Abstract
CYP3A is an enzyme subfamily in the cytochrome P450 (CYP) superfamily and includes isoforms CYP3A4, CYP3A5, CYP3A7, and CYP3A43. CYP3A enzymes are indiscriminate toward substrates and are unique in that these enzymes metabolize both endogenous compounds and diverse xenobiotics (including drugs); almost the only common characteristic of these compounds is lipophilicity and a relatively large molecular weight. CYP3A enzymes are widely expressed in human organs and tissues, and consequences of these enzymes’ activities play a major role both in normal regulation of physiological levels of endogenous compounds and in various pathological conditions. This review addresses these aspects of regulation of CYP3A enzymes under physiological conditions and their involvement in the initiation and progression of diseases.
1. Introduction
The CYP3A subfamily is affiliated with the cytochrome P450 (CYP) superfamily, which represents monooxygenases that catalyze the breakdown of various substances via hydroxylation and epoxidation with the participation of an electron donor (NADPH) and molecular oxygen [1]. CYP enzymes function as the first line of defense against exogenous chemical agents [2]. CYP enzymes are responsible for approximately three-quarters of all drug metabolism reactions in the human body [3,4]. CYP enzymes are involved in many critical metabolic reactions, including the metabolism of steroid hormones, bile acids, polyunsaturated fatty acids, leukotrienes, and eicosanoids [3].
Genes of CYP enzymes have been found in the genetic material of representatives of all kingdoms of living organisms, including plants. There are 57 known functional CYP genes in the human genome, aside from 58 pseudogenes whose protein products are enzymes metabolizing a wide range of endogenous and exogenous chemical compounds [2,5,6]. The genes of CYP enzymes are categorized into 18 families and 43 subfamilies based on the percentage of amino acid sequence homology. Just 3 families—CYP2, CYP3, and CYP4—contain more genes than the other 15 families combined [3,7]. The human CYP3 family consists of a single subfamily, CYP3A, which contains four genes (CYP3A4, CYP3A5, CYP3A7, and CYP3A43) encoding four functional enzymes [5,6,8,9,10].
CYP3A is a major subfamily in the cytochrome P450 superfamily. CYP3A enzymes are involved in the metabolism of more than 30% [11] and according to other reports 45–60% [12,13] of all pharmaceutical drugs currently on the market. CYP3A enzymes also metabolize some endogenous substrates, including hormones and bile acids, as well as nonpharmaceutical xenobiotics [11,12,14].
Expression of CYP3A enzymes is regulated and varies under the influence of various exogenous (drugs, chemicals, and diets) and endogenous factors (fatty acids, hormones, cytokines, and microRNAs [miRs or miRNAs]) [11].
CYP3A enzymes’ activity can be influenced by anthropogenic environmental chemicals: organophosphates, carbamates, parabens, benzotriazole UV stabilizers, and plasticizers [11,12]. Natural compounds present in foods—e.g., flavonoids found in fruits and vegetables, coffee, tea, chocolate, and wine—can alter CYP3A enzymes’ expression [15]. A prime example is the inhibition of CYP3A enzymes’ expression by components of grapefruit juice [12,16]. There is experimental evidence that retinoids can regulate the expression of CYP3A genes [17]. Certain diets, such as high-fat diets, can alter the expression of CYP3A genes [18], and it is likely that human dietary habits can affect basal expression of these genes [11].
Many of these substances are in turn metabolized by induced CYP3A enzymes, and this feedback mechanism implements detoxification of potentially harmful compounds [12].
Members of the CYP3A Subfamily: Localization of Genes in the Genome and of Enzymes in Tissues of the Body
To date, four functional enzymes belonging to the CYP3A subfamily have been identified in humans: CYP3A4, CYP3A5, CYP3A7, and CYP3A43 [5,6,8,9]. This subfamily is encoded by a 231 kbp cluster of four CYP3A genes in chromosomal region 7q21.1 (CYP3A4, CYP3A5, CYP3A7, and CYP3A43) and of several pseudogenes (Figure 1) [10,19,20,21]. Each gene contains 13 exons with conserved exon–intron boundaries [10,19]. The organization of the CYP3A locus indicates that it has arisen from duplications of an ancestral CYP3A cassette [12].

Figure 1.
An outline of the human cytochrome P450 3A (CYP3A) locus on chromosome 7. Arrows indicate the orientation of open reading frames of genes. P: CYP3A pseudogenes [22].
According to mass spectrometric analysis, proportions of proteins CYP3A4, CYP3A5, CYP3A7, and CYP3A43 in the total amount of CYP3A proteins are on average 85.4%, 5.4%, 3.4% and 5.8%, respectively [23]. CYP3A isoenzymes are expressed mainly in the liver and small intestine as well as in the kidneys, adrenal glands, lungs, brain, prostate, testes, placenta, pancreas, and skeletal muscles (Table 1) [5,6,8,9,11,14,20,24,25,26]. Expression of CYP3A enzymes in enterocytes is comparable to or may even exceed that in hepatocytes [12]. It is believed that CYP3A isoenzymes taken together constitute most of the protein amount of CYP enzymes in the liver and small intestine, and CYP3A4 accounts for the bulk of the CYP3A protein amount [12,23].
In the CYP3A subfamily, CYP3A4 is the major member participating in drug metabolism and is the predominant form of CYP3A in the liver (10–50%) and small intestine (40%) of adult humans. CYP3A4 shows the largest interindividual differences, by a factor of several tens to hundreds, in terms of mRNA and protein expression in the liver [23,27].
CYP3A4 in the liver is expressed more weakly in women than in men, as reported in ref. [28] or more strongly than in men, according to another report [29]. CYP3A4 is also expressed in the esophagus, duodenum, and colon (Table 1) [26]. Fetal CYP3A4 protein levels are extremely low in the first trimester but increase rapidly in the second and third trimesters of pregnancy [14]. CYP3A4 mRNA expression shows 10-fold variation and increases with age after conception [30,31].
CYP3A5 is the most abundant and best studied among minor isoforms of CYP3A [6,8]. Unlike CYP3A4, functional CYP3A5 is expressed in approximately 70% of Africans and Afro-Americans and only in 20% of Eurasians [8,12,13]. Relatively large amounts of CYP3A5 are found in the intestines, kidneys, adrenal glands, prostate, and lungs [9,25]. CYP3A5 is the predominant CYP3A protein in human kidneys, lungs, blood, and pituitary and is also present in liver and intestinal tissues [5,26]. CYP3A5 also shows interindividual differences—from severalfold to hundreds of times—in terms of protein expression in the liver [23,32,33].
CYP3A5 expression is detectable in enterocytes in approximately 70% of adults [12]. CYP3A5 is expressed during the secretory phase in the endometrium, while CYP3A4 and CYP3A43 expression in this tissue is repressed by estrogen [28]. In the lungs, CYP3A5 is most abundant in bronchial and alveolar epithelia, bronchial glands, and alveolar macrophages [9]. CYP3A5 expression appears to vary throughout development [6].
CYP3A7 and CYP3A43 are underexpressed as compared to CYP3A4/5 [6,8]. Protein expression of CYP3A43 in human liver microsomes is ~15 times lower than that of CYP3A4 [23].
CYP3A7, a predominantly fetoplacental enzyme, is highly expressed in the liver and intestines of the embryo and fetus as well as in the endometrium and placenta, although it is detectable in the liver and small intestine of some adults [5,6,8,12,26,34]. Expression of CYP3A7 varies several-hundred-fold among individuals [30]. CYP3A7 is highly expressed during the first trimester of pregnancy and then its expression gradually declines [14].
Among the CYP3A genes, CYP3A43 was the last to be identified. CYP3A43 expression is highest in the prostate (the organ with intensive metabolism of steroids) and in the brain: 170 times higher than CYP3A4 expression [35]. CYP3A43 is also found in the testes, liver, kidneys, placenta, and pancreas [5,6,8,9,14,20,24]. In the liver, CYP3A43 mRNA levels vary up to 1000-fold among whites [14].

Table 1.
Hepatic and extrahepatic expression profiles of human CYP3A4 and CYP3A5.
Table 1.
Hepatic and extrahepatic expression profiles of human CYP3A4 and CYP3A5.
| CYP3A4 | CYP3A5 | |||
|---|---|---|---|---|
| Internal | ||||
| Liver | Abundance 68–155 pmol/mg RNA sequencing (RNA-seq): very high expression Microarray and RNA-seq: over-expressed | [23,36,37,38,39,40,41,42] [43] [44] | Abundance 2–5 (CYP3A5*3 allele) or 60–291 (CYP3A5*1 allele) pmol/mg Microarray and RNA-seq: overex-pressed RNA-seq: high expression | [45] [44] [43] |
| Small intestine | RNA detected by real-time PCR; protein detected; enzyme activity detected | [46,47,48] | RNA detected by real-time PCR; protein detected; enzyme activity detected | [41,47,48] |
| Microarray and RNA-seq: overexpressed | [44] | Microarray and RNA-seq: overexpressed | [44] | |
| RNA-seq: very high expression | [43] | RNA-seq: high expression | [43] | |
| Duodenum | RNA-seq: very high expression | [43] | RNA-seq: high expression | [43] |
| Colon | Microarray and RNA-seq: detected RNA-seq: low expression | [44] [43] | Microarray and RNA-seq: detected RNA-seq: high expression | [44] [43] |
| Esophagus | RNA-seq detected RNA-seq: low expression | [44] [43] | RNA-seq detected RNA-seq: moderate expression | [44] [43] |
| Stomach | RNA-seq detected RNA-seq: low expression | [44] [43] | RNA-seq detected RNA-seq: high expression | [44] [43] |
| Gall bladder | RNA-seq: low expression | [43] | RNA-seq: high expression | [43] |
| Kidney | RNA detected by real-time PCR; protein detected; enzyme activity detected | [43,44,46,49] | RNA detected by real-time PCR; protein detected; enzyme activity detected | [46,49] |
| Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: low expression | [43] | RNA-seq: moderate expression | [43] | |
| Lung | RNA detected by real-time PCR | [46] | RNA detected by real-time PCR | [46] |
| Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: low expression | [43] | RNA-seq: moderate expression | [43] | |
| Adipocyte, adipose tissue | Microarray and RNA-seq: detected RNA-seq: low expression | [44] [43] | Microarray and RNA-seq: detected RNA-seq: low expression | [44] [43] |
| Spleen | RNA-seq detected RNA-seq: extremely low expression | [44] [43] | RNA-seq detected RNA-seq: low expression | [44] [43] |
| Bladder | RNA-seq: low expression | [43] | RNA-seq: moderate expression | [43] |
| Secretory | ||||
| Pancreas | RNA-seq detected RNA-seq: low expression | [44] [43] | RNA-seq: moderate expression | [43] |
| Adrenal gland | RNA detected by real-time PCR | [46] | RNA detected by real-time PCR | [46] |
| Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: moderate expression | [43] | RNA-seq: moderate expression | [43] | |
| Pituitary | RNA-seq detected | [44] | RNA-seq detected | [44] |
| Thyroid gland | Microarray and RNA-seq: detected RNA-seq: extremely low expression | [44] [43] | Microarray and RNA-seq: detected RNA-seq: low expression | [44] [43] |
| Salivary gland | Microarray and RNA-seq: detected RNA-seq: extremely low expression | [44] [43] | Microarray and RNA-seq: detected RNA-seq: low expression | [44] [43] |
| Breast | RNA-seq detected | [44] | RNA-seq detected | [44] |
| Skin/keratinocytes | RNA detected by real-time PCR Microarray and RNA-seq: detected RNA-seq: low expression | [50] [44] [43] | Microarray and RNA-seq: detected RNA-seq: high expression | [44] [43] |
| Nervous | ||||
| Brain (cortex) | Not detectable | [51] | RNA detected by real-time PCR; protein detected | [52,53] |
| RNA-seq detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: extremely low expression | [43] | RNA-seq: low expression | [43] | |
| Cerebellum | Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] |
| Retina | Microarray: detected | [44] | Microarray: detected | [44] |
| Spinal cord | Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] |
| Tibial nerve | RNA-seq detected | [44] | RNA-seq detected | [44] |
| Muscle | ||||
| Heart | Not detectable | [54] | Not detectable | [54] |
| Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: extremely low expression | [43] | RNA-seq: low expression | [43] | |
| Artery | RNA-seq detected | [44] | RNA-seq detected | [44] |
| Smooth muscle | Microarray: detected | [44] | Microarray: detected | [44] |
| Skeletal muscle | Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] |
| Reproductive | ||||
| Ovary | Not quantifiable | [43] | RNA detected by real-time PCR | [46] |
| Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: low expression | [43] | RNA-seq: low expression | [43] | |
| Uterus | Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] |
| Endometrium | RNA-seq: low expression | [43] | RNA-seq: moderate expression | [43] |
| Placenta | Not detectable | [46] | RNA detected by real-time PCR | [46] |
| Microarray and RNA-seq: detected | [44] | Microarray: detected | [44] | |
| RNA-seq: not detectable | [43] | RNA-seq: low expression | [43] | |
| Prostate | RNA detected by real-time PCR | [46] | RNA detected by real-time PCR | [46] |
| Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: low expression | [43] | RNA-seq: moderate expression | [43] | |
| Testis | RNA detected by real-time PCR | [46] | RNA detected by real-time PCR | [46] |
| Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] | |
| RNA-seq: low expression | [43] | RNA-seq: low expression | [43] | |
| Immune | ||||
| Lymph node | Microarray: detected RNA-seq: low expression | [44] [43] | Microarray and RNA-seq: detected RNA-seq: low expression | [44] [43] |
| Bone marrow | Microarray: detected RNA-seq: extremely low expression | [44] [43] | Microarray: detected RNA-seq: low expression | [44] [43] |
| Whole blood | Microarray and RNA-seq: detected | [44] | Microarray and RNA-seq: detected | [44] |
| White blood cells | Not detected | [44] | RNA-seq: detected | [44] |
| Thymus | Microarray: detected | [44] | Microarray: detected | [44] |
| Appendix | RNA-seq: low expression | [43] | RNA-seq: moderate expression | [43] |
2. Mechanisms of CYP3A Regulation
2.1. Constitutive Regulation of CYP3A4 Transcription
Constitutive regulation of CYP3A4 transcription, both positive and negative, is mediated by hepatocyte nuclear factor 4α (HNF4α) and other hepatic transcription factors, including HNF1α and HNF3γ [55,56,57,58,59], CCAAT/enhancer-binding proteins alpha and beta (C/EBPα and C/EBPβ), and upstream transcription factor 1 (USF1) [10,60] via binding to three major cis-acting modules: constitutive liver enhancer module 4 (CLEM4) (positions −11.4 to −10.5 kbp), the distal xenobiotic-responsive enhancer module (XREM) (−7.2 to −7.8 kbp) and the proximal promoter (prP) (Figure 2) [10].
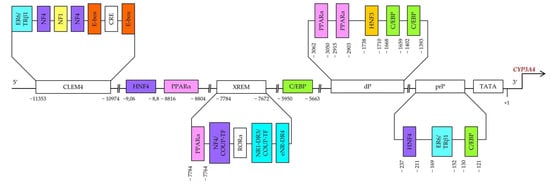
Figure 2.
Binding sites in the upstream part of CYP3A4. prP: proximal promoter, dP: distal promoter, XREM: a distal element called xenobiotic-responsive enhancer module, CLEM4: a distal element called constitutive liver enhancer module 4.
When bound to DNA, HNF4α attracts transcription coactivators and other accessory proteins and positively regulates the expression of target genes. In the liver, HNF4α is located exclusively in the nucleus and regulates the constitutive expression of a large number of target genes, including CYP3A4 [61].
HNF4α is required for the active epigenetic state of the enhancers that have been shown to increase gene transcription in mouse hepatocytes. In hepatocytes, HNF4α binds almost exclusively to active enhancers marked by histone modifications [lysine 4 histone 3 monomethylation (H3K4me1) and histone 3 lysine 27 acetylation (H3K27ac)] and DNA hydroxymethylation (5hmC), indicating a major role for HNF4α in transcription activation. Mice lacking HNF4α protein in hepatocytes show a decrease in the amounts of both H3K27ac and 5hmC in HNF4α-associated DNA regions. In terms of the mechanism, when bound to HNF4α, 5hmC requires an interaction of this protein with a ten-eleven translocation protein 3 (TET3) responsible for the oxidation of 5mC to 5hmC. Moreover, HNF4α regulates TET3 expression in the liver by direct binding to an enhancer region [62].
Functional binding sites for HNF4α [direct repeat 1 (DR1)] are located in an XREM spanning positions −7783 to −7771 bp [57], in the far distal region at −9.06/−8.8 kbp, and in the proximal promoter region at the −237/−211 bp site [58]. HNF4α determines pregnane X receptor (PXR)-mediated and constitutive androstane receptor (CAR)-mediated induction of CYP3A4 by xenobiotics. Disruption of the HNF4α-binding site in XREM has been found to reduce basal and inducible CYP3A4 expression in mice [57].
A more recent study on primary human hepatocytes and on the HepG2 cell line revealed that selective disruption of the DR1 element in XREM causes only minor changes in the level of CYP3A4 transactivation [63]. Furthermore, the relevance of this HNF4α-binding site has been further questioned by research article indicating that the XREM region does not promote HNF4α activation in the context of the natural 5′ regulatory upstream region of CYP3A4.
It has been shown that an interaction of two regions (at −9.06/−8.8 kbp and −237/−211 bp) is required for maximal expression activation by HNF4α. The effect of HNF4α is counteracted by chicken ovalbumin upstream promoter transcription factor (COUP-TF) II upstream of the promoter; this protein binds to one of the DR1 motifs. Furthermore, the activation of CYP3A4 via the DR1 element in the proximal promoter is dependent on an additional factor that binds near position −189 bp. Physiological significance of this position for HNF4α activation in vivo is supported by the presence of binding activity in the small intestine similar to that in LS174T cells. These results support the hypothesis that HNF4α directly regulates basal CYP3A4 expression, at least in the gut [58].
Hepatocyte nuclear factor 4α antisense RNA 1 (HNF4A-AS1), which is a long noncoding RNA (lncRNA), is reported to be a negative regulator of basal and rifampicin-induced expression of nuclear receptors and downstream P450 enzymes. In Huh7 cells, a knockdown of HNF4A-AS1 results in increased expression of HNF4α, PXR, and P450 enzymes (including CYP3A4) both at baseline and in the context of rifampicin-induced expression. By contrast, overexpression of HNF4A-AS1 decreases baseline expression of CAR, aryl hydrocarbon receptor, PXR, and some P450 enzymes. Of note, substantially attenuated induction of PXR, CYP1A2, CYP2C8, CYP2C19, and CYP3A4 by rifampicin was also observed in Huh7 cells transfected with an HNF4A-AS1–expressing plasmid. In addition, negative feedback of HNF4α on HNF4A-AS1–mediated gene expression was confirmed by a loss-of-function experiment. The CYP3A4 promoter enhances the expression of CYP3A4 after the HNF4A-AS1 knockdown. In general, histone modifications promote downregulation of nuclear receptors and some P450 enzymes by HNF4A-AS1 at basal and drug-induced levels [64].
The region spanning positions from −11.4 to −10.5 kbp (CLEM4) plays an important part in constitutive activation of the CYP3A4 gene. HNF1α, HNF4α, USF1, and activating protein-1 (AP-1) have been demonstrated to interact with CLEM4. Additionally, introduction of mutations into their binding sites showed that almost all these sites are required for maximal enhancer activity [65]. HNF3 may facilitate the association of other transcription factors with their binding sites through chromatin remodeling [66].
C/EBPs are key transcription factors involved in constitutive expression of several cytochrome P450 genes in the liver [67]. Genes C/EBPα and C/EBPβ can produce several N-terminally truncated isoforms of the protein as a result of post-transcriptional mechanisms [68]. CYP3A4 is modulated by the following factors: C/EBPα, a C/EBPβ isoform called liver-enriched transcriptional activator protein (LAP, ~35 kDa), and a C/EBPβ isoform called liver-enriched transcriptional inhibitory protein (LIP, ~20 kDa) [69,70].
LAP is a transcriptional activator in many genetic systems, whereas LIP is considered a functional antagonist of LAP. Because the low-molecular-weight LIP isoform lacks most of the transactivation domain but contains DNA-binding domains and dimerization domains, it has been suggested that it acts as a dominant-negative regulator of the full-length C/EBP protein and of LAP [71,72,73]. The LAP/LIP ratio controls constitutive and inducible expression of CYP3A4 and may contribute to various CYP3A4 phenotypes in the human population [60].
It has been demonstrated in HepG2 cells that functional C/EBP-binding sites are present in the proximal promoter of CYP3A4 at positions −121/−130 and in the distal promoter of CYP3A4 at positions −1393/−1402 and −1659/−1668 [59]. There is also a C/EBP-binding distal enhancer site between positions −5950 and −5663 bp in the 5′ flanking region of CYP3A4. A strong competitive effect between LAP and LIP has been found on a distal CYP3A4 sequence. C/EBP–LAP and LIP interact with a 288 bp distal enhancer site in the region near −5.95 kbp and modulate CYP3A4 expression in hepatic and nonhepatic cells [60].
2.2. Regulation of CYP3A4 Transcription
CYP3A4 expression is modulated by various mechanisms involving nuclear receptors, hormones, xenobiotics, and signaling molecules. CYP3A4 is regulated by a large number of xenobiotics, including many drugs, endogenous compounds, and many hormones, such as triiodothyronine, dexamethasone, and growth hormone [74].
Xenobiotic- and endobiotic-mediated CYP3A4 induction is indirect and entails activation of such ligand-dependent nuclear receptors as PXR, CAR, VDR, glucocorticoid receptor (GR) α, estrogen receptor (ER) α, bile acid receptor (farnesoid X receptor; FXR), oxysterol receptor (liver X receptor; LXR), and peroxisome proliferator-activated receptor alpha (PPARα) [10,11,14,27,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89] as well as by binding to the three major cis-acting modules: CLEM4, distal XREM, and prPXRE (Figure 3) [10].
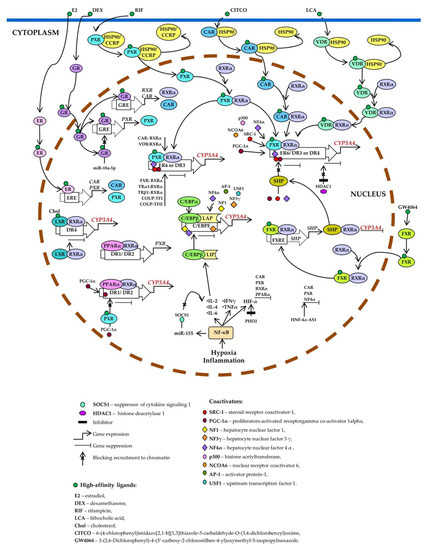
Figure 3.
