Dandekar, T.; Dangwal, S. Progeria and Aging—Omics Based Comparative Analysis. Biomedicines 2022, 10, 2440. https://doi.org/10.3390/biomedicines10102440
Caliskan A, Crouch SAW, Giddins S, Dandekar T, Dangwal S. Progeria and Aging—Omics Based Comparative Analysis. Biomedicines. 2022; 10(10):2440. https://doi.org/10.3390/biomedicines10102440
Chicago/Turabian StyleCaliskan, Aylin, Samantha A. W. Crouch, Sara Giddins, Thomas Dandekar, and Seema Dangwal. 2022. "Progeria and Aging—Omics Based Comparative Analysis" Biomedicines 10, no. 10: 2440. https://doi.org/10.3390/biomedicines10102440
APA StyleCaliskan, A., Crouch, S. A. W., Giddins, S., Dandekar, T., & Dangwal, S. (2022). Progeria and Aging—Omics Based Comparative Analysis. Biomedicines, 10(10), 2440. https://doi.org/10.3390/biomedicines10102440