Transcriptional regulation of CYP3A4. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and vitamin D receptor (VDR) control basal and inducible expression of CYP3A4 through competitive binding to the same set of response elements (everted repeats 6, ER6; direct repeats DR3, and DR4). PXR, CAR, or VDR unbound by a ligand is located in the cytoplasm as a complex with heat shock protein 90 (HSP90) or cytoplasmic constitutive active/androstane receptor retention protein (CCRP). When activated by a ligand, each of them forms a heterodimer with retinoid X receptor α (RXRα), relocates to the nucleus, binds to a response element, recruits coactivators, and activates CYP3A4 transcription. Estrogen receptor (ER) and glucocorticoid receptor (GR) raise CYP3A4 expression by enhancing the expression of CAR, RXRα, and PXR. Ligand-activated farnesoid X receptor (FXR) upregulates small heterodimer partner (SHP), which prevents the recruitment of coactivators to chromatin and/or forms heterodimers with RXRα, thereby inhibiting CYP3A4 expression. Histone deacetylase 1 (HDAC1) inhibition by carbamazepine downregulates CYP3A4. Liver X receptor (LXR) forms a heterodimer with RXRα that then binds to DR4 in the target gene, thus repressing its expression. After the binding of a ligand to LXR or RXR, the heterodimer changes its conformation, which leads to a release of corepressors and the recruitment of coactivators. This event causes transcription of a target gene (peroxisome proliferator-activated receptor alpha; PPARα), its protein product binds as a PPARα–RXRα heterodimer to motifs DR1 and DR2 and enhances the transcription of CYP3A4 and PXR. Ligand-activated PXR suppresses PPARα-dependent gene expression by inhibiting peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α) recruitment. Hypoxia and inflammation induce the activity of nuclear factor kappa B (NF-κB) and promote a release of the cytokines that increase the transcription of CCAAT enhancer-binding protein beta (C/EBPβ) and the translation of C/EBPβ-LIP mRNA. C/EBPβ-LIP competes with C/EBPα and C/EBPβ-LAP for binding to response elements in the promoter of CYP3A4, thus lowering its expression. NF-κB activates miR-155, which directly targets mRNAs of suppressors of cytokine signaling proteins (especially suppressor of cytokine signaling 1: SOCS1) thereby inhibiting obligatory negative feedback regulation of inflammatory responses. Abbreviations. C/EBPβ-LAP: a C/EBPβ isoform called liver-enriched activator protein; C/EBPβ-LIP: a C/EBPβ isoform called liver-enriched inhibitory protein; COUP-TFI: chicken ovalbumin upstream promoter transcription factor I; COUP-TFII: chicken ovalbumin upstream promoter transcription factor II; DR1, DR2, DR3, DR4, and ER6: AG(G/T)TCA-like direct repeats separated by 1, 2, 3, or 4 bases, respectively, and an inverted repeat separated by 6 bases; ERE: ER-responsive element; FXRE: FXR-responsive element, GRE: GR-responsive element; HIF-1α: hypoxia-inducible factor 1-α; HNF-4α-AS1: hepatocyte nuclear factor 4α-antisense-RNA 1; IL-2, -4, or -6: interleukin 2, 4 or 6; INFγ: interferon γ; PHD2: prolyl hydroxylase domain-containing protein 2; TNF: tumor necrosis factor; TRα1: thyroid hormone receptor-α1; TRβ1: thyroid hormone receptor-β1.
Nuclear receptors participating in the regulation of CYP3A4 share protein partners, ligands, DNA-sensing elements, and target genes, thereby forming a complex regulatory network by which the cell adapts to changes in its chemical environment [9,14].
PXR is considered the most important and critical factor that determines the activity and expression of the hepatic enzyme CYP3A4 [90,91]. PXR binds to the ligand and is then translocated into the nucleus. There, it heterodimerizes with the retinoid X receptor (RXR) and enhances CYP3A transcription by binding to AG(G/T) TCA-like direct repeats separated by 3 or 4 bases (DR3 and DR4, respectively) and to everted repeats separated by 6 bases (ER6) [90,91,92]. Heterodimer PXR–RXRα binds to ER6 in the proximal promoter [93], to DR3 in XREM [76], to ER6 in a far distal enhancer module [91], and to a DR4 motif [94,95].
RXR plays a central role in the regulation of many endobiotic and xenobiotic metabolic pathways and represents an additional node of control over PXR-mediated regulation of CYP3A4 (especially in RXR-deficient tissues [6]) by forming a heterodimer with numerous nuclear receptors for the modulation of enzymes.
CAR is a nuclear receptor known primarily to mediate CYP2B induction. PXR and CAR can be activated by the same sets of compounds such as phenobarbital and TCPOBOP [96,97,98]. Although PXR binds strongly to the DR4, DR3, or ER6 motif in the CYP3A4 promoter [99,100], CAR binds only weakly to the proximal ER6 motif in CYP3A4 [100,101], resulting in a preference of CAR for CYP2B6 over CYP3A4 in humans.
VDR is a member of the nuclear receptor superfamily of transcription regulators and mediates various biological effects of 1,25-dihydroxyvitamin D3 by modulating the transcription of target genes [102].
PXR, CAR, and VDR use the same set of binding sites in the 5′ regulatory region of CYP3A4 gene [12,14] and control CYP3A4 expression through competitive interaction with the same response elements in DNA (ER6, DR3, and DR4) [103]. In the absence of a ligand, CAR, DVR and PXR are located in the cytosol in complex with chaperones. After ligand binding, the PXR or CAR are released from the complex and translocated to the nucleus, where they form a heterodimer with RXRα. VDR is located in the cytosol of vitamin D–sensitive cells, and upon binding to 1,25(OH)2D3, the complex forms the VDR–RXR heterodimer, which relocates to the cell nucleus [12,14].
Aside from PXR, CAR, and VDR, RXRα forms heterodimers with other nuclear receptors that affect the expression and induction of CYP3A4. VDR, FXR, LXRα, CAR, and thyroid hormone receptors TRα1 and TRβ1 bind to PXR-binding sites ER6 in the proximal region and to DR3 in XREM. COUP-TFI, which is a nuclear receptor of steroid hormone receptors, binds as a homodimer to both PXR-specific motifs but preferentially binds to distal DR3. On the contrary, COUP-TFII is able to bind only as a homodimer to the distal element DR3. TRβ1 binds as a homodimer exclusively to element ER6 [104].
FXR is a primary bile acid receptor expressed in hepatic and intestinal tissues, where it regulates the uptake, metabolism, and removal of bile acids. In the absence of a ligand, FXR is in complex with a corepressor. After activation by the ligand, FXR forms a heterodimer with RXR and binds to the DNA sequence (FXR response element—FXRE) in the promoter of target genes and activates transcription [105].
There are two types of FXR’s participation in CYP3A4 expression. Bile acid–activated FXR induces CYP3A4 expression and phase I hydroxylation reactions (xenobiotic metabolism) [84,106,107]. On the other hand, FXR activation represses CYP3A4 expression by inducing FXR target genes that repress the activation of transcription: SHP (small heterodimeric partner) in the liver [108] and fibroblast growth factor in the small intestine [109].
LXRα and LXRβ (liver X-receptors α and β) form heterodimers with RXRα to function as transcriptional regulators [110]. Heterodimers LXR–RXRα bind to an LXR-responsive DNA element consisting of a direct repeat of the core AGGTCA sequence separated by four nucleotides (DR4), thereby enhancing target gene expression [111].The specific involvement of LRX in the activation or suppression of gene transcription depends on the type of cells and genes. In the absence of a ligand, the LXR heterodimer with RXR inhibits transcription due to the N-CoR corepressor (nuclear receptor corepressor) and SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) [110,111,112,113]. After ligand activation and dissociation with corepressors, coactivators are recruited and transcription is stimulated [114].
PPARα is an important transcription factor regulating genes encoding endo- and xenobiotic-metabolizing and lipid-metabolizing enzymes. As heterodimer PPARα–RXRα, PPARα binds to motifs DR1 and DR2 [direct repeats of the AG(G/T)TCA half-site separated by 1 and 2 nucleotides, respectively]. PPARα–RXRα specifically binds to three DR1-like motifs and one DR1/DR2 motif (PBR-I, -II, and -III) at positions −2915/−2903 and −3062/−3050 as well as −7784/−7764 and −8816/−8804 [79].
PPARα activates PXR expression by binding to the region −1514 to −1321 bp. Moreover, forced overexpression of ERα or GRα exerts a positive effect on the activity of a PXR reporter construct carrying a 2.2 kbp proximal promoter sequence [115,116,117].
Experimental data point to redundancy as well as cooperativity of at least three functional PPARα-dependent sites in an CYP3A4 enhancer that mediate both constitutive and inducible transactivation. Presumably, the binding regions upstream of CYP3A4 are permanently occupied by PPARα, and a cofactor environment determines whether the repression or activation of CYP3A4 transcription takes place after ligand binding. Therefore, the addition of a chemical ligand does not lead to an enhancement of the binding but rather to a greater release of corepressors and thus an increase in transcription [79].
Less is known about the regulation of the CYP3A5 gene, although it is likely that genes CYP3A4 and CYP3A5 share a common HNF4α-dependent pathway regulating constitutive expression [8,9,11,78]. Unlike CYP3A4, the CYP3A5 gene sequence does not contain a distal region of XREM (from position −7836 to −7208 bp), and this may be the reason for the difference in expression regulation between CYP3A4 and CYP3A5 [14].
It has been shown that CAR and PXR activate the CYP3A5 promoter in hepatocytes and intestines of carriers of allele CYP3A5*1 [118]. According to other data, basal expression of CYP3A5 in extrahepatic tissues (intestines, kidneys, lungs, and prostate) does not depend on PXR [11,118]. CYP3A5 is the main enzyme of the CYP3A subfamily in the lungs, and its induction in this organ, unlike in the liver, is triggered only by glucocorticoids (not by typical CYP3A inducers) [6,12]. In lung adenocarcinoma A549 cells, CYP3A5 is activated by glucocorticoids via a GR [9].
Expression of CYP3A5 in organs devoid of PXR appears to be mediated by the loss of a suppressive YY1-binding element in the CYP3A5 promoter [25]. YY1 is a ubiquitous transcription factor belonging to a class of proteins having clusters of GLI-Kruppel zinc finger domains taking part in the modulation of transcription of many genes; YY1 can function either as a transcriptional activator or as a repressor, as seems to be the case for CYP3A genes [25,119]. The loss of YY1-mediated transcriptional repression may contribute to the expansion of tissue specificity of CYP3A5 in the absence of induction. For example, the expression of CYP3A5 in the kidneys promotes salt and water retention mediated by cortisol metabolite 6β-hydroxycortisol, which arises under the action of CYP3A5; this effect can be useful in hot climates. This phenomenon may explain the high prevalence of polymorphism-dependent CYP3A5 expression in the kidneys of most Africans [25,120].
Nuclear receptors CAR and PXR play a key role in the induction of CYP3A7 [121]. When the proximal ER6 motif of CYP3A7 was compared with this motif of CYP3A4, two nucleotide mutations (−169G → −168T and −161A → −160T) were found, which may account for the reduced ability of complexes PXR–RXRα and VDR–RXRα to recognize and bind the CYP3A7 promoter and to activate this gene’s transcription [122]. In addition, compared to CYP3A4 and CYP3A5, the ligand-binding affinity and catalytic ability of CYP3A7 are significantly weaker due to reduced structural plasticity of the CYP3A7 protein [34].
There are virtually no data on the mechanisms of regulation of CYP3A43. Four cis-regulatory elements are known that control CYP3A4 transcription and interact with the CYP3A4 promoter, one of which (R2) overlaps with known enhancers CLEM4 or XREM, and deletion of R4 increases CYP3A4 expression while decreasing CYP3A43 expression. The single-nucleotide polymorphism (SNP) rs62471956 in the R4 region correlates with higher expression of CYP3A43 and lower expression of CYP3A4 [123].
Thus, most CYP3A inducers act through transcriptional activation [9,11,14]. CYP3A isoforms and nuclear receptors involved in their regulation are subject (as part of post-transcriptional regulation) to ubiquitination (CYP3A4 and CYP3A5) and phosphorylation (CYP3A4 and PXR) [11], whereas post-translational regulation of CYP3A enzymes consists of the stabilization of CYP3A mRNAs and proteins [6,11].
Molecular mechanisms of induction may differ among the four major human CYP3A genes and among their polymorphic variants owing to differences in their structure, and the mechanisms can also differ among different tissues, possibly because of different ratios of crucial protein factors. This complexity is a consequence of the wide range of CYP3A ligands and of nuclear receptors mediating the induction of CYP3A genes [9,14].
2.3. Negative Regulation of CYP3A Enzymes
Suppression of CYP3A genes is mediated by the activation of nuclear factor kappa B (NF-κB) [124]. In addition, tumor necrosis factor (TNF) attenuates PXR-mediated transcriptional activation induced by rifampicin in vitro [124,125]. Proteins p53, NF-κB, and C/EBP-LIP are involved in the repression of CYP3A4 gene activity [9]. NF-κB activation plays an important role in the suppression of CYP3A genes by disrupting the binding of the PXR–RXRα complex to DNA [126]. Cytokine-mediated downregulation of CYP3A4 during the inflammatory response via the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway is of great importance [10].
Major cytokines—interleukin (IL)-1β, IL-1α, IL-6, and TNF—downregulate genes CYP3A4 and CYP3A5 in human hepatocytes and Cyp3a genes in murine and rat hepatocytes [127,128,129]. The level of CYP3A4 mRNA is significantly reduced by IL-1β, IL-6, and TNF in a 3D culture of human hepatocarcinoma FLC-4 cells [128,130].
TNF attenuates PXR-mediated transcriptional activation induced by rifampicin. Induction of CYP3A4-luciferase activity of a cotransfected CYP3A4-luciferase construct and of a PXR expression plasmid by rifampicin is significantly weakened by TNF [131]. IL-6 appears to be a key factor for downregulation of the activity of CYP3A enzymes during inflammation [130,132,133]. It has been shown that IL-6 negatively regulates CYP3A4 with the participation of glycoprotein receptor gp130 and of the C/EBPβ signaling pathway [127].
Endoplasmic-reticulum stress is reported to decrease PXR expression and CYP3A4 induction in primary human hepatocytes and human HepG2 cells. Apparently, the reason is the suppression of HNF-4α and LAP during endoplasmic-reticulum stress, whereas overexpression of HNF-4α or LAP restores PXR expression. An analyzed sequence—located in the PXR promoter region 2 adjacent elements (recognized by HNF-4α and C/EBPs)—is also responsive to IL-6 (as detected via luciferase activity), indicating a functional relation between endoplasmic-reticulum stress and proinflammatory cytokine signaling [134].
Another mechanism of CYP genes’ suppression involving the serotonergic system has been described in a rat model of diethylnitrosamine-induced liver failure [135]. Namely, serotonergic-system dysfunction caused by a tryptophan-free diet lowered the levels of proinflammatory cytokines TGF-β and IL-1β but increased the amount of anti-inflammatory cytokine IL-4. Triggering of the repressive mechanism IL-4–JAK1–STAT6– suppressor of cytokine signaling 1 (SOCS1) in the JAK2–STAT5b-mediated signal transduction pathway and proline-rich, extensin-like receptor kinase (pERK)1/2–GR–STAT6 signal transduction pathway led to the repression of CYP2C11 and of CYP3A genes [135].
2.4. An Additional Level of Regulation of CYP3A Genes
2.4.1. Epigenetic Mechanisms of CYP3A Gene Regulation
Expression of CYP genes is influenced by epigenetic factors including histone modifications, DNA methylation, and regulation by noncoding RNAs [136].
Histone proteins undergo post-translational modifications such as acetylation, methylation, phosphorylation, ubiquitination, and some others, which can affect gene expression by changing chromatin architecture and DNA accessibility for transcription [136]. For instance, histone modifications may participate in the CYP3A4 and CYP3A7 expression changes that occur in the embryonic period and in the first 2 years after birth. This mechanism seems to be important for CYP3A4, although other mechanisms may play a greater role in the regulation of CYP3A7 during ontogenesis [137].
DNA methylation at CpG sites in a promoter region results in transcriptional repression and has numerous effects on the expression of CYP genes in various organs [136]. For example, a DNA methylation inhibitor called 5-aza-2-deoxycytidine increases the expression of PXR and CYP3A4 in HepG2 cells in a concentration- and time-dependent manner. Conversely, methylation of a CYP3A4 enhancer inhibits the PXR-mediated transcriptional activity of the CYP3A4 gene and the binding of PXR to the CYP3A4 promoter. As a result of methylation of the CYP3A4 promoter and enhancer, rifampicin stops enhancing CYP3A4 expression [138].
Noncoding RNAs—miRNAs and lncRNAs—have been identified as epigenetic modulators of expression of CYP genes [136]. Non-coding microRNAs can mediate transcriptional and post-transcriptional regulation of CYP3A in two ways. Firstly, by directly acting on the 3′-untranslated region (3′UTR) of the CYP3A4 mRNA or, secondly, by binding to the 3′UTR mRNA of nuclear receptor genes involved in the regulation of the CYP gene, thus acting on CYP3A4 activators or repressors [139,140].
For example, it has been shown that in non-alcoholic fatty liver disease, changes in the levels of miR-150-5p and miR-200a-3p [141] can post-transcriptionally suppress CYP3A4 [138,140,141,142], whereas miR-34a, miR-30c-1-3p and miR-27b suppress CYP3A4, affecting RXRa, PXR and VDR [136,140]. In a small clinical study, miR-27b expression was found to correlate with blood plasma levels of 4β-hydroxycholesterol, thus revealing an association between CYP3A and miR-27b in vivo [142]. Furthermore, in a study on 105 male patients treated with alprazolam, a significant association was found between the urinary metabolic ratio of 6β-hydroxycortisol to cortisol and miR-27b in plasma [143]. Indirect regulation of CYP3A4 by miR-27b may be due to miR-27b binding to VDR mRNA, resulting in a decrease in CYP3A4 protein expression [140].
MiR-148a has been found to influence inducible and constitutive expression of CYP3A4 by downregulating PXR [144]. From data on the impact of miRNAs on CYP3A5 expression, it is known that miR-543 directly binds to the 3′UTR of CYP3A5 mRNA and reduces the expression of the CYP3A5 protein [145]. Recently, in a study on the impact of basal CYP3A5 expression in donor liver transplants on the metabolism of an immunosuppressant (tacrolimus), it was demonstrated that miR-26b-5p directly regulates CYP3A5 expression, whereas miR-29a-5p, miR-99a-5p, and miR-532-5p target regulatory molecules HNF4α, NR1I3, and NR1I2, respectively [146].
There is research suggesting that lncRNAs may also contribute to the regulation of drug metabolism by acting on miRNAs [147]. The most common mechanism of CYP gene regulation mediated by lncRNAs is based on interactions between a lncRNA and mRNA of regulatory proteins. LncRNAs expressed near genomic loci of nuclear-receptor genes can modulate CYP gene expression along with expression of the corresponding nuclear receptors. Genes of antisense lncRNAs HNF1A-AS1 and HNF4A-AS1 are located adjacent to the genes that regulate CYP gene expression, i.e., genes of transcription factors HNF1α and HNF4α, respectively, thereby acting as coregulators of transcription of their target genes, including CYP3A4 [136,148].
2.4.2. Genetic Polymorphisms of CYP3A Genes and Their Influence on the Activity of CYP3A Enzymes
Although members of the CYP3A subfamily share high identity of amino acid and DNA sequences, their tissue- and age-specific expression patterns and substrate specificity differ considerably [137]. Genetic polymorphisms are a major cause of inter-individual differences in CYP3A expression and function [3,149].
Most CYP3A4 polymorphic variants are SNPs [26]. To date, 9815 SNPs have been found in the human CYP3A gene as documented in the National Center for Biotechnology Information (NCBI) SNP database (http://www.ncbi.nlm.nih.gov/snp). The following is known about the functional effects of SNPs of CYP3A genes.
CYP3A4, which codes for CYP3A4, the main enzyme of the CYP3A subfamily, has several SNPs correlating with metabolic activity of CYP3A4 [150,151], for example, allele CYP3A4*1B (rs2740574, −392A>G) located in the promoter [152] and allele CYP3A4*1G (rs2242480, 20230C>T) both increasing the activity of the CYP3A4 enzyme [153,154,155] and allele CYP3A4*22 (rs35599367, 15389 C>T) situated in intron 6 and reducing CYP3A4 enzymatic activity [153,155,156]. SNP CYP3A4*22 is known to double the production of a nonfunctional CYP3A4 variant [26,149,157,158]. Most other CYP3A4 polymorphisms have very low frequencies and their phenotypic effects are weak and often inconsistent [12,26,149]. On the other hand, CYP3A4*6, *17, *20, and *26 have been shown to correlate with reduced enzymatic activity due to a loss of function [159,160,161,162].
Another common allele that affects drug metabolism and hormone metabolism is CYP3A5*3 (rs776746) [157]. Hepatic CYP3A5 protein levels are low or undetectable in most whites. The reason is a common A>G mutation in intron 3 of the CYP3A5 gene (allele CYP3A5*3) [12]. The low CYP3A5 protein expression in CYP3A5*3 carriers is attributed to defective mRNA splicing and reduced synthesis of the functional protein [163]. Individuals with at least one CYP3A5*1 allele are known to express CYP3A5, whereas individuals with the CYP3A5*3/*3 genotype are known to not express CYP3A5 [164].
CYP3A7 is expressed in the liver at a much lower level in 90% of adults than in the fetus, but a high-expression phenotype is seen in 10% of adults. Two-thirds of the latter carry a promoter allele called CYP3A7*1C (rs45446698, located in the promoter) or (less commonly) CYP3A7*1B. Allele CYP3A7*1C induces overexpression of the fetal CYP3A7 gene in adults, influences sex hormone levels [165] and is an exclusive marker of high CYP3A7 expression in the gut [26].
For CYP3A43, genetic variant rs472660 in the intron is known to be associated with a response to antipsychotic drug olanzapine, and this gene’s functional SNP marker rs680055 correlates with this treatment response significantly [166]. The CYP3A43 Ala340Ala genotype is strongly associated with biomarkers of vitamin D metabolism [35].
Many SNPs at the CYP3A locus show large differences in allele frequency across populations [167]. The CYP3A5*3 allele frequency ranges from ~50–55% in African Americans to 91% in Europeans [9,26]. The nonfunctional form is present in 85% of the Japanese, 65% of the Chinese, 67% of Mexicans, and 40% of American Indians [26]. Northern Europeans have signatures of positive selection in CYP3A4 and CYP3A43 promoter regions, which play a key part in drug metabolism [168]. Differences in genotype frequencies at loci of CYP3A subfamily genes between African American and white American populations have been described [163]. Genes CYP3A4 and CYP3A5 possess high diversity of haplotypes having different frequencies in different ethnic groups [5].
2.4.3. CYP3A Inducers
Ligands of nuclear receptors modulating CYP3A expression are presented in Figure 4. After binding to a ligand, PXR regulates the expression of genes whose products participate in cholesterol metabolism and bile acid metabolism [169]. Typical ligands of PXR are steroids [169]. These are diverse compounds including pregnanes, progesterones, corticosterones, testosterone, pregnenolone, bile acids, intermediate sterol compounds (such as 5-cholestanoic acid 3,7,12-triols, 7α-hydroxy-4-cholesten-3-one, and 4-cholesten-3-one), bile acid precursors, dexamethasone, and 17β-estradiol [76,93,170,171,172,173,174,175,176]. Endogenous activators of PXR also include metabolites of microbial origin, such as metabolites of bile acids and of tryptophan, e.g., indole-3-propionic acid [177].
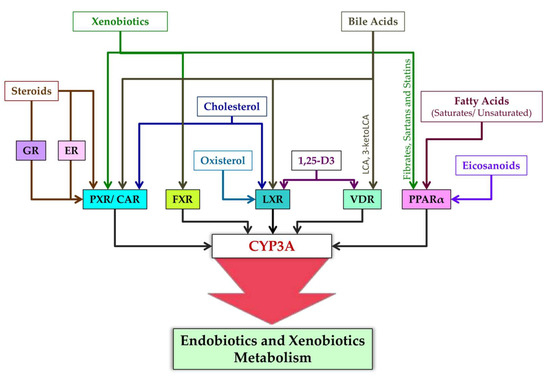
Figure 4.
Ligands of nuclear receptors modulating CYP3A expression.
CAR is constitutively active in the absence of ligands, takes part in physiological and pathophysiological pathways regulating glucose, lipid, and bile acid metabolism, and promotes cell proliferation, tissue regeneration, and cancer progression [178,179]. This constitutive activity, however, is inhibited by inverse agonist ligands [178]. Such endogenous ligands of CAR are testosterone metabolites androstanol and androstenol [180,181,182], estradiol [183], and cholesterol [184].
VDR is activated by 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], which is an active metabolite of vitamin D and is involved in bone metabolism and maintenance of calcium-and-phosphorus homeostasis [176,185,186]. Furthermore, CYP11A1-produced hydroxyvitamin D derivatives 20S(OH)D3 and 20,23(OH)2D3 can act as “partial” or “biased” agonists (ligands) on VDR [187]. VDR is also engaged by bile acid metabolites: lithocholic acid (LCA) and 3-keto LCA [85,188].
FXR was originally identified as a receptor triggered by farnesol, which is an intermediate of cholesterol synthesis [189]. Later, bile acids were found to be endogenous ligands of FXR [190], and FXR target genes were identified that contribute to the modulation of bile acid biosynthesis, secretion, and reabsorption [191].
PPARs (all isoforms) are activated by fatty acids and their derivatives, for example, by eicosanoid and prostaglandin derivatives as products of lipoxygenase and cyclooxygenase pathways [192]. Saturated fatty acids have weaker binding affinity for PPAR, whereas polyunsaturated fatty acids have stronger affinity [193]. More specific ligands of PPARα are eicosanoids, 8(S)-hydroxyeicosatetraenoic acid, and leukotriene B4; of PPARγ are 15-deoxy-Δ12,14-prostaglandin, J2,15-hydroxyeicosatetraenoic acid, and 9- and 13-hydroxyoctadecadienoic acids; and of PPARβ/δ is 8(S)-hydroxyeicosatetraenoic acid [193]. PPAR activation is cell type specific, e.g., a ligand of PPAR called 15-keto-prostaglandin E2 is produced in lung and colon epithelial cells, whereas 15-deoxy-Δ12,14-prostaglandin J2 predominantly in macrophages [192,194]. Other ligands of PPAR are xenobiotic drugs fibrates, sartans, and statins [195,196].
Ligands of LXR are some endogenous compounds: cholesterol and the oxysterols (oxidized derivatives of cholesterol) called 22(R)-hydroxycholesterol and 24,25(S)-epoxycholesterol [193,197,198] as well as 1,25(OH)2D3 [199].
2.4.4. CYP3A Inhibitors
CYP3A enzymes metabolize a variety of compounds among which almost the only common feature is lipophilicity and relatively large size; therefore, it is not surprising that many of these compounds act as inhibitors of CYP3A enzymes [200].
For CYP3A enzymes, there are weak inhibitors, which increase the area under the concentration–time curve (AUC) by ≥1.25-fold to <2-fold, moderate inhibitors, which enlarge AUC by ≥2-fold to <5-fold, and strong inhibitors, which increase AUC by ≥5-fold. AUC is a graphical representation of a concentration or dose, of drug in plasma during certain period of time. The AUC gives insight into the bioavailability variables of the drug and its clearance rate from the body [201]. Strong inhibitors of CYP3A enzymes include such antifungals as itraconazole, ketoconazole, and voriconazole; anticancer agents ceritinib, idelalisib, ribociclib, and tucatinib; macrolide antibiotics clarithromycin and telithromycin; antivirals (used to treat HIV infection) indinavir, nelfinavir, ritonavir, and saquinavir; an atypical antidepressant called nefazodone; and the drug mibefradil for hypertension and angina pectoris [202,203].
Several mechanisms of inhibition of CYP3A enzymes are possible, as shown in in vitro studies, and one compound can act through several mechanisms [204,205]. Rapidly reversible inhibition is mediated by direct rapidly reversible binding of an inhibitor or its metabolite to an CYP3A enzyme, resulting in competitive or noncompetitive inhibition, the extent of which is determined by the enzyme’s relative binding constants of the inhibiting ligand (a substrate or an inhibitor), and by inhibitor concentration. The most potent reversible inhibitors of CYP3A enzymes include azole antifungals and first-generation inhibitors of the HIV protease [204].
N-alkyl–substituted compounds can often inhibit CYP3A enzymes reversibly, and in vitro, this effect increases after preincubation. The reason is the oxidation of the inhibitor giving rise to a nitrosoalkane, which forms a slowly reversible complex with the reduced heme in a CYP3A enzyme’s molecule. These compounds include macrolides, such as erythromycin; some antidepressants; and other drugs, e.g., diltiazem, lidocaine, and tamoxifen [204].
The mechanism of irreversible inhibition is thought to involve CYP3A-mediated formation of reactive metabolites that covalently bind to the enzyme thereby causing its inactivation. Irreversible inhibition of CYP3A enzymes is characteristic of some 17α-ethynyl–substituted steroids (ethinylestradiol and levonorgestrol), furanopyridine, an HIV protease inhibitor [204,206], and possibly other furans; for example, it is known that furanocoumarin—a component of grapefruit juice—can markedly inhibit the metabolism of substrates of CYP3A enzymes [12,16,207]. Naringenin is also considered as a CYP3A inhibitor of grapefruit juice but there are data that neither naringin nor naringenin are primarily responsible for this effect [208].
Owing to their nonspecific detergent effect, bile acids can suppress the activity of CYP3A enzymes [209,210].
In principle, knowledge about enzyme kinetics helps to extrapolate in vitro data to in vivo conditions, but such modeling requires the assumptions many of which are difficult to verify [211]. Furthermore, it is necessary to take into account the functioning of intestinal and hepatic CYP3A enzymes, the contribution of different isoforms to the formation of metabolites, and the fact that most inhibitors of CYP3A enzymes are also substrates of such an enzyme, and at a high turnover rate of an inhibitor, its rapid loss can take place [211,212]. For getting knowledge on ligand interactions with different CYP3A enzymes, recently it became possible to use the free available AlphaFold Protein Structure Database [213] containing three-dimensional structures of all CYP3A P450 enzymes from all organisms.
It should also be mentioned that CYP3A inducers usually affect the efficacy of co-administered drugs, whereas CYP3A inhibitors affect their safety; therefore, CYP3A inhibition is considered clinically more important [12].
3. Involvement of CYP3A Enzymes in Biological Processes
CYP3A enzymes have very broad substrate specificity and metabolize a wide range of compounds in terms of chemical and biological properties. They catalyze reactions of hydroxylation, N-demethylation, O-dealkylation, S-oxidation, deamination, and epoxidation [26] of endogenous and exogenous compounds. CYP3A perform physiological functions by taking part in such endogenous processes as steroid catabolism, bile acid metabolism, and lipid and vitamin D metabolism. CYP3A enzymes metabolize a wide variety of therapeutics and may play an important role in alterations of biological activities of drugs or in enhanced clearance of drugs as well as in drug interactions. For instance, CYP3A enzymes’ substrates are such endogenous compounds as hormones, cholesterol, bile acids, arachidonic acid, and vitamin D as well as the vast majority of drugs and of xenobiotics that are not pharmaceuticals [11,12,14].
3.1. Metabolism of Endogenous Compounds
3.1.1. Biotransformation of Cholesterol and Bile Acids by CYP3A Enzymes
CYP3A enzymes are involved in cholesterol and bile acid metabolic pathways [214,215,216]. Cholesterol is metabolized to 4β-hydroxycholesterol [13] and to 25-hydroxycholesterol by CYP3A4 and CYP3A5 [13,217]. CYP3A4 has shown little contribution to cholesterol 22-, 24-, 26-, and 27-hydroxylation [218]. Excess cholesterol is converted to bile acids by various enzymes such as CYP7A1, CYP27A1, and CYP8B1, including CYP3A4 and CYP3A5.
In humans, most bile acid synthesis takes place in the liver via a pathway initiated by the rate-limiting enzyme cholesterol 7-hydroxylase (CYP7A1) and involving sterol 12-hydroxylase (CYP8B1) and sterol 27-hydroxylase (CYP27A1) [106,219,220,221]. Primary bile acids—cholic and chenodeoxycholic—are formed in the liver and are then converted in the intestine into secondary ones: deoxycholic and lithocholic [106,220].
CYP3A enzymes participate in bile acid biotransformation [214,215,216]. CYP3A enzymes are associated with a minor pathway for the biosynthesis of primary cholate of bile acids. 25-Hydroxylation of 5β-cholestan-3α,7α,12α-triol, which is an intermediate in the biosynthesis of cholic acid, is catalyzed by CYP3A4 and CYP3A5 [222]. In bile acid homeostasis, CYP3A4 catalyzes the 6α-hydroxylation of both taurochenodeoxycholic acid and LCA [223]. CYP3A activity is the main route of elimination of the toxic secondary metabolite of bile acids: LCA. This acid is metabolized to its 3-OH derivative by CYP3A4 and CYP3A5 [224]. Additionally, deoxycholic acid is metabolized into 1β-, 3β-, 4β-, 5β-, 6α-, 6β-, and 19-oxidized derivatives by CYP3A4 and CYP3A7 [215,216].
3.1.2. Biotransformation of Hormones by CYP3A Enzymes
CYP3A enzymes play an important part in hormonal homeostasis. With regard to steroid hormones, the CYP3A subfamily plays an important role in the metabolism of androgens (testosterone, androstenedione, dehydroepiandrosterone, and dihydrotestosterone), progesterone, cortisol, and their metabolites [10,12,14]. CYP3A metabolizes testosterone, progesterone, and cortisol via the 6β-hydroxylation reaction unique to this P450 subfamily. The highest 6β-hydroxylation activity is observed in CYP3A4, followed by CYP3A5 and CYP3A7 [225,226]. In this context, testosterone stimulates progesterone 6β-hydroxylation, whereas progesterone inhibits CYP3A-mediated 6β-hydroxylation of testosterone [226]. Cortisone is also metabolized to 6β-hydroxycortisone by CYP3A enzymes [227,228].
2β-, 15β- and 16β-hydroxylation of testosterone is mediated by CYP3A4 [227,229]. The formation of 6β- and 2β-hydroxytestosterone is catalyzed by CYP3A5 and CYP3A7, while 2α-hydroxytestosterone generation is catalyzed only by CYP3A7 [228,230]. Extrahepatic CYP3A4 is more often responsible for the metabolism of hormones in situ, for example, it takes part in irreversible oxidation of testosterone in the prostate, thereby terminating its androgenic effect [14].
CYP3A4, CYP3A5, and CYP3A7 mediates 7β- and 16α-hydroxylation of dehydroepiandrosterone. The formation of 7β-hydroxydehydroepiandrosterone is catalyzed mainly by CYP3A4, and 16α-hydroxydehydroepiandrosterone by CYP3A7 [5,229,231]. Therefore, the profiles of metabolites of 16α- and 7β-hydroxylation of dehydroepiandrosterone can be useful for differentiation between CYP3A4 and CYP3A7 activities [231].
CYP3A7 performs a function in estriol synthesis [5]. Estradiol is metabolized by CYP3A4 giving rise to 2-, 4-, and 16β-hydroxylation products [232]. The formation of 2- and 4-hydroxylated estradiol metabolites is also mediated by CYP3A5 and CYP3A7, although their contribution is smaller than that of CYP3A4 [233]. In addition, CYP3A4, CYP3A5 and CYP3A7 metabolize estrone to form 2-, 4- and 16α-hydroxylated metabolites [234,235,236,237]. Progesterone is metabolized by CYP3A4 to form 2β-, 6β-, 16α- and 21-hydroxylated metabolites, as well as CYP3A5 and CYP3A7 to form 6β-hydroxyprogesterone [226,227].
3.1.3. Biotransformation of Vitamin D by the CYP3A Subfamily
In the liver and small intestine, CYP3A4 and CYP3A5 are involved in vitamin D metabolism [25]. CYP3A4 contributes to 24-hydroxylation and 25-hydroxylation of vitamins D3. The active form of vitamin D—1,25-dihydroxycholecalciferol [1,25-(OH)2D3]—is a CYP3A4 substrate [14,85]. CYP3A4 participates in tissue-specific conversion of vitamin D3 metabolites to their respective inactive metabolites by performing the 24- or 25-hydroxylation of 1-hydroxyvitamin D3 and the 4β-hydroxylation of 25-hydroxyvitamin D3 [14]. CYP3A5 catalyzes 23- or 24-hydroxylation of 1,25-(OH)2D3 [238]. Although vitamin D is inactivated by other extrarenal hydroxylases such as CYP24A1, it is primarily biotransformed by CYP3A4.
3.1.4. Biotransformation of Arachidonic Acid by CYP3A Enzymes
Arachidonic acid is the precursor to various physiologically active molecules such as epoxyeicosatrienoic acids (EETs), which are a class of functionally bioactive lipid mediators derived from the metabolism of long-chain polyunsaturated fatty acids under the action of multiple enzymes of three main families, including cyclooxygenases, lipoxygenases, and cytochromes P450 [239,240]. The role of eicosanoids—produced by cyclooxygenases and lipoxygenases—in the control of physiological and pathological processes is well known at present. CYP epoxygenase metabolites of arachidonic acid, i.e., EETs, act as lipid mediators inducing numerous biological responses in both cardiac and noncardiac tissues. EETs play an important part in blood pressure regulation, inflammation, and cell proliferation [239,240].
In the relevant human CYP-mediated pathway, the main CYP enzymes that form EETs are CYP2C8, CYP2C9, CYP1A2, and CYP3A4 in the liver [239,241] and CYP2J2 in the heart [242]. CYP4F2 is the main CYP subfamily that forms hydroxyeicosatetraenoic acids in the liver and kidneys [239,243]. CYP3A4 is involved in the biosynthesis of 5,6-, 8,9-, 11,12-, and 14,15-EETs in the liver [239]. Little is known about the participation of CYP3A5 and CYP3A7 in the production of EETs [11].
3.2. Biotransformation of Exogenous Compounds
The substrates of CYP3A enzymes are a wide range of prescription drugs as well as xenobiotics such as carcinogens benzo[a]pyrene, 7,8-dihydrodiol, and atlatoxin B [244,245]. In the lungs, CYP3A4 and CYP3A5 metabolize procarcinogens of tobacco smoke to active carcinogens, which are polycyclic aromatic hydrocarbons such as benzo(a)pyrene [246], whereas CYP3A4 participates in the activation of tobacco-specific carcinogen N′-nitrosonornicotine (NNN) in the liver [247].
CYP3A enzymes can metabolize many structurally and functionally distinct classes of drugs (Table 2) [74,248,249,250,251,252,253,254,255,256,257,258].
Table 2.
Classes of drugs that are metabolized by CYP3A enzymes.
The involvement of CYP3A enzymes in the metabolism of most drugs underlies their main biomedical significance as enzymes that influence drug kinetics and as participants in drug interactions.
3.3. Endogenous and Exogenous Biomarkers of Activity of CYP3A Enzymes
Accurate predictions of the activity of CYP3A enzymes are needed to provide effective pharmacotherapy to predict treatment outcomes or potential adverse effects of various therapeutic agents and to assess the development and progression of diseases.
Research is underway for identifying optimal endogenous biomarkers of the activity of CYP3A enzymes, e.g., derivatives of cholesterol, of bile acids, or of steroid hormones, which can be quantified by measuring the concentration of certain compounds in a urine or blood sample without the introduction of xenobiotics into the human body [259]. Appropriate combinations of such markers should make it possible to predict the activity of CYP3A enzymes while taking into account contributions of different isoforms [11]. For example, in clinical studies, the metabolic ratio of 6β-hydroxycortisol to cortisol in urine has already been characterized as an endogenous biomarker of CYP3A activities [260].
Many metabolites of endogenous compounds are used as markers of the activity of CYP3A enzymes both in vitro and in clinical studies [11,13,222,260,261,262,263,264]. The metabolic ratio of 1β-hydroxydeoxycholic acid to deoxycholic acid in urine has been proposed as a potential endogenous biomarker of CYP3A activities [214]. 19-Hydroxylated deoxycholic acid, which is specifically produced by CYP3A7, has been suggested as a marker of CYP3A7 activity in vitro, although the usefulness of this marker in vivo is unclear [263]. Profiles of 16α- and 7β-hydroxylation derivatives of dehydroepiandrosterone can be employed to discriminate the activities of CYP3A4 and CYP3A7, whereas the contribution of CYP3A5 to these products is negligible [231].
Furthermore, lately, plasma 4β-hydroxycholesterol—a cholesterol metabolite that is a substrate of CYP3A4 and CYP3A5—has been regarded as an endogenous biomarker of CYP3A activities. Although little is known about the isoform specificity of 6β-hydroxylation, the metabolic ratio of urinary 6β-hydroxycortisone to urinary cortisone has also been proposed as a biomarker for the activity of CYP3A enzymes [265], whereas the ratio of urinary 6β-hydroxycortisol to urinary cortisol (6β-OH-C/C) and plasma 4β-OH-cholesterol (4β-OH-C) is currently used to evaluate the activity of CYP3A enzymes [266,267]. In one study, a statistical predictive model was built by combining the ratios 7β-hydroxy-dehydroepiandrosterone/dehydroepiandrosterone, 16α-hydroxy-dehydroepiandrosterone/dehydroepiandrosterone, and 6β-hydroxy-cortisol/cortisol [264].
Predicting the activity of CYP3A enzymes by means of an exogenous compound is central to research on interactions between drugs that have similar metabolic pathways involving CYP enzymes or that are used simultaneously in the treatment of diseases [259,268]. Over the past few decades, a number of marker compounds have been used for this purpose. Among them, midazolam—a benzodiazepine derivative used in more than 70% of phenotyping cocktails—has become the “gold standard” [13,266,268]. This drug can be administered orally and intravenously for characterization of metabolism by CYP3A enzymes by means of its 1′-hydroxylation in the intestines and liver, respectively. It has a short half-life (based on excretion) allowing for the detection of acute metabolic changes, and it and possesses a favorable safety profile as well as a lack of transport by P-glycoprotein and other transporters [13]. Other substrates for phenotyping of the activity of CYP3A enzymes are alprazolam and nifedipine [269,270]. The activity assay of CYP3A enzymes in the liver by a 14C-erythromycin phenotypic breath test also use [271].
Among the problems with exogenous probes (e.g., midazolam and alfentanil), safety is often cited, especially for children and cancer patients. Nonetheless, phenotyping with midazolam can be carried out by means of a single dose or microdose to minimize adverse effects, whereas phenotyping with alfentanil can be performed by pupillary miosis measurement; in general, it is believed that the above-mentioned problem can be solved [13].
Some results of studies comparing exogenous with endogenous biomarkers of the activity of CYP3A enzymes are contradictory [260,272]. The lack of consensus is especially true for gender-related and ethnic differences in the activity of CYP3A enzymes [265,269]. Nevertheless, midazolam clearance appears to be superior to 4β-hydroxycholesterol in terms of the accuracy of predicting drug interactions. As a biomarker of CYP3A4, 4β-hydroxycholesterol is questionable because its concentration can be affected for example by the patient’s diet, by the presence of liver and kidney diseases, by the drugs used for their treatment, and by medications influencing cholesterol levels. Some authors believe that 4β-hydroxycholesterol is not suitable as a phenotypic marker of CYP3A activities [13].
4. CYP3A Involvement in Pathological Processes
4.1. Diseases Related to the Participation of CYP3A Enzymes in Bile Acid Metabolism
Bile acids are necessary for bile production and lipid absorption from the small intestine. They help with the excretion of cholesterol, hydrophobic metabolites of xenobiotics, toxins, and metals from the human body as well as play the role of signaling molecules in energy metabolism [74,106,221], participate in the regulation of lipid and glucose homeostasis, thermoregulation, and immune responses [221], and have anti-inflammatory properties under normal physiological conditions [107]. But the accumulation of large amounts of bile acids causes inflammation and damage to the liver [107]. High concentrations of bile acids are toxic to hepatocytes, enterocytes, and cholangiocytes and raise the risk of digestive-tract diseases, including cholestasis and cancers [74,106,273,274].
Bile acids undergo enterohepatic circulation: ~95% of their amount is reabsorbed and returned to the liver, but in each cycle they are synthesized de novo by hepatocytes to replace the bile acids that are eliminated [106]. The regulation of bile acid synthesis and metabolism is very complex and has to be tightly regulated to maintain nondangerous concentrations sufficient to perform a physiological function [107,221].
In humans, most bile acid synthesis occurs in the liver via a pathway initiated by the rate-limiting enzyme cholesterol 7-hydroxylase (CYP7A1) and involving sterol 12-hydroxylase (CYP8B1) and sterol 27-hydroxylase (CYP27A1) [106,107,219,220,221]. Primary bile acids—cholic and chenodeoxycholic—arise in the liver and are then converted in the intestine into secondary ones: deoxycholic and lithocholic [106,220].
The CYP3A subfamily participates in cholesterol and bile acid metabolic pathways: regulation of the conversion of cholesterol to 4β-hydroxycholesterol is performed by CYP3A4 and CYP3A5 [13], and bile acid biotransformation is performed by CYP3A enzymes [214,215,216]. Primary and secondary bile acids in turn alter the expression of CYP3A enzymes in the relevant organs: CYP3A4 and CYP3A5 in the liver and intestines and CYP3A7 and CYP3A43 only in the liver [220].
Cholestasis is a pathological condition where normal flow of bile is low or disturbed, and bile acids accumulate in the liver. The reason is either mechanical obstruction of bile ducts or defects in hepatic transporters [275]. The outcome is inflammation and damage to the bile ducts, followed by exposure of hepatocytes to high concentrations of bile acids, which can result in hepatocyte death [275,276]. For control over processes in the gastrointestinal tract in cholestatic conditions, the composition of bile acids and the size of their pool are important, and hydrophobic bile acids are especially cytotoxic [107,276].
In vitro and in vivo studies show a decrease in CYP3A activity in cholestatic liver disease. Nevertheless, CYP3A4 activity can be stimulated early in cholestasis because bile acids can induce CYP3A4 expression indirectly through FXR [76,277] and PXR [277,278,279]. Furthermore, LCA activates CYP3A4 mainly through VDR in the colon [85]. There are reports of early activation of CYP3A enzymes in animal models of cholestasis induced by bile duct ligation [278,280]. CYP3A subfamily activation as an in vivo adaptive response to cholestasis has been confirmed in experiments on humanized CYP3A4/lacZ transgenic mice carrying a genetic construct containing an upstream regulatory region of the human CYP3A4 gene [278]. In rat models of cholestasis, when cholestasis is moderate, mRNA and protein expression of CYP3A1 is significantly increased while the expression of CYP3A2 is unchanged; the expression and activity of both enzymes are sharply lower in severe cholestasis; CYP3A1 and CYP3A2 are the metabolically most important isoforms of CYP3A in male rats [280].
On the other hand, at later stages of the disease, CYP3A4 activity is downregulated due to elevated levels of estrogen and bile acids in the blood [74]. As the concentration of bile acids increases, the activity of several CYP enzymes in rats, e.g., CYP3A2, diminishes because bile acids exert nonspecific detergent effects on these enzymes [209]. As for estrogen, cholestasis is associated with its elevated levels (in mice) [281], whereas estrogen is involved in the regulation of CYP3A4 and suppresses its expression in humans [28].
The inhibitory effect of bile acids on the activity of CYP enzymes correlates with hydrophobicity of these acids. The most toxic and having a strong inhibitory effect is LCA; deoxycholic and chenodeoxycholic acids are less toxic, and cholic acid is the least toxic bile acid [74]. Among bile acids, LCA is the most active PXR ligand [221]. A protective role of PXR against bile acid toxicity is known: CYP7A1 expression is lower compared to a control in human hepatocytes treated with a PXR agonist: rifampicin [282].
The difference in the effects of early and late cholestasis on CYP3A enzymes’ activity is likely due to differences in the activation of PXR and CAR. It is known that PXR interacts with nuclear receptors CAR and FXR, which control homeostasis of cholesterol, bile acids, bilirubin, glucose, and lipids [283]. FXR is an important player in processes of bile acid detoxification. FXR activated by them induces CYP3A4 expression and phase I hydroxylation reactions [106,107]. In addition, bile acids activate a membrane bile acid receptor: TGR5. Activation of FXR and of a G protein–coupled bile acid receptor (GPBAR1, i.e., TGR5) by agonists improves insulin and glucose sensitivity and stimulates energy metabolism thus preventing diabetes mellitus, obesity, and nonalcoholic fatty liver disease, which is one of the leading causes of liver diseases, because of its association with obesity, type 2 diabetes mellitus, and dyslipidemia [107,141,284].
Nonalcoholic fatty liver disease is reported to correlate with reduced expression and function of CYP3A4 [141]. Enzymatic activity and mRNA expression of CYP3A4 are low in patients with nonalcoholic fatty liver disease, and this observation is confirmed by experiments on mice in vivo and on cultured cells in vitro [284,285,286,287,288,289]. There is evidence that changes in hepatic levels of some miRNAs, in particular miR-150-5p and miR-200a-3p [141] in nonalcoholic fatty liver disease can post-transcriptionally modulate the expression of CYP3A4 [140,290,291,292].
Cardiac dysfunction is associated with an increase in concentrations of bile acids in the blood during liver diseases. It is possible that bile acids as a ligand can activate FXR, which can play a part in the development of atherosclerosis [293,294].
FXR, CAR, and PXR have many regulatory effects in common, and their functions—in the context of regulation of liver detoxification and bile acid metabolism—overlap substantially. An increase in bile acid clearance can be caused by CYP3A4 upregulation after the stimulation of vitamin D receptor by vitamin D and LCA [221]. Clinically significant outcomes of cholestatic diseases (and of treatment with bile acids or their derivatives) may be affected by the activity of some drug-metabolizing CYP enzymes regulated by the same nuclear receptors as CYP3A enzymes are [221]. In theory, polymorphisms in CYP3A4 and PXR genes may affect the degree of bile acid detoxification [74], but evidence is not yet available. The function of PPARα as a key transcription factor in the regulation of bile acid metabolism and lipid metabolism is also important for proper liver functioning and for the pathogenesis of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis [295,296].
Stimulation of CYP3A4 activity in cholestasis may be an effective therapeutic approach to such diseases [74]. In clinical practice, rifampicin and ursodeoxycholic acid are used to treat cholestasis, consistently with this acid’s ability to reduce the toxicity of more hydrophobic bile acids [74]. Ursodeoxycholic acid can induce CYP3A enzymes and reduce the concentration of their substrates [221].
It must be mentioned that several mechanisms of therapeutic action of ursodeoxycholic acid have been proposed [275]. Aside from inducing CYP3A enzymes, ursodeoxycholic acid can inhibit intestinal absorption of hydrophobic bile acids thereby raising the hydrophilicity of the bile acid pool [58,297]. Ursodeoxycholic acid also contributes to the restoration of the “bicarbonate umbrella”: biliary secretion of bicarbonate ions, which protect cholangiocytes from damage by bile acids [298]. Although the therapeutic benefits of ursodeoxycholic acid are well documented, many patients do not adequately respond to treatment with this compound, whereas high doses of ursodeoxycholic acid are associated with a high risk of esophageal and gastric varicose veins and a need for a liver transplant [275,299].
New therapeutic approaches are being developed for the treatment of cholestatic pathologies, including agonists of FXR, which mediates the induction of CYP3A4 expression by bile acids [106]. Furthermore, other derivatives of natural bile acids have been proposed: “bile mimetics,” such as nor-ursodeoxycholic acid and obeticholic acid, which is a potent FXR agonist [300].
4.2. Diseases Associated with the Participation of CYP3A Enzymes in Arachidonic Acid Metabolism
Arachidonic acid metabolites known as eicosanoids (EETs), a subclass of oxylipins, have a wide range of physiological effects in cardiovascular homeostasis and regulation of cell growth, inflammation, and immune responses. EETs function as regulators of cardiac, vascular [301,302,303,304], and renal physiology [305,306,307]. In the cardiovascular system, EETs serve as vascular relaxation factors independent of nitric oxide and prostacyclin I2 [308].
Eicosanoids (EETs), whose emergence depends on CYP3A-mediated metabolism, are implicated in the pathophysiology of various diseases. EETs promote the growth of a primary tumor and metastasis as well as the exit of a tumor from an indolent state [309]. The functions of EETs are associated with carcinogenesis, metastasis of various cancers (including colon, liver, kidney, breast, and prostate cancers [309,310,311,312,313]), with angiogenesis [314,315,316,317], and restoration of cardiac tissue after ischemic stroke [318].
The CYP3A4 epoxygenase, responsible for the production of EETs, is overexpressed in breast cancer and is linked with the initiation and progression of breast cancer [319]. CYP3A4 activity can accelerate tumor progression that is independent of activation of oncogenes. This notion is supported by the results of a study on breast cancer cell lines in which 8,9-, 11,12-, and 14,15-EETs are synthesized with the participation of highly enzymatically active CYP3A4 [313]. In that work, silencing of CYP3A4 blocked the cell cycle at the G2–M checkpoint and induced apoptosis of MCF7 cells by inhibiting STAT3 (Tyr-705) phosphorylation, thereby inhibiting the growth and survival of the tumor cells; a knockdown of CYP3A5 suppressed the proliferation of cell lines MCF7, T47D, and MDA-MB-231 to various degrees [313]. CYP3A4 takes part in breast cancer progression by stimulating angiogenesis through the activation of vascular endothelial growth factor (VEGF) [309,310,320] and in cancer cell proliferation through the activation of phosphatidylinositol 3-kinase (PI3K)–protein kinase B (AKT) and STAT3 pathways partially because of the synthesis of (+/−)-14,15-EET [313].
Mechanisms have been discovered by which CYP monooxygenases specific for cancer cells promote tumor progression and are associated with breast cancer mitochondria, whereas EETs stimulate the electron transport chain/respiration and inhibit AMPKα (AMP-activated protein kinase) [321]. A CYP3A4 knockdown or biguanide inhibition activates AMPKα, promotes autophagy, and prevents breast tumor formation [321]. These findings indicate that arachidonic acid, CYP epoxygenases metabolizing it, and EETs are connected with mitochondrial function and oxidative stress in cancer cells; this phenomenon may be another potential mechanism underlying antiapoptotic actions of these substances. In addition, EETs promote epithelial–mesenchymal transition (EMT) and drug resistance via STAT and AKT signaling pathways [313,322,323,324,325].
CYP3A4 is necessary for tumor formation in ER+HER2− breast cancer because this enzyme suppresses autophagy, in part by inhibiting AMPK activation [326]. Although the production of an EET via CYP3A has been proven to correlate with the progression of breast cancer, little is known about the role of this enzyme in the development of chemoresistant breast cancer. A study on tamoxifen-resistant breast cancer [327] revealed that CYP3A4 expression and its epoxide product, 11,12-EET, are upregulated in tamoxifen-resistant MCF-7 cells (TAMR-MCF-7) compared to control MCF-7 cells. In that work, treatment of TAMR-MCF-7 cells with ketoconazole and azamulin (selective CYP3A4 inhibitors) or 14,15-epoxyeicosa-5(Z)-enoic acid (an antagonist of epoxyeicosatrienoic acid) inhibited cell proliferation and restored sensitivity to 4-hydroxy tamoxifen. It was found there that the molecular mechanism underlying this process is the suppression of E2F1-dependent transcription of the peptidyl prolyl isomerase gene (Pin1); it codes for a major regulator of angiogenesis and of characteristics of the epithelial–mesenchymal transition of TAMR-MCF-7 cells. Suppression of Pin1 via inhibition of EETs blocked proliferation and migration of TAMR-MCF-7 cells and angiogenesis mediated by these cells.
In human hepatoma cell line Hep3B, overexpression of CYP3A4 also promotes cell growth and cell cycle transition from the G1 to S phase; these effects are attenuated by a putative EET receptor antagonist: 14,15-epoxyeicosa-5(Z)-enoic acid [328].
EETs are known to exert anti-inflammatory actions in human and mouse tissues [329], thereby complicating the biological effects of EETs on cancer. Experimental studies suggest that 11(12)-EET can reduce the expression of TNF [329], whereas 14(15)-EET can downregulate TNF-α and IL-1β [330].
Arachidonic acid metabolism is reported to be one of the metabolic pathways most affected by SARS-CoV-2 infection [331]. EETs have been shown to take part in kidney damage [332]. Therefore, it seems that a therapeutic strategy based on specific inhibitors or inducers of CYP3A, depending on circumstances, may be useful against COVID-19.
4.3. Roles of CYP3A Enzymes in Diseases Associated with the CYP3A Involvement in the Metabolism of Sex Steroids
The role of CYP3A enzymes in the metabolism of sex steroid hormones implies an association with the development of hormone-dependent diseases. It has long been known that CYP3A enzymes are expressed in normal and tumorous tissues of the breast [333,334,335] and of the prostate [336,337], in cells of the endometrium and cervix [338,339], and in ovarian tumors [340].
According to [341], the concentration of CYP3A4 in breast cancer tissue varies from 0.5 to 63 pmol/(mg protein). Overexpression of CYP3A4 in a cancerous tissue compared to the normal tissue in most cases correlates with predisposition to breast cancer [319,342,343] and ovarian cancer [344]. On the contrary, in prostate cancer, CYP3A4 is underexpressed [345,346].
CYP3A4 participates in the conversion of 17-β-estradiol to potentially genotoxic 16-α-hydroxyestrone. It has been demonstrated that high expression of CYP3A4/5 at mRNA and protein levels in tumor tissues of the mammary gland correlates with lymph node metastasis (lymph node status) and may be a key sign of an unfavorable prognosis of breast cancer. In this context, CYP3A4/5 expression is not associated with the presence of ER and progesterone receptor [342].
The CYP3A4 protein is expressed in both the tumor and surrounding normal breast tissue but is overexpressed in the tumor, according to data obtained by an immunohistochemical method in tissue samples from women with infiltrating ductal carcinoma; the staining was positive in all tumor samples but only in 68% of normal breast tissue samples [347]. Additionally, according to some data [319] obtained by immunohistochemistry, in the stroma and glandular tissue of a mammary gland containing a tumor, CYP3A4 is overexpressed as compared to the healthy tissue. By contrast, according to other data (obtained by immunoblotting), CYP3A4 expression is lower in breast tumor tissue compared to the morphologically normal adjacent tissue [348].
Immunohistochemical analysis of primary epithelial ovarian cancer tissue has revealed high protein levels of several P450 enzymes, namely CYP3A5, CYP3A7, and CYP3A43, as compared to normal ovarian tissues [344].
Androgen receptor-regulated signaling pathways play a key role in the progression of the prostate cancer in which androgen-metabolizing enzymes CYP3A4, CYP3A5, and CYP3A43 are expressed. Several CYP enzymes, including CYP3A43, can be considered prognostic and diagnostic markers of prostate cancer [349]. In particular, CYP3A hydroxylates testosterone, which is essential for the normal functioning of prostate cells and is crucial for the progression of prostate cancer, to less active metabolites. Measurement of mRNA levels of CYP3A genes in prostate samples has shown that basolateral cells of the prostate normally express CYP3A enzymes abundantly, but in the tumor tissue, mRNA and protein levels of CYP3A5 are markedly lower [346].
Similar results have been obtained by immunohistochemical analysis in surgical tissue samples from patients with prostate cancer. CYP3A4 immunoreactivity proved to be significantly lower in tumor tissues than in a benign epithelium; diminished immunoreactivity of CYP3A4 correlated with a poor prognosis of the disease [345]. In contrast, the authors of ref. [350] have demonstrated androgenic induction of CYP3A5 mRNA in prostate cancer LNCaP cells.
PXR and CAR are overexpressed in tumor tissues, for example in prostate cancer [351], breast cancer [352], endometrial cancer [353], and ovarian cancer [354]. Various levels of expression of PXR, which regulates the expression of CYP3A genes, have been found in endometrial cancer tissues in contrast to normal tissues. As compared to tissues with PXR downregulation, tissues with high expression of PXR also featured overexpression of CYP3A4/7 and underexpression of estrogen receptor. Among endometrial cancer cell lines, HEC-1 cells, which overexpress PXR and underexpress ER and progesterone receptor, show a stronger transcriptional response of the PXR–CYP3A pathway to ligands of PXR [355]. Moreover, ligands of PXR enhance PXR-mediated transcription in a ligand- and promoter-dependent manner, resulting in differential regulation of the expression of PXR’s individual target genes, especially CYP3A4 and MDR1, in endometrial cancer cells [356]. These data suggest that steroid/xenobiotic metabolism in tumor tissue via the PXR–CYP3A pathway may play an important part in the pathogenesis of endometrial cancer [355,357].
Several miRNAs potentially regulated by CAR and capable of activating CYP3A4 were identified by [353]. The discovered miRNAs are tumor suppressors, and their level is low in malignant tumors of the endometrium.
Expression of PXR has been found in human prostate tumor tissues, and treatment of PC-3 cells with SR12813 (a selective agonist of PXR) stimulates the expression of multidrug resistance 1 (MDR1) and CYP3A4 and enhances the resistance of these prostate cancer cells to chemotherapy. In the normal prostate, the expression of PXR turned out to be the highest in the stratum basale; at earlier stages of the disease, PXR immunoreactivity is higher in prostate tumor tissues than in normal tissues, whereas with progression to later stages, the PXR expression tends to decline [351].
4.4. Diseases Related to the Participation of CYP3A Enzymes in Vitamin D Metabolism
Vitamin D has multiple effects on the biological processes that regulate the metabolism of calcium and phosphorus and also affects proliferation, differentiation, and apoptosis of cells as well as immune regulation. Epidemiological studies and experimental laboratory data show that vitamin D deficiency correlates with the onset and progression of many common chronic diseases such as bone metabolism disorders, tumors, cardiovascular diseases, infectious diseases, autoimmune diseases, diabetes mellitus, and others [358,359,360,361,362,363,364,365].
The proportion of people with vitamin D deficiency is high among patients with chronic infection caused by hepatitis C virus, non-alcoholic fatty liver disease, multiple sclerosis, psoriasis, osteoarthritis or chronic kidney disease [358]. A recent study showed vitamin D deficiency in 82% of hospitalized patients with COVID-19 [366]. CYP3A4, by taking part in the inactivation of an active vitamin D metabolite [1.25(OH)2D3], may have a significant impact on circulating vitamin D levels and hence calcium homeostasis, which in turn may influence bone and immune-system health [367,368] by downregulating the overexpression of inflammatory cytokines such as IL-1α, IL-1β, and TNF [369,370]. Pathologies such as vitamin D–dependent rickets and osteoporosis can progress because of the inactivating effect of CYP3A4 on the active form of vitamin D [361,365,371].
It is possible that the progression of cardiovascular diseases may be due to the inactivating effect of CYP3A4 on the active form of vitamin D because a low concentration of 1,25(OH)2D3 in blood serum is strongly associated with the initiation of cardiovascular diseases [372] and with the incidence of arterial hypertension [373].
Genetic factors have been recognized as some of the main causes of vitamin D deficiency [374]. For instance, in patients with rickets, there is a decreased serum concentration of vitamin D metabolites owing to mutations in CYP3A4; in particular, the CYP3A4 I301T mutation results in a stronger activity inactivating vitamin D metabolites, thereby reducing levels of active vitamin D [375]; in other words, CYP3A4 polymorphisms can worsen some pathological conditions. Numerous studies indicate that the risk and prevalence of type 2 diabetes mellitus correlate with vitamin D deficiency [374,376,377,378,379].
Recent research articles have elucidated the relation between CYP3A4 polymorphisms and type 2 diabetes mellitus by showing that people with some CYP3A4 polymorphisms are at a higher risk of type 2 diabetes mellitus [380]. Nonetheless, regarding the possible association of CYP3A4 with diabetes mellitus, experimental clinical data are inconsistent. For example, it has been reported that the functional activity and expression of CYP3A4 are higher in experimental diabetes [381]. Fatty acids (palmitic, oleic, stearic, and linoleic) participate in the mechanism of CYP3A4 induction in experimental diabetes. It is these acids, not insulin, that raise the activity and mRNA and protein expression of CYP3A4, as demonstrated in vitro on HepG2 and Fa2N-4 cells that are incubated with the serum of rats with streptozotocin-induced diabetes [381].
During investigation into the expression of hepatic CYP enzymes in patients with diabetes mellitus, the results revealed that this disease correlates with a significant decrease in hepatic CYP3A4 enzymatic activity and protein levels. For instance, after an assay of the activity of seven major CYP enzymes in 38 patients with type 2 diabetes mellitus and 35 patients without diabetes (oral administration of the midazolam as a probe drug), it turned out that the activity of CYP3A enzymes in patients with diabetes mellitus is lower by approximately 38% [382]. It is also reported that hydroxylation of marker substrates—midazolam (at the 1′- or 4-position) and testosterone (6β-hydroxylation)—by CYP3A enzymes is notably reduced in liver microsomes of diabetic patients, regardless of the genotype of CYP3A genes [383]. A significant decrease in the activity and protein level of CYP3A4 has been registered in diabetic patients with nonalcoholic fatty degeneration of the liver or nonalcoholic steatohepatitis. Furthermore, this decrease in activity continued with growing disease severity as it progressed from nonalcoholic fatty degeneration of the liver to nonalcoholic steatohepatitis [289].
The ability of CYP3A4 to catalyze the inactivation of 1,25(OH)2D3 and of other active forms of vitamin D has an influence on cancer cell proliferation and may be clinically important in various common cancers, mostly breast, prostate, and colorectal malignant tumors [362,363,384,385,386]. A possible reason is that vitamin D takes part in cell proliferation by affecting the expression of genes p21 and p27, by promoting apoptosis and autophagy, by regulating angiogenesis, by upregulating antioxidants, and by exerting anti-inflammatory actions via downregulation of genes for multiple inflammatory mediators (interleukins; e.g., IL-12, IL-2, TNF, and interferon gamma) as well as by regulating various inflammatory cascades such as the NF-κB (nuclear factor kappa light chain enhancer of activated B cells) pathway, mitogen-activated protein kinase (MAPK) signal transduction, and cyclooxygenase-2 (COX-2) signaling [362].
An in vitro analysis of human liver microsomes has revealed the ability of CYP3A4 to inactivate other active forms of vitamin D, its 20(OH)D3 derivative, and other relevant metabolites that may also have a physiological effect on cancer cell proliferation [386]. There are also abundant data from various studies indicating that vitamin D reduces the risk of developing of numerous types of cancer, including lung, ovarian, prostate, breast, colon, non-Hodgkin’s lymphoma [371]. There is also convincing evidence that vitamin D reduces the risk of autoimmune diseases, such as multiple sclerosis and type 1 diabetes. Less convincing evidences exist for reducing the risk of type 2 diabetes, rheumatoid arthritis, osteoarthritis, hypertension and stroke [365,371,383].
4.5. Changes in the Expression and Activity of CYP3A Enzymes in Various Pathological Conditions
It is now clear that the expression and activity of CYP enzymes are affected by such pathological conditions as infection, inflammation, and cancer [10,387,388].
4.5.1. Inflammation-Dependent Changes in the Expression and Activity of CYP3A Enzymes
The influence of inflammation on the expression of CYP3A enzymes is an important topic because an alteration of these enzymatic activities leads to a change in pharmacokinetics of prescription drugs. Inflammation is a common sign of many diseases and is implicated in the pathogenesis of such illnesses as infectious diseases, cancer, diabetes mellitus, rheumatoid arthritis, and inflammatory bowel disease as well as age-related processes such as normal aging and metabolic aberrations [389]. Sources of inflammation are infections, e.g., hepatitis, human immunodeficiency virus (HIV) infection, and COVID-19 [390]. Aside from infections, other possible sources of inflammation in the human body are diseases of organs (the kidneys, liver, lungs, and heart), diabetes mellitus, autoimmune diseases (ankylosing spondylitis, psoriasis, systemic lupus erythematosus, Crohn’s disease, and celiac disease), and cancer. Additionally, possible sources of inflammation are vaccination, a surgical procedure, a critical condition of a patient, and treatment with immunomodulatory agents, anti-TNF antibodies, or monoclonal antibodies [390].
In such disease states, the regulation of CYP enzymes is connected with inflammation status [389]. It is recognized that changes related to CYP enzymes are a common consequence of the immunostimulation following infection and inflammation [391,392]. The source of modulation of CYP enzymes’ activities is endogenous inflammation markers: cytokines, adipokines, lipid metabolites of nitric oxide, proteases, and reactive oxygen species [132]. Inflammation is accompanied by suppression of the CYP enzymes that metabolize xenobiotics, including medical drugs [393]. The most studied CYP subfamily in inflammation is CYP3A [390].
In rodent and cell models, transcription of CYP genes is reported to be downregulated by inflammatory signals, e.g., cytokines. Such studies have shown that IL-1β, IL-1α, IL-6, and TNF indirectly lower the activity of multiple P450 enzymes [129,394]. Several clinical studies have revealed low activity of CYP3A4 in inflammatory conditions [271,395]. Some clinical research indicates that inhibitors of IL-6 enhance drug metabolism via CYP3A4, implying that IL-6 is an important regulator of this enzyme [396]. It has been demonstrated that CYP3A4 activity is suppressed in adenovirus-infected primary hepatocytes and that adenovirus-induced modification of PXR expression may be responsible for the alterations of CYP3A4 activity in the liver [397].
Infections
In patients without a clinically significant liver disease, investigation into the effect of chronic hepatitis C on pharmacokinetics of a probe of CYP3A enzymes called midazolam has uncovered weaker CYP3A4 activity in patients with chronic hepatitis C than in healthy volunteers [398,399]. In HIV-infected patients, a study on the activity of CYP enzymes—as assessed by phenotypic tests based on caffeine, dextromethorphan, and midazolam—has revealed a lower activity of CYP3A4 (18%) in HIV-infected adults compared with a control group [400]. Another study involving phenotypic tests based on midazolam, dextromethorphan, and digoxin showed that the activity of CYP3A enzymes is approximately 50% lower in HIV-infected patients than in healthy volunteers [401].
The most vivid pathophysiological feature of COVID-19 is the state of an excessive inflammatory response that may affect the expression of CYP enzymes [402]. The impact of a release of immunogenic proteins in COVID-19 on the activity of CYP enzymes is still largely unexplored. The first research article in this field addressed the effect of moderate-to-severe COVID-19 on enzymatic activity of six major CYP proteins in patients with SARS-CoV-2 infection by a phenotyping-cocktail method [403]. In that report, the activity of CYP enzymes and subsequent phenotypic classification were based on metabolic ratios, and evaluation of the metabolic ratios uncovered a decrease in the activity of CYP3A enzymes by 22.8%. Mean serum levels of C-reactive protein, IL-6, and TNF were significantly higher during SARS-CoV-2 infection than at 3 months after the illness [403].
Furthermore, research has been conducted on the influence of COVID-19 on substrates of CYP enzymes [404]. For instance, it has been documented that plasma concentrations of some substrates of CYP3A enzymes (lopinavir, darunavir, and direct oral anticoagulants) are significantly higher in patients with COVID-19 [405,406,407,408]. In particular, lopinavir concentrations correlated with levels of C-reactive protein and IL-6 because these concentrations diminished after administration of an anti–IL-6 antibody (tocilizumab) [407,409].
It has also been reported that CYP3A enzymes can have an effect on COVID-19 pathophysiology through arachidonic acid metabolites and vitamin D. For example, arachidonic acid metabolism is one of the most affected metabolic pathways during SARS-CoV-2 infection [331], and EETs (whose production is catalyzed by CYP3A enzymes) play a role in kidney damage in this context [332]. The level of an active form of vitamin D, which attenuates overexpression of such inflammatory cytokines as IL-1α, IL-1β, and TNF [369], is determined by CYP3A4 activity. Serum vitamin D levels are low in most of critically ill COVID-19 patients [410]. Thus, CYP3A4, by participating in the metabolism of vitamin D and eicosanoids, may be implicated in COVID-19 pathogenesis. On the other hand, because the expression of CYP3A enzymes can be significantly altered by inflammatory factors in patients with COVID-19, drug pharmacokinetics may be differ too among these patients.
Inflammatory Conditions
There are several relevant articles about patients with metabolic disorders. Research papers about the liver tissue of patients with diabetes mellitus point to low expression and activity of CYP3A enzymes as compared to controls [383]. There is evidence of reduced clearance of lidocaine (primarily metabolized by CYP3A enzymes) in patients with type 1 diabetes mellitus [411].
Type 2 diabetes mellitus is associated with upregulation of inflammation markers such as IL-6 and TNF. High levels of IL-6 and TNF correlate with suppression of some drug-metabolizing enzymes, especially isoforms of CYP3A proteins [412]. A recent study based on a test involving a cocktail of probe drugs (including midazolam) for CYP3A enzymes revealed an approximately 38% decline of the mean metabolic activity of CYP3A enzymes in patients with type 2 diabetes mellitus in comparison with controls [382]. In that report, IL-6 concentration significantly negatively correlated with the activity of CYP3A enzymes in a univariate analysis of data from all diabetic and nondiabetic patients. These results suggest that the deficient CYP-mediated clearance of drugs in type 2 diabetes mellitus may be related to inflammatory processes.
Research on a wide range of cytokines in patients with rheumatoid arthritis in relation to the phenotype of CYP3A4—quantified as the concentration of an endogenous metabolite (4β-hydroxycholesterol) in the serum of patients treated with inhibitors of TNF—has detected significant associations between cytokine levels and the CYP3A4 phenotype. The activity of CYP3A4 has been shown to correlate with levels of proinflammatory cytokines, in particular IL-1Ra (IL-1 receptor antagonist), IL-6, and C-X-C motif chemokine ligand 8 (CXCL8), and to negatively correlate with the CYP3A4 phenotype during the treatment with inhibitors of TNF. These findings indicate that the immune response linked with elevated levels of IL-1Ra, IL-6, and CXCL8 can suppress CYP3A4-mediated metabolism [413]. In another work, patients with rheumatoid arthritis also manifested lower plasma concentrations of 4-β-hydroxycholesterol (an endogenous metabolite of CYP3A4) than did healthy controls [414]. The role of IL-6 in the diminished CYP3A-mediated clearance of drugs in rheumatoid arthritis has been documented in clinical trials of monoclonal antibodies targeting IL-6 [415,416,417]. These studies have shown that pharmacological inhibition of IL-6 restores the activity of CYP3A enzymes and significantly reduces the therapeutic action of simvastatin.
Ulcerative colitis and Crohn‘s disease are two widespread types of inflammatory bowel diseases that are characterized by chronic and progressive inflammation in intestines, e.g., in the colon. In colon biopsy samples from patients with ulcerative colitis and patients with Crohn’s disease, a DNA microarray analysis has uncovered broad suppression of genes involved in drug metabolism; this dysregulation is accompanied by pronounced downregulation of PXR [418]. Some authors have compared serum levels of proinflammatory cytokines and pharmacokinetic parameters of drugs–substrates of CYP3A enzymes between healthy individuals and patients with ulcerative colitis or Crohn’s disease [419,420]; it has been demonstrated there that higher baseline serum levels of proinflammatory cytokines (TNF, IL-1β, IL-6, and IL-8) in the patients do not cause a change in CYP3A4 activity, as evidenced by the pharmacokinetic parameters of the drugs–substrates of CYP3A enzymes in such patients. Those researchers concluded that inflammatory bowel disease does not elevate the production of proinflammatory cytokines to the clinically significant levels that could alter pharmacokinetics of drugs–substrates of CYP3A enzymes [419,420]. Nonetheless, examination of intestinal biopsy samples from people with and without Crohn’s disease and quantification of CYP3A4 expression by Western blotting indicate a significant decrease in protein expression of CYP3A4 in the ileum (by 45%) and colon (by 78%) in subjects with Crohn’s disease relative to subjects without it [421].
In biopsy samples, investigators have demonstrated a decrease in CYP3A4 and PXR mRNA expression in inflamed small-intestinal tissue versus noninflamed duodenum within individual subjects (children) with Crohn’s disease but not in controls [422]. Significant underexpression of CYP3A proteins has been confirmed in the duodenum during active inflammation in children with Crohn’s disease [423]. Research into hepatic and intestinal CYP3A4 activities by means of intravenous and oral midazolam and budesonide in patients with Crohn’s disease and celiac disease has detected a marked drop of CYP3A4 activity in vivo in patients with celiac disease [424]. One of the limitations of that study is a relatively small sample size, which makes the results less generalizable. The findings about downregulation of CYP3A4 mRNA in the intestine in celiac disease has been reproduced in biopsy samples from the duodenum and ileum [425].
Cancer
Inflammatory responses play a crucial part in various stages of carcinogenesis, including initiation, promotion, malignant transformation, invasion, and metastasis. Immune cells that infiltrate tumors participate in extensive and dynamic interactions and antagonistic and synergistic effects with cancer cells. Components of cancerous inflammation include the chemokines, prostaglandins, and cytokines that indirectly suppress the activity of CYP enzymes [426]. In an activity assay of CYP3A enzymes in the liver by a 14C-erythromycin phenotypic breath test in cancer patients with a C-reactive-protein level >10 mg/L, a decline of CYP3A enzymes’ activity by 30% was recorded as compared with patients without the acute phase response [271]. Analysis of phenotypic activity of CYP3A enzymes (the omeprazole sulfone/omeprazole ratio) in advanced ovarian cancer suggests that reduced CYP3A activity correlates with elevated serum concentrations of C-reactive protein, IL-6, IL-8, and TNF [427]. In patients experiencing cancer progression, the expression and activity of CYP3A4 is significantly lower, which can be explained by a greater plasma concentration of inflammation mediators, in particular, IL-6 [428,429].
The course of inflammatory responses can be influenced by such CYP3A substrates as bile acids because the latter interact with certain receptors, e.g., FXR, VDR, and LXR [293]. These receptors are expressed in immune cells such as monocytes, macrophages, natural killer cells, dendritic cells, T- and B-lymphocytes [426]. For instance, a launch of the SHP–FXR pathway blocks the JNK cascade and prevents the binding of NF-κB to promoters of genes encoding proinflammatory cytokines. FXR stimulates the binding of nuclear receptor corepressor 1 (NCOR1) to promoters of proinflammatory genes, thereby hindering the interaction of NF-κB with them [430]. The VDR signaling pathway performs an important function in the regulation of inflammatory responses—especially in inflammatory bowel disease—by transmitting signals of bile acids via VDR within the adaptive immune system [431,432]. LXR is relevant and important for macrophage biology in the context of atherosclerosis, which is currently regarded as a chronic inflammatory disease [433].
4.5.2. Aberrant Intratumoral Expression of CYP3A Enzymes
Overexpression of CYP3A4 in a malignant tissue compared to a respective normal tissue is usually associated with predisposition to breast [319,342,343] and ovarian [344] cancers and may play an important role in the initiation of endometrial cancer [355,357]. We already mentioned that CYP3A4 participates in breast cancer progression by stimulating angiogenesis through VEGF activation [309,310,320] and by promoting cancer cell proliferation through the activation of PI3 kinase–AKT and STAT3 pathways, in part via the synthesis of (+/−)-14,15-EET [313]. CYP3A4 is also required for tumor formation in ER+HER2− breast cancer because this enzyme suppresses autophagy, in part by inhibiting AMPK (AMP-activated protein kinase) activation [326].
Higher expression of CYP3A4 and CYP3A5 than in adjacent normal tissues has been reported in studies on rhabdomyosarcoma, which is the most common soft tissue sarcoma in children [434]. In tumor samples in that study, mRNA expression levels of CYP3A family members proved to be elevated compared to the corresponding normal neighboring-tissue samples; similar differences in protein levels were found, too.
In another research article, analysis of CYP3A4 and CYP3A5 expression in tumor tissue of children with nonrhabdomyosarcoma soft tissue sarcomas also showed that in such patients, mRNA and protein expression levels of these genes are significantly higher in the tumor tissue than in nonmalignant neighboring tissue; estimated 5-year relative survival of the treated patients is ~50% because they typically present with tumor progression, recurrence, metastasis, and/or resistance to chemotherapy [435]. It has been demonstrated that CYP3A4 overexpression may correlate with metastasis of Ewing’s sarcoma [436].
The main clinical consequences of aberrant intratumoral expression of CYP3A4 may be mediated by administration of anticancer drugs that are CYP3A substrates during the treatment of Ewing’s sarcoma. Local expression of CYP3A enzymes in malignant tissue may contribute to the development of multidrug resistance or toxicity observed in this type of tumor.
Recent investigation into the aberrant expression of CYP3A5 in cancer, depending on the malignancy and ability to metastasize, uncovered a role of CYP3A5 in cancer progression, metastasis, and invasion.
Data from early studies pointed to underexpression of CYP3A4 and/or -5 by a tumorous tissue in primary and secondary liver tumors. A study by R. Tsunedomi revealed that CYP3A5 expression is dramatically reduced during venous invasion in patients with hepatitis C–associated hepatocellular carcinoma (hepatitis C–associated HCC) [437].
In contrast to CYP3A4, aberrantly low CYP3A5 expression in HCC and a negative association between CYP3A5 expression and HCC malignant characteristics have been detected in a large HCC cohort; several reports show that CYP3A5 can act as an HCC suppressor and can counteract the malignant tumor phenotype [438,439].
For instance, CYP3A5 expression is often low in tumor tissues at the level of transcripts and proteins in hepatocellular hepatoma; in particular, this underexpression contributes to worse overall survival of patients and to tumor metastasis [438,439]. Ref. [438] indicates that CYP3A5 expression negatively correlates with several malignant characteristics and poor prognosis of HCC; e.g., lower levels of CYP3A5 are associated with more aggressive vascular invasion, poor differentiation, shorter time to disease recurrence after treatment, and worse overall survival of the patients. In the same study, it was found that CYP3A5 can produce reactive oxygen species (ROS) when it metabolizes its substrates, and ROS are known to serve as cellular secondary messengers activating or inhibiting signaling cascades. There is evidence that forced expression of CYP3A5 dramatically suppresses migration and invasion of HCC cells in vitro via inhibition of ROS–mTORC2–p-AKT signaling, and consequently CYP3A5-induced ROS accumulation is a key regulator of the activity of mTORC2: a serine/threonine kinase that translates external stimuli into processes related to cell growth. The reason is that CYP3A5 selectively inhibits the phosphorylation of kinase AKT at Ser473, thus blocking its activity [438].
While examining CYP3A5 expression in many cancers, the authors of ref. [440] discovered that CYP3A5 is aberrantly underexpressed in lung cancer. They demonstrated by RT-PCR, Western blotting, and immunohistochemistry that CYP3A5 is usually downregulated in lung adenocarcinoma (LUAD) tissues and cell lines. Low expression of CYP3A5 is significantly associated with poor prognosis among patients with LUAD. External stimulation of CYP3A5 expression significantly inhibits LUAD cell migration and invasion in vitro and dramatically suppresses metastatic capacity in vivo. A subsequent study has revealed that CYP3A5 significantly reduces the phosphorylation of a TGF-β signaling protein called SMAD1 in LUAD cells [440].
SMAD1 is a central mediator of TGF-β signaling and is involved in a wide range of biological activities including cell growth, apoptosis, development, and immune responses. Several research articles highlight an important stimulatory effect of SMAD1 on tumor cell invasion and metastasis in various types of cancer [441].
It has been shown that a decrease in SMAD1 phosphorylation in LUAD cells leads to the suppression of LUAD metastasis by means of the ATOH8–SMAD1 axis, namely, CYP3A5 interacts with transcription factor ATOH8, and this interaction in turn mediates SMAD1 pathway inactivation [440].
CYP3A5 is the main extrahepatic isoform of CYP3A expressed in the prostate. Androgen receptor (AR) signal transduction is crucial for the growth and progression of prostate cancer. Intratumoral activation of CYP3A5 in prostate cancer is reported to mediate the growth of prostate cancer cells by facilitating nuclear translocation of AR [442]. The observed influence on AR signaling is due to the change in CYP3A5 activity, not in CYP3A4 activity [443].
Thus, the mechanism of regulation of AR expression by CYP3A5 is related to the fact that CYP3A5 is a component of the feedback loop that modulates AR sensitivity to androgens. A decrease in the amount of AR in the nucleus as a result of inhibition of CYP3A5 causes suppression of growth in LNCaP and C4-2 cell lines. On the contrary, the CYP3A inducer rifampicin enhances the nuclear localization of AR. Accordingly, CYP3A5 regulates the translocation of AR into the nucleus and downstream signaling, which leads to tumor [442].
Proof that CYP3A5 inducers promote AR nuclear translocation, downstream signaling, and cell growth whereas CYP3A5 inhibitors reverse these actions is also offered by a study on African Americans, who often carry wild-type CYP3A5 and overexpress the CYP3A5 protein. The observed alterations of AR activity turned out to be specific to changes in CYP3A5 activity because the effects were attenuated by a CYP3A5 knockout in MDAPCa2b cells [443].
Thus, taken together, the presented studies provide a new insight into the participation of CYP3A5 in carcinogenesis. Firstly, CYP3A5 has a protective effect against HCC progression by acting as a suppressor of the pathogenesis and metastasis; secondly, prometastatic signal transduction in LUAD depends on CYP3A5; and finally, CYP3A5 promotes AR nuclear translocation in prostate cancer thereby playing a crucial role in AR signaling. Table 3 summarizes information about the role of CYP3A in the conditions of the diseases.
Table 3.
CYP3A Involvement in Pathological Processes.
5. Concluding Remarks
This review is aimed at highlighting the main roles of CYP3A enzymes along with their unique characteristics in the metabolism of biologically active endogenous compounds and numerous xenobiotics that are important in clinical pharmacology as well as the involvement of these enzymes in a wide range of physiological and pathological phenomena. The scientific literature cited in this review attests to remarkable efforts and advances in the understanding how the CYP3A family of phase I biotransformation enzymes is integrated into the vast and complex network of physiological processes detoxifying endo- and xenobiotics. The function of CYP3A enzymes is complex because the effects of activation their genes are determined by a wide range of endogenous and exogenous ligands and by a unique regulatory system that involves CYP3A enzymes in many physiological and pathological processes in cells and tissues of the body (Figure 5).
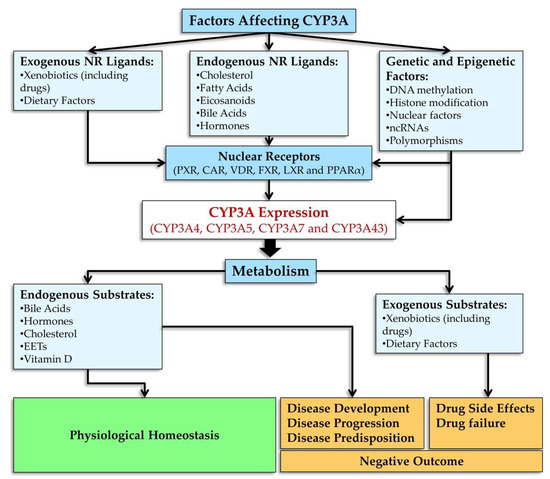
Figure 5.
Effects of CYP3A enzymes on the metabolism of endo- and xenobiotics have an influence on a wide range of physiological and pathophysiological processes in the body.
The totality of evidence indicates that the activation of CYP3A genes can be either beneficial or detrimental during diseases of various organs and tissues. The ultimate effects depend both on the context of a disease and on the nature of ligands of the nuclear receptors that control CYP3A genes’ transcription.
Currently, the molecular mechanisms by which CYP3A enzymes take part in pathogenesis are well understood only for a few diseases; in particular, a role of CYP3A5 in carcinogenesis has been demonstrated. There are more reports of (i) diseases associated with the participation of CYP3A enzymes in the metabolism of endogenous compounds and (ii) pathological conditions affecting the expression and activity of CYP3A enzymes. The consequence of an alteration of these enzymes’ activities is a change in the pharmacokinetics of the drugs used for treatment. Much basic research has been conducted on the role of CYP3A enzymes in pathological processes, but clinical studies that are aimed at influencing the mechanisms of signaling pathways regulating CYP3A genes in various diseases are still insufficient, and further investigation is needed.
Author Contributions
Conceptualization, A.Y.G.; writing—original draft preparation, L.S.K., M.L.P. and A.Y.G.; writing—review and editing, L.S.K., M.L.P. and A.Y.G.; supervision, A.Y.G.; project administration, A.Y.G.; funding acquisition, A.Y.G. and L.S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Foundation for Basic Research, project No. 19-34-90129, and by a government-funded project FGMU-2022-0004, N1021050601082-2-1.6.4;3.1.6.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Shevchuk N.A. for comments and for proofreading the article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 3′UTR | 3′-untranslated region |
| AMPKα | AMP-activated protein kinase |
| AP-1 | activating protein-1 |
| AR | androgen receptor |
| C/EBP | CCAAT/enhancer-binding protein |
| CAR | constitutive androstane receptor |
| CCRP | cytoplasmic constitutive active/androstane receptor retention protein |
| CLEM4 | constitutive liver enhancer module 4 |
| COUP-TF | chicken ovalbumin upstream promoter transcription factor |
| CXCL8 | C-X-C motif chemokine ligand 8 |
| CYP | cytochrome P450 |
| DR | direct repeat |
| EET | epoxyeicosatrienoic acids |
| ER | estrogen receptor |
| ER6 | everted repeats 6 |
| FXR | farnesoid X receptor |
| FXRE | FXR response element |
| GR | glucocorticoid receptor |
| HCC | hepatocellular carcinoma |
| Histone deacetylase 1 | histone deacetylase 1 |
| HIV | human immunodeficiency virus |
| HNF | hepatocyte nuclear factor |
| HNF4A-AS1 | hepatocyte nuclear factor 4α antisense RNA 1 |
| HSP90 | heat shock protein 90 |
| IL | interleukin |
| JAK | Janus kinase |
| LAP | liver-enriched transcriptional activator protein |
| LCA | lithocholic acid |
| LIP | liver-enriched transcriptional inhibitory protein |
| lncRNA | long noncoding RNA |
| LUAD | lung adenocarcinoma |
| LXR | liver X receptor |
| miRNA, miR | microRNA |
| NF-κB | nuclear factor kappa B |
| pERK | proline-rich, extensin-like receptor kinase-1 |
| PGC-1α | peroxisome proliferator-activated receptor-gamma coactivator 1 alpha |
| PPAR | peroxisome proliferator-activated receptor |
| prP | proximal promoter |
| PXR | pregnane X receptor |
| RNA-seq | RNA sequencing |
| ROS | reactive oxygen species |
| SHP | small heterodimeric partner |
| SNP | single-nucleotide polymorphism |
| SOCS1 | suppressor of cytokine signaling 1 |
| STAT | signal transducer and activator of transcription |
| TET | ten-eleven translocation protein |
| TNF | tumor necrosis factor |
| USF1 | upstream transcription factor 1 |
| VDR | vitamin D receptor |
| VEGF | vascular endothelial growth factor |
| XREM | xenobiotic-responsive enhancer module |
References
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Annalora, A.J.; Marcus, C.B.; Iversen, P.L. Alternative Splicing in the Cytochrome P450 Superfamily Expands Protein Diversity to Augment Gene Function and Redirect Human Drug Metabolism. Drug Metab. Dispos. 2017, 45, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Waring, R.H. Cytochrome P450: Genotype to phenotype. Xenobiotica 2020, 50, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.S.; Chaudhry, A.S.; Prasad, B.; Thummel, K.E.; Schuetz, E.G.; Zhong, X.B.; Tien, Y.C.; Jeong, H.; Pan, X.; Shireman, L.M.; et al. Interindividual Variability in Cytochrome P450-Mediated Drug Metabolism. Drug Metab. Dispos. 2016, 44, 343–351. [Google Scholar] [CrossRef]
- Chen, X.; Wang, H.; Zhou, G.; Zhang, X.; Dong, X.; Zhi, L.; Jin, L.; He, F. Molecular population genetics of human CYP3A locus: Signatures of positive selection and implications for evolutionary environmental medicine. Environ. Health Perspect. 2009, 117, 1541–1548. [Google Scholar] [CrossRef][Green Version]
- Gibson, G.G.; Plant, N.J.; Swales, K.E.; Ayrton, A.; El-Sankary, W. Receptor-dependent transcriptional activation of cytochrome P4503A genes: Induction mechanisms, species differences and interindividual variation in man. Xenobiotica 2002, 32, 165–206. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef]
- Daly, A.K. Significance of the minor cytochrome P450 3A isoforms. Clin. Pharmacokinet. 2006, 45, 13–31. [Google Scholar] [CrossRef]
- Raunio, H.; Hakkola, J.; Pelkonen, O. Regulation of CYP3A genes in the human respiratory tract. Chem. Biol. Interact. 2005, 151, 53–62. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Fujino, C.; Sanoh, S.; Katsura, T. Variation in Expression of Cytochrome P450 3A Isoforms and Toxicological Effects: Endo- and Exogenous Substances as Regulatory Factors and Substrates. Biol. Pharm. Bull. 2021, 44, 1617–1634. [Google Scholar] [CrossRef] [PubMed]
- Burk, O.; Wojnowski, L. Cytochrome P450 3A and their regulation. Naunyn Schmiedeberg’s Arch. Pharmacol. 2004, 369, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Penzak, S.R.; Rojas-Fernandez, C. 4β-Hydroxycholesterol as an Endogenous Biomarker for CYP3A Activity: Literature Review and Critical Evaluation. J. Clin. Pharmacol. 2019, 59, 611–624. [Google Scholar] [CrossRef]
- Qin, X.; Wang, X. Role of vitamin D receptor in the regulation of CYP3A gene expression. Acta Pharm. Sin. B 2019, 9, 1087–1098. [Google Scholar] [CrossRef]
- Tsujimoto, M.; Horie, M.; Honda, H.; Takara, K.; Nishiguchi, K. The structure-activity correlation on the inhibitory effects of flavonoids on cytochrome P450 3A activity. Biol. Pharm. Bull. 2009, 32, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K. Food-drug interactions via human cytochrome P450 3A (CYP3A). Drug Metab. Drug Interact. 2004, 20, 195–217. [Google Scholar] [CrossRef]
- Chen, S.; Wang, K.; Wan, Y.J. Retinoids activate RXR/CAR-mediated pathway and induce CYP3A. Biochem. Pharmacol. 2010, 79, 270–276. [Google Scholar] [CrossRef]
- Li, X.; Wang, Z.; Klaunig, J.E. Modulation of xenobiotic nuclear receptors in high-fat diet induced non-alcoholic fatty liver disease. Toxicology 2018, 410, 199–213. [Google Scholar] [CrossRef]
- Finta, C.; Zaphiropoulos, P.G. Intergenic mRNA molecules resulting from trans-splicing. J. Biol. Chem. 2002, 277, 5882–5890. [Google Scholar] [CrossRef]
- Gellner, K.; Eiselt, R.; Hustert, E.; Arnold, H.; Koch, I.; Haberl, M.; Deglmann, C.J.; Burk, O.; Buntefuss, D.; Escher, S.; et al. Genomic organization of the human CYP3A locus: Identification of a new, inducible CYP3A gene. Pharmacogenetics 2001, 11, 111–121. [Google Scholar] [CrossRef]
- Ramos, K.N.; Gregornik, D.; Ramos, K.S. Pharmacogenomics insights into precision pediatric oncology. Curr. Opin. Pediatr. 2021, 33, 564–569. [Google Scholar] [CrossRef]
- Ensembl. Available online: https://www.ensembl.org/ (accessed on 10 October 2022).
- Ohtsuki, S.; Schaefer, O.; Kawakami, H.; Inoue, T.; Liehner, S.; Saito, A.; Ishiguro, N.; Kishimoto, W.; Ludwig-Schwellinger, E.; Ebner, T.; et al. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: Comparison with mRNA levels and activities. Drug Metab. Dispos. 2012, 40, 83–92. [Google Scholar] [CrossRef]
- Domanski, T.L.; Finta, C.; Halpert, J.R.; Zaphiropoulos, P.G. cDNA cloning and initial characterization of CYP3A43, a novel human cytochrome P450. Mol. Pharmacol. 2001, 59, 386–392. [Google Scholar] [CrossRef]
- Nem, D.; Baranyai, D.; Qiu, H.; Godtel-Armbrust, U.; Nestler, S.; Wojnowski, L. Pregnane X receptor and yin yang 1 contribute to the differential tissue expression and induction of CYP3A5 and CYP3A4. PLoS ONE 2012, 7, e30895. [Google Scholar] [CrossRef]
- Ince, I.; Knibbe, C.A.; Danhof, M.; de Wildt, S.N. Developmental changes in the expression and function of cytochrome P450 3A isoforms: Evidence from in vitro and in vivo investigations. Clin. Pharmacokinet. 2013, 52, 333–345. [Google Scholar] [CrossRef]
- Achour, B.; Barber, J.; Rostami-Hodjegan, A. Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: A meta-analysis. Drug Metab. Dispos. 2014, 42, 1349–1356. [Google Scholar] [CrossRef]
- Williams, E.T.; Leyk, M.; Wrighton, S.A.; Davies, P.J.; Loose, D.S.; Shipley, G.L.; Strobel, H.W. Estrogen regulation of the cytochrome P450 3A subfamily in humans. J. Pharmacol. Exp. Ther. 2004, 311, 728–735. [Google Scholar] [CrossRef]
- Zhang, Y.; Klein, K.; Sugathan, A.; Nassery, N.; Dombkowski, A.; Zanger, U.M.; Waxman, D.J. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. PLoS ONE 2011, 6, e23506. [Google Scholar] [CrossRef]
- Leeder, J.S.; Gaedigk, R.; Marcucci, K.A.; Gaedigk, A.; Vyhlidal, C.A.; Schindel, B.P.; Pearce, R.E. Variability of CYP3A7 expression in human fetal liver. J. Pharmacol. Exp. Ther. 2005, 314, 626–635. [Google Scholar] [CrossRef]
- Fanni, D.; Fanos, V.; Ambu, R.; Lai, F.; Gerosa, C.; Pampaloni, P.; Van Eyken, P.; Senes, G.; Castagnola, M.; Faa, G. Overlapping between CYP3A4 and CYP3A7 expression in the fetal human liver during development. J. Matern. Fetal Neonatal. Med. 2015, 28, 1291–1295. [Google Scholar] [CrossRef]
- Huang, W.; Lin, Y.S.; McConn, D.J., 2nd; Calamia, J.C.; Totah, R.A.; Isoherranen, N.; Glodowski, M.; Thummel, K.E. Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab. Dispos. 2004, 32, 1434–1445. [Google Scholar] [CrossRef]
- Vyhlidal, C.A.; Pearce, R.E.; Gaedigk, R.; Calamia, J.C.; Shuster, D.L.; Thummel, K.E.; Leeder, J.S. Variability in Expression of CYP3A5 in Human Fetal Liver. Drug Metab. Dispos. 2015, 43, 1286–1293. [Google Scholar] [CrossRef]
- Sevrioukova, I.F. Structural Basis for the Diminished Ligand Binding and Catalytic Ability of Human Fetal-Specific CYP3A7. Int. J. Mol. Sci. 2021, 22, 5831. [Google Scholar] [CrossRef]
- Wilson, R.T.; Masters, L.D.; Barnholtz-Sloan, J.S.; Salzberg, A.C.; Hartman, T.J. Ancestry-Adjusted Vitamin D Metabolite Concentrations in Association With Cytochrome P450 3A Polymorphisms. Am. J. Epidemiol. 2018, 187, 754–766. [Google Scholar] [CrossRef]
- An, X.X.; Yu, Y.; Li, G.F.; Yu, G. Abundance and Associated Variations of Cytochrome P450 Drug-Metabolizing Enzymes in the Liver of East Asian Adults: A Meta-Analysis. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 225–233. [Google Scholar] [CrossRef]
- Montanha, M.C.; Cottura, N.; Booth, M.; Hodge, D.; Bunglawala, F.; Kinvig, H.; Granana-Castillo, S.; Lloyd, A.; Khoo, S.; Siccardi, M. PBPK Modelling of Dexamethasone in Patients With COVID-19 and Liver Disease. Front. Pharmacol. 2022, 13, 814134. [Google Scholar] [CrossRef]
- Westlind-Johnsson, A.; Malmebo, S.; Johansson, A.; Otter, C.; Andersson, T.B.; Johansson, I.; Edwards, R.J.; Boobis, A.R.; Ingelman-Sundberg, M. Comparative analysis of CYP3A expression in human liver suggests only a minor role for CYP3A5 in drug metabolism. Drug Metab. Dispos. 2003, 31, 755–761. [Google Scholar] [CrossRef]
- Kawakami, H.; Ohtsuki, S.; Kamiie, J.; Suzuki, T.; Abe, T.; Terasaki, T. Simultaneous absolute quantification of 11 cytochrome P450 isoforms in human liver microsomes by liquid chromatography tandem mass spectrometry with in silico target peptide selection. J. Pharm. Sci. 2011, 100, 341–352. [Google Scholar] [CrossRef]
- Wolbold, R.; Klein, K.; Burk, O.; Nussler, A.K.; Neuhaus, P.; Eichelbaum, M.; Schwab, M.; Zanger, U.M. Sex is a major determinant of CYP3A4 expression in human liver. Hepatology 2003, 38, 978–988. [Google Scholar] [CrossRef]
- Lin, Y.S.; Dowling, A.L.; Quigley, S.D.; Farin, F.M.; Zhang, J.; Lamba, J.; Schuetz, E.G.; Thummel, K.E. Co-regulation of CYP3A4 and CYP3A5 and contribution to hepatic and intestinal midazolam metabolism. Mol. Pharmacol. 2002, 62, 162–172. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 2014, 13, 397–406. [Google Scholar] [CrossRef]
- GeneCards. Available online: https://www.genecards.org (accessed on 10 October 2022).
- Klees, T.M.; Sheffels, P.; Thummel, K.E.; Kharasch, E.D. Pharmacogenetic determinants of human liver microsomal alfentanil metabolism and the role of cytochrome P450 3A5. Anesthesiology 2005, 102, 550–556. [Google Scholar] [CrossRef]
- Bieche, I.; Narjoz, C.; Asselah, T.; Vacher, S.; Marcellin, P.; Lidereau, R.; Beaune, P.; de Waziers, I. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharm. Genom. 2007, 17, 731–742. [Google Scholar] [CrossRef]
- Thelen, K.; Dressman, J.B. Cytochrome P450-mediated metabolism in the human gut wall. J. Pharm. Pharmacol. 2009, 61, 541–558. [Google Scholar] [CrossRef]
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The human intestinal cytochrome P450 “pie”. Drug Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef]
- Aleksa, K.; Matsell, D.; Krausz, K.; Gelboin, H.; Ito, S.; Koren, G. Cytochrome P450 3A and 2B6 in the developing kidney: Implications for ifosfamide nephrotoxicity. Pediatr. Nephrol. 2005, 20, 872–885. [Google Scholar] [CrossRef]
- Du, L.; Neis, M.M.; Ladd, P.A.; Lanza, D.L.; Yost, G.S.; Keeney, D.S. Effects of the differentiated keratinocyte phenotype on expression levels of CYP1-4 family genes in human skin cells. Toxicol. Appl. Pharmacol. 2006, 213, 135–144. [Google Scholar] [CrossRef]
- Dutheil, F.; Dauchy, S.; Diry, M.; Sazdovitch, V.; Cloarec, O.; Mellottee, L.; Bieche, I.; Ingelman-Sundberg, M.; Flinois, J.P.; de Waziers, I.; et al. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: Regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab. Dispos. 2009, 37, 1528–1538. [Google Scholar] [CrossRef]
- Agarwal, V.; Kommaddi, R.P.; Valli, K.; Ryder, D.; Hyde, T.M.; Kleinman, J.E.; Strobel, H.W.; Ravindranath, V. Drug metabolism in human brain: High levels of cytochrome P4503A43 in brain and metabolism of anti-anxiety drug alprazolam to its active metabolite. PLoS ONE 2008, 3, e2337. [Google Scholar] [CrossRef]
- Booth Depaz, I.M.; Toselli, F.; Wilce, P.A.; Gillam, E.M. Differential expression of human cytochrome P450 enzymes from the CYP3A subfamily in the brains of alcoholic subjects and drug-free controls. Drug Metab. Dispos. 2013, 41, 1187–1194. [Google Scholar] [CrossRef]
- Michaud, V.; Frappier, M.; Dumas, M.C.; Turgeon, J. Metabolic activity and mRNA levels of human cardiac CYP450s involved in drug metabolism. PLoS ONE 2010, 5, e15666. [Google Scholar] [CrossRef]
- Biggs, J.S.; Wan, J.; Cutler, N.S.; Hakkola, J.; Uusimaki, P.; Raunio, H.; Yost, G.S. Transcription factor binding to a putative double E-box motif represses CYP3A4 expression in human lung cells. Mol. Pharmacol. 2007, 72, 514–525. [Google Scholar] [CrossRef]
- Jover, R.; Moya, M.; Gomez-Lechon, M.J. Transcriptional regulation of cytochrome p450 genes by the nuclear receptor hepatocyte nuclear factor 4-alpha. Curr. Drug Metab. 2009, 10, 508–519. [Google Scholar] [CrossRef]
- Tirona, R.G.; Lee, W.; Leake, B.F.; Lan, L.B.; Cline, C.B.; Lamba, V.; Parviz, F.; Duncan, S.A.; Inoue, Y.; Gonzalez, F.J.; et al. The orphan nuclear receptor HNF4α determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat. Med. 2003, 9, 220–224. [Google Scholar] [CrossRef]
- Tegude, H.; Schnabel, A.; Zanger, U.M.; Klein, K.; Eichelbaum, M.; Burk, O. Molecular mechanism of basal CYP3A4 regulation by hepatocyte nuclear factor 4α: Evidence for direct regulation in the intestine. Drug Metab. Dispos. 2007, 35, 946–954. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Bort, R.; Jover, R.; Tindberg, N.; Ingelman-Sundberg, M.; Gomez-Lechon, M.J.; Castell, J.V. Transcriptional regulation of human CYP3A4 basal expression by CCAAT enhancer-binding protein α and hepatocyte nuclear factor-3γ. Mol. Pharmacol. 2003, 63, 1180–1189. [Google Scholar] [CrossRef]
- Martinez-Jimenez, C.P.; Gomez-Lechon, M.J.; Castell, J.V.; Jover, R. Transcriptional regulation of the human hepatic CYP3A4: Identification of a new distal enhancer region responsive to CCAAT/enhancer-binding protein β isoforms (liver activating protein and liver inhibitory protein). Mol. Pharmacol. 2005, 67, 2088–2101. [Google Scholar] [CrossRef]
- Gonzalez, F.J. Regulation of hepatocyte nuclear factor 4α-mediated transcription. Drug Metab. Pharmacokinet. 2008, 23, 2–7. [Google Scholar] [CrossRef]
- Thakur, A.; Wong, J.C.H.; Wang, E.Y.; Lotto, J.; Kim, D.; Cheng, J.C.; Mingay, M.; Cullum, R.; Moudgil, V.; Ahmed, N.; et al. Hepatocyte Nuclear Factor 4-Alpha Is Essential for the Active Epigenetic State at Enhancers in Mouse Liver. Hepatology 2019, 70, 1360–1376. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y. Rifampicin induction of CYP3A4 requires pregnane X receptor cross talk with hepatocyte nuclear factor 4α and coactivators, and suppression of small heterodimer partner gene expression. Drug Metab. Dispos. 2006, 34, 756–764. [Google Scholar] [CrossRef]
- Wang, P.; Chen, S.; Wang, Y.; Wang, X.; Yan, L.; Yang, K.; Zhong, X.B.; Han, S.; Zhang, L. The Long Noncoding RNA Hepatocyte Nuclear Factor 4alpha Antisense RNA 1 Negatively Regulates Cytochrome P450 Enzymes in Huh7 Cells via Histone Modifications. Drug Metab. Dispos. 2021, 49, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Saito, T.; Takahashi, Y.; Ozeki, T.; Kiyotani, K.; Fujieda, M.; Yamazaki, H.; Kunitoh, H.; Kamataki, T. Identification of a novel polymorphic enhancer of the human CYP3A4 gene. Mol. Pharmacol. 2004, 65, 326–334. [Google Scholar] [CrossRef]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Wedel, A.; Ziegler-Heitbrock, H.W. The C/EBP family of transcription factors. Immunobiology 1995, 193, 171–185. [Google Scholar] [CrossRef]
- An, M.R.; Hsieh, C.C.; Reisner, P.D.; Rabek, J.P.; Scott, S.G.; Kuninger, D.T.; Papaconstantinou, J. Evidence for posttranscriptional regulation of C/EBPalpha and C/EBPbeta isoform expression during the lipopolysaccharide-mediated acute-phase response. Mol. Cell. Biol. 1996, 16, 2295–2306. [Google Scholar] [CrossRef]
- Welm, A.L.; Timchenko, N.A.; Darlington, G.J. C/EBPα regulates generation of C/EBPβ isoforms through activation of specific proteolytic cleavage. Mol. Cell. Biol. 1999, 19, 1695–1704. [Google Scholar] [CrossRef]
- Descombes, P.; Schibler, U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 1991, 67, 569–579. [Google Scholar] [CrossRef]
- Raught, B.; Liao, W.S.; Rosen, J.M. Developmentally and hormonally regulated CCAAT/enhancer-binding protein isoforms influence beta-casein gene expression. Mol. Endocrinol. 1995, 9, 1223–1232. [Google Scholar] [CrossRef][Green Version]
- Zahnow, C.A.; Cardiff, R.D.; Laucirica, R.; Medina, D.; Rosen, J.M. A role for CCAAT/enhancer binding protein beta-liver-enriched inhibitory protein in mammary epithelial cell proliferation. Cancer Res. 2001, 61, 261–269. [Google Scholar]
- Huber, R.; Pietsch, D.; Panterodt, T.; Brand, K. Regulation of C/EBPβ and resulting functions in cells of the monocytic lineage. Cell. Signal. 2012, 24, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, K.N.; Chen, C. The role of CYP3A4 in the biotransformation of bile acids and therapeutic implication for cholestasis. Ann. Transl. Med. 2014, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Quattrochi, L.C.; Guzelian, P.S. Cyp3A regulation: From pharmacology to nuclear receptors. Drug Metab. Dispos. 2001, 29, 615–622. [Google Scholar] [PubMed]
- Goodwin, B.; Hodgson, E.; Liddle, C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol. Pharmacol. 1999, 56, 1329–1339. [Google Scholar] [CrossRef]
- Goodwin, B.; Hodgson, E.; D’Costa, D.J.; Robertson, G.R.; Liddle, C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol. Pharmacol. 2002, 62, 359–365. [Google Scholar] [CrossRef]
- Jover, R.; Bort, R.; Gomez-Lechon, M.J.; Castell, J.V. Cytochrome P450 regulation by hepatocyte nuclear factor 4 in human hepatocytes: A study using adenovirus-mediated antisense targeting. Hepatology 2001, 33, 668–675. [Google Scholar] [CrossRef]
- Thomas, M.; Burk, O.; Klumpp, B.; Kandel, B.A.; Damm, G.; Weiss, T.S.; Klein, K.; Schwab, M.; Zanger, U.M. Direct transcriptional regulation of human hepatic cytochrome P450 3A4 (CYP3A4) by peroxisome proliferator—Activated receptor alpha (PPARα). Mol. Pharmacol. 2013, 83, 709–718. [Google Scholar] [CrossRef]
- Pascussi, J.M.; Drocourt, L.; Gerbal-Chaloin, S.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur. J. Biochem. 2001, 268, 6346–6358. [Google Scholar] [CrossRef]
- Duniec-Dmuchowski, Z.; Ellis, E.; Strom, S.C.; Kocarek, T.A. Regulation of CYP3A4 and CYP2B6 expression by liver X receptor agonists. Biochem. Pharmacol. 2007, 74, 1535–1540. [Google Scholar] [CrossRef]
- Wang, D.; Lu, R.; Rempala, G.; Sadee, W. Ligand-Free Estrogen Receptor α (ESR1) as Master Regulator for the Expression of CYP3A4 and Other Cytochrome P450 Enzymes in the Human Liver. Mol. Pharmacol. 2019, 96, 430–440. [Google Scholar] [CrossRef]
- Carazo, A.; Hyrsova, L.; Dusek, J.; Chodounska, H.; Horvatova, A.; Berka, K.; Bazgier, V.; Gan-Schreier, H.; Chamulitrat, W.; Kudova, E.; et al. Acetylated deoxycholic (DCA) and cholic (CA) acids are potent ligands of pregnane X (PXR) receptor. Toxicol. Lett. 2017, 265, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Gnerre, C.; Blattler, S.; Kaufmann, M.R.; Looser, R.; Meyer, U.A. Regulation of CYP3A4 by the bile acid receptor FXR: Evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics 2004, 14, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Adachi, R.; Honma, Y.; Masuno, H.; Kawana, K.; Shimomura, I.; Yamada, S.; Makishima, M. Selective activation of vitamin D receptor by lithocholic acid acetate, a bile acid derivative. J. Lipid Res. 2005, 46, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Pascussi, J.M.; Gerbal-Chaloin, S.; Duret, C.; Daujat-Chavanieu, M.; Vilarem, M.J.; Maurel, P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: Crosstalk and consequences. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Yoshinari, K.; Aoyama, K.; Sugawara, M.; Sekiya, Y.; Nagata, K.; Yamazoe, Y. Role of vitamin D receptor in the lithocholic acid-mediated CYP3A induction in vitro and in vivo. Drug Metab. Dispos. 2008, 36, 2058–2063. [Google Scholar] [CrossRef]
- Schroder, A.; Wollnik, J.; Wrzodek, C.; Drager, A.; Bonin, M.; Burk, O.; Thomas, M.; Thasler, W.E.; Zanger, U.M.; Zell, A. Inferring statin-induced gene regulatory relationships in primary human hepatocytes. Bioinformatics 2011, 27, 2473–2477. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Hooiveld, G.; Muller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARα between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef]
- Kojima, K.; Nagata, K.; Matsubara, T.; Yamazoe, Y. Broad but distinct role of pregnane x receptor on the expression of individual cytochrome p450s in human hepatocytes. Drug Metab. Pharmacokinet. 2007, 22, 276–286. [Google Scholar] [CrossRef]
- Liu, F.J.; Song, X.; Yang, D.; Deng, R.; Yan, B. The far and distal enhancers in the CYP3A4 gene co-ordinate the proximal promoter in responding similarly to the pregnane X receptor but differentially to hepatocyte nuclear factor-4α. Biochem. J. 2008, 409, 243–250. [Google Scholar] [CrossRef]
- Taneja, G.; Maity, S.; Jiang, W.; Moorthy, B.; Coarfa, C.; Ghose, R. Transcriptomic profiling identifies novel mechanisms of transcriptional regulation of the cytochrome P450 (Cyp)3a11 gene. Sci. Rep. 2019, 9, 6663. [Google Scholar] [CrossRef]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Toriyabe, T.; Nagata, K.; Takada, T.; Aratsu, Y.; Matsubara, T.; Yoshinari, K.; Yamazoe, Y. Unveiling a new essential cis element for the transactivation of the CYP3A4 gene by xenobiotics. Mol. Pharmacol. 2009, 75, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Makkonen, H.; Dunlop, T.W.; Matilainen, M.; Vaisanen, S.; Carlberg, C. Identification of pregnane X receptor binding sites in the regulatory regions of genes involved in bile acid homeostasis. J. Mol. Biol. 2005, 346, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef]
- Wang, Y.M.; Ong, S.S.; Chai, S.C.; Chen, T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin. Drug Metab. Toxicol. 2012, 8, 803–817. [Google Scholar] [CrossRef]
- Chai, X.; Zeng, S.; Xie, W. Nuclear receptors PXR and CAR: Implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin. Drug Metab. Toxicol. 2013, 9, 253–266. [Google Scholar] [CrossRef]
- Faucette, S.R.; Sueyoshi, T.; Smith, C.M.; Negishi, M.; Lecluyse, E.L.; Wang, H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J. Pharmacol. Exp. Ther. 2006, 317, 1200–1209. [Google Scholar] [CrossRef]
- Xie, W.; Barwick, J.L.; Simon, C.M.; Pierce, A.M.; Safe, S.; Blumberg, B.; Guzelian, P.S.; Evans, R.M. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000, 14, 3014–3023. [Google Scholar] [CrossRef]
- Kakizaki, S.; Yamamoto, Y.; Ueda, A.; Moore, R.; Sueyoshi, T.; Negishi, M. Phenobarbital induction of drug/steroid-metabolizing enzymes and nuclear receptor CAR. Biochim. Biophys. Acta Gen. Subj. 2003, 1619, 239–242. [Google Scholar] [CrossRef]
- Pike, J.W.; Meyer, M.B.; Lee, S.M.; Onal, M.; Benkusky, N.A. The vitamin D receptor: Contemporary genomic approaches reveal new basic and translational insights. J. Clin. Investig. 2017, 127, 1146–1154. [Google Scholar] [CrossRef]
- Drocourt, L.; Ourlin, J.C.; Pascussi, J.M.; Maurel, P.; Vilarem, M.J. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J. Biol. Chem. 2002, 277, 25125–25132. [Google Scholar] [CrossRef]
- Istrate, M.A.; Nussler, A.K.; Eichelbaum, M.; Burk, O. Regulation of CYP3A4 by pregnane X receptor: The role of nuclear receptors competing for response element binding. Biochem. Biophys. Res. Commun. 2010, 393, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Stanimirov, B.; Stankov, K.; Mikov, M. Pleiotropic functions of bile acids mediated by the farnesoid X receptor. Acta Gastro Enterol. Belg. 2012, 75, 389–398. [Google Scholar]
- Garcia, M.; Thirouard, L.; Sedes, L.; Monrose, M.; Holota, H.; Caira, F.; Volle, D.H.; Beaudoin, C. Nuclear Receptor Metabolism of Bile Acids and Xenobiotics: A Coordinated Detoxification System with Impact on Health and Diseases. Int. J. Mol. Sci. 2018, 19, 3630. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017, 1, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Pan, X.; Jeong, H. GW4064, an agonist of farnesoid X receptor, represses CYP3A4 expression in human hepatocytes by inducing small heterodimer partner expression. Drug Metab. Dispos. 2015, 43, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ding, X.; Fang, C.; Zhang, Q.Y. Regulation of intestinal cytochrome P450 expression by hepatic cytochrome P450: Possible involvement of fibroblast growth factor 15 and impact on systemic drug exposure. Mol. Pharmacol. 2014, 85, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef]
- Inoue, J.; Satoh, S.; Kita, M.; Nakahara, M.; Hachimura, S.; Miyata, M.; Nishimaki-Mogami, T.; Sato, R. PPARalpha gene expression is up-regulated by LXR and PXR activators in the small intestine. Biochem. Biophys. Res. Commun. 2008, 371, 675–678. [Google Scholar] [CrossRef]
- Hu, X.; Li, S.; Wu, J.; Xia, C.; Lala, D.S. Liver X receptors interact with corepressors to regulate gene expression. Mol. Endocrinol. 2003, 17, 1019–1026. [Google Scholar] [CrossRef]
- Codina, A.; Love, J.D.; Li, Y.; Lazar, M.A.; Neuhaus, D.; Schwabe, J.W. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2005, 102, 6009–6014. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.G.; Phillips, A.; Aouabdi, S.; Plant, K.; Plant, N. Transcriptional regulation of the human pregnane-X receptor. Drug Metab. Rev. 2006, 38, 31–49. [Google Scholar] [CrossRef]
- Aouabdi, S.; Gibson, G.; Plant, N. Transcriptional regulation of the PXR gene: Identification and characterization of a functional peroxisome proliferator-activated receptor α binding site within the proximal promoter of PXR. Drug Metab. Dispos. 2006, 34, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Smutny, T.; Hyrsova, L.; Braeuning, A.; Ingelman-Sundberg, M.; Pavek, P. Transcriptional and post-transcriptional regulation of the pregnane X receptor: A rationale for interindividual variability in drug metabolism. Arch. Toxicol. 2021, 95, 11–25. [Google Scholar] [CrossRef]
- Burk, O.; Koch, I.; Raucy, J.; Hustert, E.; Eichelbaum, M.; Brockmoller, J.; Zanger, U.M.; Wojnowski, L. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). J. Biol. Chem. 2004, 279, 38379–38385. [Google Scholar] [CrossRef]
- Shi, Y.; Seto, E.; Chang, L.S.; Shenk, T. Transcriptional repression by YY1, a human GLI-Kruppel-related protein, and relief of repression by adenovirus E1A protein. Cell 1991, 67, 377–388. [Google Scholar] [CrossRef]
- Thompson, E.E.; Kuttab-Boulos, H.; Witonsky, D.; Yang, L.; Roe, B.A.; Di Rienzo, A. CYP3A variation and the evolution of salt-sensitivity variants. Am. J. Hum. Genet. 2004, 75, 1059–1069. [Google Scholar] [CrossRef]
- Bertilsson, G.; Berkenstam, A.; Blomquist, P. Functionally conserved xenobiotic responsive enhancer in cytochrome P450 3A7. Biochem. Biophys. Res. Commun. 2001, 280, 139–144. [Google Scholar] [CrossRef]
- Hara, H.; Yasunami, Y.; Adachi, T. Loss of CYP3A7 gene induction by 1,25-dihydroxyvitamin D3 is caused by less binding of VDR to the proximal ER6 in CYP3A7 gene. Biochem. Biophys. Res. Commun. 2004, 321, 909–915. [Google Scholar] [CrossRef]
- Collins, J.M.; Wang, D. Cis-acting regulatory elements regulating CYP3A4 transcription in human liver. Pharm. Genom. 2020, 30, 107–116. [Google Scholar] [CrossRef]
- Gu, X.; Ke, S.; Liu, D.; Sheng, T.; Thomas, P.E.; Rabson, A.B.; Gallo, M.A.; Xie, W.; Tian, Y. Role of NF-κB in regulation of PXR-mediated gene expression: A mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J. Biol. Chem. 2006, 281, 17882–17889. [Google Scholar] [CrossRef]
- Okamura, M.; Shizu, R.; Hosaka, T.; Sasaki, T.; Yoshinari, K. Possible involvement of the competition for the transcriptional coactivator glucocorticoid receptor-interacting protein 1 in the inflammatory signal-dependent suppression of PXR-mediated CYP3A induction in vitro. Drug Metab. Pharmacokinet. 2019, 34, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, N.; Ji, X.; Mai, J.; Li, Q. Organotin compound DBDCT induces CYP3A suppression through NF-κB-mediated repression of PXR activity. Metallomics 2019, 11, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Bort, R.; Gomez-Lechon, M.J.; Castell, J.V. Down-regulation of human CYP3A4 by the inflammatory signal interleukin-6: Molecular mechanism and transcription factors involved. FASEB J. 2002, 16, 1799–1801. [Google Scholar] [CrossRef]
- Mimura, H.; Kobayashi, K.; Xu, L.; Hashimoto, M.; Ejiri, Y.; Hosoda, M.; Chiba, K. Effects of cytokines on CYP3A4 expression and reversal of the effects by anti-cytokine agents in the three-dimensionally cultured human hepatoma cell line FLC-4. Drug Metab. Pharmacokinet. 2015, 30, 105–110. [Google Scholar] [CrossRef]
- Aitken, A.E.; Morgan, E.T. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab. Dispos. 2007, 35, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Dickmann, L.J.; Patel, S.K.; Wienkers, L.C.; Slatter, J.G. Effects of interleukin 1β (IL-1β) and IL-1β/interleukin 6 (IL-6) combinations on drug metabolizing enzymes in human hepatocyte culture. Curr. Drug Metab. 2012, 13, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Ghose, R.; Mallick, P.; Taneja, G.; Chu, C.; Moorthy, B. In Vitro Approaches to Study Regulation of Hepatic Cytochrome P450 (CYP) 3A Expression by Paclitaxel and Rifampicin. Methods Mol. Biol. 2016, 1395, 55–68. [Google Scholar] [CrossRef] [PubMed]
- de Jong, L.M.; Jiskoot, W.; Swen, J.J.; Manson, M.L. Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes 2020, 11, 1509. [Google Scholar] [CrossRef]
- Dickmann, L.J.; Patel, S.K.; Rock, D.A.; Wienkers, L.C.; Slatter, J.G. Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab. Dispos. 2011, 39, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Vachirayonsti, T.; Ho, K.W.; Yang, D.; Yan, B. Suppression of the pregnane X receptor during endoplasmic reticulum stress is achieved by down-regulating hepatocyte nuclear factor-4α and up-regulating liver-enriched inhibitory protein. Toxicol. Sci. 2015, 144, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Kot, M.; Daujat-Chavanieu, M. Altered cytokine profile under control of the serotonergic system determines the regulation of CYP2C11 and CYP3A isoforms. Food Chem. Toxicol. 2018, 116, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tolleson, W.H.; Yu, D.; Chen, S.; Guo, L.; Xiao, W.; Tong, W.; Ning, B. Regulation of cytochrome P450 expression by microRNAs and long noncoding RNAs: Epigenetic mechanisms in environmental toxicology and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2019, 37, 180–214. [Google Scholar] [CrossRef]
- Giebel, N.L.; Shadley, J.D.; McCarver, D.G.; Dorko, K.; Gramignoli, R.; Strom, S.C.; Yan, K.; Simpson, P.M.; Hines, R.N. Role of Chromatin Structural Changes in Regulating Human CYP3A Ontogeny. Drug Metab. Dispos. 2016, 44, 1027–1037. [Google Scholar] [CrossRef]
- Wang, X.; Wei, L.; Yang, J.; Wang, Y.; Chen, S.; Yang, K.; Meng, X.; Zhang, L. DNA methylation determines the regulation of pregnane X receptor on CYP3A4 expression. Clin. Exp. Pharmacol. Physiol. 2021, 48, 250–259. [Google Scholar] [CrossRef]
- Kugler, N.; Klein, K.; Zanger, U.M. MiR-155 and other microRNAs downregulate drug metabolizing cytochromes P450 in inflammation. Biochem. Pharmacol. 2020, 171, 113725. [Google Scholar] [CrossRef]
- Pan, Y.Z.; Gao, W.; Yu, A.M. MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab. Dispos. 2009, 37, 2112–2117. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, M.; Liu, L.; Peng, J.; Guo, C.; Chen, X.; Huang, L.; Tan, J.; Yang, G. Transcriptional Repression of CYP3A4 by Increased miR-200a-3p and miR-150-5p Promotes Steatosis in vitro. Front. Genet. 2019, 10, 484. [Google Scholar] [CrossRef]
- Ekstrom, L.; Skilving, I.; Ovesjo, M.L.; Aklillu, E.; Nylen, H.; Rane, A.; Diczfalusy, U.; Bjorkhem-Bergman, L. miRNA-27b levels are associated with CYP3A activity in vitro and in vivo. Pharmacol. Res. Perspect. 2015, 3, e00192. [Google Scholar] [CrossRef]
- Zastrozhin, M.S.; Skryabin, V.Y.; Smirnov, V.V.; Petukhov, A.E.; Pankratenko, E.P.; Zastrozhina, A.K.; Grishina, E.A.; Ryzhikova, K.A.; Bure, I.V.; Golovinskii, P.A.; et al. Effects of plasma concentration of micro-RNA Mir-27b and CYP3A4*22 on equilibrium concentration of alprazolam in patients with anxiety disorders comorbid with alcohol use disorder. Gene 2020, 739, 144513. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Nakajima, M.; Mohri, T.; Yokoi, T. Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J. Biol. Chem. 2008, 283, 9674–9680. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, W.; Zha, J.; Yan, Y.; Wei, Y.; Chen, X.; Zhu, X.; Ge, L. miR543 acts as a novel oncogene in oral squamous cell carcinoma by targeting CYP3A5. Oncol. Rep. 2019, 42, 973–990. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.Q.; Tang, D.; Wan, P.; Qin, T.; Yang, T.H.; Wu, J.; Ji, H.; Liu, J.C.; Xue, F.; Tang, Y.J.; et al. Multiple microRNAs regulate tacrolimus metabolism through CYP3A5. Pharmacol. Res. 2021, 164, 105382. [Google Scholar] [CrossRef]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Wang, X.; Lyu, Y.; Chen, X.; Liu, K.; et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef]
- Chen, L.; Bao, Y.; Piekos, S.C.; Zhu, K.; Zhang, L.; Zhong, X.B. A Transcriptional Regulatory Network Containing Nuclear Receptors and Long Noncoding RNAs Controls Basal and Drug-Induced Expression of Cytochrome P450s in HepaRG Cells. Mol. Pharmacol. 2018, 94, 749–759. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharm. J. 2011, 11, 274–286. [Google Scholar] [CrossRef]
- Hsieh, K.P.; Lin, Y.Y.; Cheng, C.L.; Lai, M.L.; Lin, M.S.; Siest, J.P.; Huang, J.D. Novel mutations of CYP3A4 in Chinese. Drug Metab. Dispos. 2001, 29, 268–273. [Google Scholar]
- Lee, S.J.; Goldstein, J.A. Functionally defective or altered CYP3A4 and CYP3A5 single nucleotide polymorphisms and their detection with genotyping tests. Pharmacogenomics 2005, 6, 357–371. [Google Scholar] [CrossRef]
- Wang, C.E.; Lu, K.P.; Chang, Z.; Guo, M.L.; Qiao, H.L. Association of CYP3A4*1B genotype with Cyclosporin A pharmacokinetics in renal transplant recipients: A meta-analysis. Gene 2018, 664, 44–49. [Google Scholar] [CrossRef]
- Miura, M.; Satoh, S.; Kagaya, H.; Saito, M.; Numakura, K.; Tsuchiya, N.; Habuchi, T. Impact of the CYP3A4*1G polymorphism and its combination with CYP3A5 genotypes on tacrolimus pharmacokinetics in renal transplant patients. Pharmacogenomics 2011, 12, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.C.; Ng, C.M.; Barrett, J.S.; Luo, A.J.; Zhang, B.K.; Deng, C.H.; Xi, L.Y.; Cheng, K.; Ming, Y.Z.; Yang, G.P.; et al. Effects of CYP3A4 and CYP3A5 polymorphisms on tacrolimus pharmacokinetics in Chinese adult renal transplant recipients: A population pharmacokinetic analysis. Pharm. Genom. 2013, 23, 251–261. [Google Scholar] [CrossRef] [PubMed]
- He, B.X.; Shi, L.; Qiu, J.; Zeng, X.H.; Zhao, S.J. The effect of CYP3A4*1G allele on the pharmacokinetics of atorvastatin in Chinese Han patients with coronary heart disease. J. Clin. Pharmacol. 2014, 54, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Murayama, N.; Shimizu, M.; Shimada, T.; Guengerich, F.P.; Yamazaki, H. CYP3A4 intron 6 C>T polymorphism (CYP3A4*22) is associated with reduced CYP3A4 protein level and function in human liver microsomes. J. Toxicol. Sci. 2013, 38, 349–354. [Google Scholar] [CrossRef]
- Sanchez Spitman, A.B.; Moes, D.; Gelderblom, H.; Dezentje, V.O.; Swen, J.J.; Guchelaar, H.J. Effect of CYP3A4*22, CYP3A5*3, and CYP3A combined genotypes on tamoxifen metabolism. Eur. J. Clin. Pharmacol. 2017, 73, 1589–1598. [Google Scholar] [CrossRef]
- Bins, S.; Huitema, A.D.R.; Laven, P.; Bouazzaoui, S.E.; Yu, H.; van Erp, N.; van Herpen, C.; Hamberg, P.; Gelderblom, H.; Steeghs, N.; et al. Impact of CYP3A4*22 on Pazopanib Pharmacokinetics in Cancer Patients. Clin. Pharmacokinet. 2019, 58, 651–658. [Google Scholar] [CrossRef]
- Lamba, J.K.; Lin, Y.S.; Schuetz, E.G.; Thummel, K.E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 2002, 54, 1271–1294. [Google Scholar] [CrossRef]
- Elens, L.; van Gelder, T.; Hesselink, D.A.; Haufroid, V.; van Schaik, R.H. CYP3A4*22: Promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics 2013, 14, 47–62. [Google Scholar] [CrossRef]
- Werk, A.N.; Lefeldt, S.; Bruckmueller, H.; Hemmrich-Stanisak, G.; Franke, A.; Roos, M.; Kuchle, C.; Steubl, D.; Schmaderer, C.; Brasen, J.H.; et al. Identification and characterization of a defective CYP3A4 genotype in a kidney transplant patient with severely diminished tacrolimus clearance. Clin. Pharmacol. Ther. 2014, 95, 416–422. [Google Scholar] [CrossRef]
- Kumondai, M.; Gutierrez Rico, E.M.; Hishinuma, E.; Ueda, A.; Saito, S.; Saigusa, D.; Tadaka, S.; Kinoshita, K.; Nakayoshi, T.; Oda, A.; et al. Functional Characterization of 40 CYP3A4 Variants by Assessing Midazolam 1’-Hydroxylation and Testosterone 6β-Hydroxylation. Drug Metab. Dispos. 2021, 49, 212–220. [Google Scholar] [CrossRef]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.G.; Wood, A.J.; Kim, R.B.; Stein, C.M.; Wilkinson, G.R. Genetic variability in CYP3A5 and its possible consequences. Pharmacogenomics 2004, 5, 243–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, M.; Luan, P.; Jia, W.; Liu, Q.; Ma, Z.; Dang, J.; Lu, H.; Ma, Q.; Wang, Y.; et al. Associations Between Cytochrome P450 (CYP) Gene Single-Nucleotide Polymorphisms and Second-to-Fourth Digit Ratio in Chinese University Students. Med. Sci. Monit. 2021, 27, e930591. [Google Scholar] [CrossRef]
- Brandl, E.J.; Chowdhury, N.I.; Tiwari, A.K.; Lett, T.A.; Meltzer, H.Y.; Kennedy, J.L.; Muller, D.J. Genetic variation in CYP3A43 is associated with response to antipsychotic medication. J. Neural. Transm. 2015, 122, 29–34. [Google Scholar] [CrossRef]
- Bhatnagar, V.; Garcia, E.P.; O’Connor, D.T.; Brophy, V.H.; Alcaraz, J.; Richard, E.; Bakris, G.L.; Middleton, J.P.; Norris, K.C.; Wright, J.; et al. CYP3A4 and CYP3A5 polymorphisms and blood pressure response to amlodipine among African-American men and women with early hypertensive renal disease. Am. J. Nephrol. 2010, 31, 95–103. [Google Scholar] [CrossRef]
- Dobon, B.; Rossell, C.; Walsh, S.; Bertranpetit, J. Is there adaptation in the human genome for taste perception and phase I biotransformation? BMC Evol. Biol. 2019, 19, 39. [Google Scholar] [CrossRef]
- Xue, Y.; Moore, L.B.; Orans, J.; Peng, L.; Bencharit, S.; Kliewer, S.A.; Redinbo, M.R. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol. Endocrinol. 2007, 21, 1028–1038. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterstrom, R.H.; et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 1998, 92, 73–82. [Google Scholar] [CrossRef]
- Goodwin, B.; Gauthier, K.C.; Umetani, M.; Watson, M.A.; Lochansky, M.I.; Collins, J.L.; Leitersdorf, E.; Mangelsdorf, D.J.; Kliewer, S.A.; Repa, J.J. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 223–228. [Google Scholar] [CrossRef]
- Jones, S.A.; Moore, L.B.; Shenk, J.L.; Wisely, G.B.; Hamilton, G.A.; McKee, D.D.; Tomkinson, N.C.; LeCluyse, E.L.; Lambert, M.H.; Willson, T.M.; et al. The pregnane X receptor: A promiscuous xenobiotic receptor that has diverged during evolution. Mol. Endocrinol. 2000, 14, 27–39. [Google Scholar] [CrossRef]
- Krasowski, M.D.; Yasuda, K.; Hagey, L.R.; Schuetz, E.G. Evolution of the pregnane x receptor: Adaptation to cross-species differences in biliary bile salts. Mol. Endocrinol. 2005, 19, 1720–1739. [Google Scholar] [CrossRef]
- Moore, L.B.; Goodwin, B.; Jones, S.A.; Wisely, G.B.; Serabjit-Singh, C.J.; Willson, T.M.; Collins, J.L.; Kliewer, S.A. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 7500–7502. [Google Scholar] [CrossRef]
- Dussault, I.; Yoo, H.D.; Lin, M.; Wang, E.; Fan, M.; Batta, A.K.; Salen, G.; Erickson, S.K.; Forman, B.M. Identification of an endogenous ligand that activates pregnane X receptor-mediated sterol clearance. Proc. Natl. Acad. Sci. USA 2003, 100, 833–838. [Google Scholar] [CrossRef]
- Zhou, C.; Verma, S.; Blumberg, B. The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl. Recept. Signal. 2009, 7, e001. [Google Scholar] [CrossRef]
- Dutta, M.; Lim, J.J.; Cui, J.Y. Pregnane X Receptor and the Gut-Liver Axis: A Recent Update. Drug Metab. Dispos. 2022, 50, 478–491. [Google Scholar] [CrossRef]
- Qatanani, M.; Moore, D.D. CAR, the continuously advancing receptor, in drug metabolism and disease. Curr. Drug Metab. 2005, 6, 329–339. [Google Scholar] [CrossRef]
- Stern, S.; Kurian, R.; Wang, H. Clinical Relevance of the Constitutive Androstane Receptor. Drug Metab. Dispos. 2022, 50, 1010–1018. [Google Scholar] [CrossRef]
- Forman, B.M.; Tzameli, I.; Choi, H.S.; Chen, J.; Simha, D.; Seol, W.; Evans, R.M.; Moore, D.D. Androstane metabolites bind to and deactivate the nuclear receptor CAR-β. Nature 1998, 395, 612–615. [Google Scholar] [CrossRef]
- Swales, K.; Negishi, M. CAR, driving into the future. Mol. Endocrinol. 2004, 18, 1589–1598. [Google Scholar] [CrossRef]
- Oliviero, F.; Lukowicz, C.; Boussadia, B.; Forner-Piquer, I.; Pascussi, J.M.; Marchi, N.; Mselli-Lakhal, L. Constitutive Androstane Receptor: A Peripheral and a Neurovascular Stress or Environmental Sensor. Cells 2020, 9, 2426. [Google Scholar] [CrossRef]
- Choi, S.Y.; Koh, K.H.; Jeong, H. Isoform-specific regulation of cytochromes P450 expression by estradiol and progesterone. Drug Metab. Dispos. 2013, 41, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Sberna, A.L.; Assem, M.; Xiao, R.; Ayers, S.; Gautier, T.; Guiu, B.; Deckert, V.; Chevriaux, A.; Grober, J.; Le Guern, N.; et al. Constitutive androstane receptor activation decreases plasma apolipoprotein B-containing lipoproteins and atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Strugnell, S.A.; DeLuca, H.F. Current understanding of the molecular actions of vitamin D. Physiol. Rev. 1998, 78, 1193–1231. [Google Scholar] [CrossRef]
- Noh, K.; Chow, E.C.Y.; Quach, H.P.; Groothuis, G.M.M.; Tirona, R.G.; Pang, K.S. Significance of the Vitamin D Receptor on Crosstalk with Nuclear Receptors and Regulation of Enzymes and Transporters. AAPS J. 2022, 24, 71. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.K.; Hobrath, J.V.; Oak, A.S.W.; Tang, E.K.Y.; Tieu, E.W.; Li, W.; Tuckey, R.C.; Jetten, A.M. Endogenously produced nonclassical vitamin D hydroxy-metabolites act as “biased” agonists on VDR and inverse agonists on RORα and RORγ. J. Steroid Biochem. Mol. Biol. 2017, 173, 42–56. [Google Scholar] [CrossRef]
- Masuno, H.; Ikura, T.; Morizono, D.; Orita, I.; Yamada, S.; Shimizu, M.; Ito, N. Crystal structures of complexes of vitamin D receptor ligand-binding domain with lithocholic acid derivatives. J. Lipid Res. 2013, 54, 2206–2213. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Discovery of farnesoid X receptor and its role in bile acid metabolism. Mol. Cell. Endocrinol. 2022, 548, 111618. [Google Scholar] [CrossRef]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef]
- Dixon, E.D.; Nardo, A.D.; Claudel, T.; Trauner, M. The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes 2021, 12, 645. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Inoue, I.; Itoh, F.; Aoyagi, S.; Tazawa, S.; Kusama, H.; Akahane, M.; Mastunaga, T.; Hayashi, K.; Awata, T.; Komoda, T.; et al. Fibrate and statin synergistically increase the transcriptional activities of PPARα/RXRα and decrease the transactivation of NFκB. Biochem. Biophys. Res. Commun. 2002, 290, 131–139. [Google Scholar] [CrossRef]
- Staels, B.; Maes, M.; Zambon, A. Fibrates and future PPARα agonists in the treatment of cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 542–553. [Google Scholar] [CrossRef]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR α. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Kliewer, S.A.; Moore, L.B.; Smith-Oliver, T.A.; Oliver, B.B.; Su, J.L.; Sundseth, S.S.; Winegar, D.A.; Blanchard, D.E.; Spencer, T.A.; et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 1997, 272, 3137–3140. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.K.; Qayyum, S.; Song, Y.; Janjetovic, Z.; Oak, A.S.W.; Slominski, R.M.; Raman, C.; Stefan, J.; Mier-Aguilar, C.A.; et al. Vitamin D and lumisterol derivatives can act on liver X receptors (LXRs). Sci. Rep. 2021, 11, 8002. [Google Scholar] [CrossRef]
- Lewis, D.F.; Eddershaw, P.J.; Goldfarb, P.S.; Tarbit, M.H. Molecular modelling of CYP3A4 from an alignment with CYP102: Identification of key interactions between putative active site residues and CYP3A-specific chemicals. Xenobiotica 1996, 26, 1067–1086. [Google Scholar] [CrossRef] [PubMed]
- Scheff, J.D.; Almon, R.R.; Dubois, D.C.; Jusko, W.J.; Androulakis, I.P. Assessment of pharmacologic area under the curve when baselines are variable. Pharm. Res. 2011, 28, 1081–1089. [Google Scholar] [CrossRef]
- Flockhart, D.A.; Thacker, D.; McDonald, C.; Desta, Z. The Flockhart Cytochrome P450 Drug-Drug Interaction Table; Updated 2021; Division of Clinical Pharmacology, Indiana University School of Medicine: Indianapolis, IN, USA, 2022. [Google Scholar]
- Food and Drug Administration. Clinical Drug Interaction Studies-Cytochrome P450 Enzyme-and Transporter-Mediated Drug Interactions Guidance for Industry; Center for Drug Evaluation and Research (CDER), US Department of Health and Human Services Food and Drug Administration: Silver Springs, MD, USA, 2020. [Google Scholar]
- Thummel, K.E.; Wilkinson, G.R. In vitro and in vivo drug interactions involving human CYP3A. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 389–430. [Google Scholar] [CrossRef]
- Liu, Y.T.; Hao, H.P.; Liu, C.X.; Wang, G.J.; Xie, H.G. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab. Rev. 2007, 39, 699–721. [Google Scholar] [CrossRef] [PubMed]
- Sahali-Sahly, Y.; Balani, S.K.; Lin, J.H.; Baillie, T.A. In vitro studies on the metabolic activation of the furanopyridine L-754,394, a highly potent and selective mechanism-based inhibitor of cytochrome P450 3A4. Chem. Res. Toxicol. 1996, 9, 1007–1012. [Google Scholar] [CrossRef]
- Bailey, D.G.; Arnold, J.M.; Spence, J.D. Grapefruit juice and drugs. How significant is the interaction? Clin. Pharmacokinet. 1994, 26, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.J.; Bernier, S.M. Naringin and naringenin are not the primary CYP3A inhibitors in grapefruit juice. Life Sci. 1996, 59, 1025–1030. [Google Scholar] [CrossRef]
- Chen, J.; Farrell, G.C. Bile acids produce a generalized reduction of the catalytic activity of cytochromes P450 and other hepatic microsomal enzymes in vitro: Relevance to drug metabolism in experimental cholestasis. J. Gastroenterol. Hepatol. 1996, 11, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, J.; Borcic, V.; Mikov, M. Bile acids and their oxo derivatives: Potential inhibitors of carbonic anhydrase I and II, androgen receptor antagonists and CYP3A4 substrates. Biomed. Chromatogr. 2017, 31, e3870. [Google Scholar] [CrossRef]
- Ito, K.; Iwatsubo, T.; Kanamitsu, S.; Nakajima, Y.; Sugiyama, Y. Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 461–499. [Google Scholar] [CrossRef]
- Bertz, R.J.; Granneman, G.R. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin. Pharmacokinet. 1997, 32, 210–258. [Google Scholar] [CrossRef]
- AlphaFold Protein Structure Database. Available online: https://alphafold.ebi.ac.uk/ (accessed on 10 October 2022).
- Hayes, M.A.; Li, X.Q.; Gronberg, G.; Diczfalusy, U.; Andersson, T.B. CYP3A Specifically Catalyzes 1β-Hydroxylation of Deoxycholic Acid: Characterization and Enzymatic Synthesis of a Potential Novel Urinary Biomarker for CYP3A Activity. Drug Metab. Dispos. 2016, 44, 1480–1489. [Google Scholar] [CrossRef]
- Lin, Q.; Tan, X.; Wang, W.; Zeng, W.; Gui, L.; Su, M.; Liu, C.; Jia, W.; Xu, L.; Lan, K. Species Differences of Bile Acid Redox Metabolism: Tertiary Oxidation of Deoxycholate is Conserved in Preclinical Animals. Drug Metab. Dispos. 2020, 48, 499–507. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, L.Z.; Chen, Y.J.; Zhu, P.P.; Yin, S.S.; Su, M.M.; Ni, Y.; Miao, J.; Wu, W.L.; Chen, H.; et al. Continuum of Host-Gut Microbial Co-metabolism: Host CYP3A4/3A7 are Responsible for Tertiary Oxidations of Deoxycholate Species. Drug Metab. Dispos. 2019, 47, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Nitta, S.I.; Hashimoto, M.; Kazuki, Y.; Takehara, S.; Suzuki, H.; Oshimura, M.; Akita, H.; Chiba, K.; Kobayashi, K. Evaluation of 4β-Hydroxycholesterol and 25-Hydroxycholesterol as Endogenous Biomarkers of CYP3A4: Study with CYP3A-Humanized Mice. AAPS J. 2018, 20, 61. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Miyazaki, T.; Ikegami, T.; Iwamoto, J.; Maeda, T.; Hirayama, T.; Saito, Y.; Teramoto, T.; Matsuzaki, Y. Cholesterol 25-hydroxylation activity of CYP3A. J. Lipid Res. 2011, 52, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Jurutka, P.W.; Thompson, P.D.; Whitfield, G.K.; Eichhorst, K.R.; Hall, N.; Dominguez, C.E.; Hsieh, J.C.; Haussler, C.A.; Haussler, M.R. Molecular and functional comparison of 1,25-dihydroxyvitamin D(3) and the novel vitamin D receptor ligand, lithocholic acid, in activating transcription of cytochrome P450 3A4. J. Cell. Biochem. 2005, 94, 917–943. [Google Scholar] [CrossRef]
- Jurica, J.; Dovrtelova, G.; Noskova, K.; Zendulka, O. Bile Acids, Nuclear Receptors and Cytochrome P450. Physiol. Res. 2016, 65, S427–S440. [Google Scholar] [CrossRef]
- Furster, C.; Wikvall, K. Identification of CYP3A4 as the major enzyme responsible for 25-hydroxylation of 5β-cholestane-3α,7α,12α-triol in human liver microsomes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1999, 1437, 46–52. [Google Scholar] [CrossRef]
- Araya, Z.; Wikvall, K. 6α-hydroxylation of taurochenodeoxycholic acid and lithocholic acid by CYP3A4 in human liver microsomes 6alpha-hydroxylation of taurochenodeoxycholic acid and lithocholic acid by CYP3A4 in human liver microsomes. Biochim. Biophys. Acta 1999, 1438, 47–54. [Google Scholar] [CrossRef]
- Wong, S.Y.; Teo, J.S.M.; Chai, S.F.; Yeap, S.L.; Lau, A.J. Vitamin E analogues differentially inhibit human cytochrome P450 3A (CYP3A)-mediated oxidative metabolism of lithocholic acid: Impact of δ-tocotrienol on lithocholic acid cytotoxicity. Toxicology 2019, 423, 62–74. [Google Scholar] [CrossRef]
- Waxman, D.J.; Chang, T.K. Thin-Layer Chromatography Analysis of Human CYP3A-Catalyzed Testosterone 6β-Hydroxylation. Methods Mol. Biol. 2006, 320, 133–141. [Google Scholar] [CrossRef]
- Niwa, T.; Okamoto, A.; Narita, K.; Toyota, M.; Kato, K.; Kobayashi, K.; Sasaki, S. Comparison of steroid hormone hydroxylation mediated by cytochrome P450 3A subfamilies. Arch. Biochem. Biophys. 2020, 682, 108283. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Toyota, M.; Kawasaki, H.; Ishii, R.; Sasaki, S. Comparison of the Stimulatory and Inhibitory Effects of Steroid Hormones and α-Naphthoflavone on Steroid Hormone Hydroxylation Catalyzed by Human Cytochrome P450 3A Subfamilies. Biol. Pharm. Bull. 2021, 44, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.C.; Templeton, I.; Thummel, K.E.; Davis, C.; Kunze, K.L.; Isoherranen, N. Evaluation of 6β-hydroxycortisol, 6β-hydroxycortisone, and a combination of the two as endogenous probes for inhibition of CYP3A4 in vivo. Clin. Pharmacol. Ther. 2011, 89, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Shimada, T. Progesterone and testosterone hydroxylation by cytochromes P450 2C19, 2C9, and 3A4 in human liver microsomes. Arch. Biochem. Biophys. 1997, 346, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kandel, S.E.; Han, L.W.; Mao, Q.; Lampe, J.N. Digging Deeper into CYP3A Testosterone Metabolism: Kinetic, Regioselectivity, and Stereoselectivity Differences between CYP3A4/5 and CYP3A7. Drug Metab. Dispos. 2017, 45, 1266–1275. [Google Scholar] [CrossRef]
- Stevens, J.C.; Hines, R.N.; Gu, C.; Koukouritaki, S.B.; Manro, J.R.; Tandler, P.J.; Zaya, M.J. Developmental expression of the major human hepatic CYP3A enzymes. J. Pharmacol. Exp. Ther. 2003, 307, 573–582. [Google Scholar] [CrossRef]
- Yu, A.M.; Fukamachi, K.; Krausz, K.W.; Cheung, C.; Gonzalez, F.J. Potential role for human cytochrome P450 3A4 in estradiol homeostasis. Endocrinology 2005, 146, 2911–2919. [Google Scholar] [CrossRef]
- Williams, J.A.; Ring, B.J.; Cantrell, V.E.; Jones, D.R.; Eckstein, J.; Ruterbories, K.; Hamman, M.A.; Hall, S.D.; Wrighton, S.A. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab. Dispos. 2002, 30, 883–891. [Google Scholar] [CrossRef]
- Yamazaki, H.; Shaw, P.M.; Guengerich, F.P.; Shimada, T. Roles of cytochromes P450 1A2 and 3A4 in the oxidation of estradiol and estrone in human liver microsomes. Chem. Res. Toxicol. 1998, 11, 659–665. [Google Scholar] [CrossRef]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17β-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef]
- Lee, A.J.; Conney, A.H.; Zhu, B.T. Human cytochrome P450 3A7 has a distinct high catalytic activity for the 16α-hydroxylation of estrone but not 17beta-estradiol. Cancer Res. 2003, 63, 6532–6536. [Google Scholar] [PubMed]
- Cribb, A.E.; Knight, M.J.; Dryer, D.; Guernsey, J.; Hender, K.; Tesch, M.; Saleh, T.M. Role of polymorphic human cytochrome P450 enzymes in estrone oxidation. Cancer Epidemiol. Biomark. Prev. 2006, 15, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hashizume, T.; Shuhart, M.C.; Davis, C.L.; Nelson, W.L.; Sakaki, T.; Kalhorn, T.F.; Watkins, P.B.; Schuetz, E.G.; Thummel, K.E. Intestinal and hepatic CYP3A4 catalyze hydroxylation of 1α,25-dihydroxyvitamin D(3): Implications for drug-induced osteomalacia. Mol. Pharmacol. 2006, 69, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Kornilov, A.M.; Kent, U.M.; Hollenberg, P.F. Anandamide metabolism by human liver and kidney microsomal cytochrome p450 enzymes to form hydroxyeicosatetraenoic and epoxyeicosatrienoic acid ethanolamides. J. Pharmacol. Exp. Ther. 2007, 321, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Colombero, C.; Cardenas, S.; Venara, M.; Martin, A.; Pennisi, P.; Barontini, M.; Nowicki, S. Cytochrome 450 metabolites of arachidonic acid (20-HETE, 11,12-EET and 14,15-EET) promote pheochromocytoma cell growth and tumor associated angiogenesis. Biochimie 2020, 171–172, 147–157. [Google Scholar] [CrossRef]
- Rifkind, A.B.; Lee, C.; Chang, T.K.; Waxman, D.J. Arachidonic acid metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: Regioselective oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid epoxygenation in human liver microsomes. Arch. Biochem. Biophys. 1995, 320, 380–389. [Google Scholar] [CrossRef]
- Wu, S.; Moomaw, C.R.; Tomer, K.B.; Falck, J.R.; Zeldin, D.C. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J. Biol. Chem. 1996, 271, 3460–3468. [Google Scholar] [CrossRef]
- Powell, P.K.; Wolf, I.; Jin, R.; Lasker, J.M. Metabolism of arachidonic acid to 20-hydroxy-5,8,11, 14-eicosatetraenoic acid by P450 enzymes in human liver: Involvement of CYP4F2 and CYP4A11. J. Pharmacol. Exp. Ther. 1998, 285, 1327–1336. [Google Scholar]
- Wolf, K.K.; Paine, M. Metabolic Barrier of the Gastrointestinal Tract. In Comprehensive Toxicology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 3, pp. 74–98. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, L.; Zhang, N.Y.; Karrow, N.A.; Krumm, C.S.; Qi, D.S.; Sun, L.H. Aflatoxin B1 metabolism: Regulation by phase I and II metabolizing enzymes and chemoprotective agents. Mutat. Res. Rev. Mutat. Res. 2018, 778, 79–89. [Google Scholar] [CrossRef]
- Piipari, R.; Savela, K.; Nurminen, T.; Hukkanen, J.; Raunio, H.; Hakkola, J.; Mantyla, T.; Beaune, P.; Edwards, R.J.; Boobis, A.R.; et al. Expression of CYP1A1, CYP1B1 and CYP3A, and polycyclic aromatic hydrocarbon-DNA adduct formation in bronchoalveolar macrophages of smokers and non-smokers. Int. J. Cancer 2000, 86, 610–616. [Google Scholar] [CrossRef]
- Patten, C.J.; Smith, T.J.; Friesen, M.J.; Tynes, R.E.; Yang, C.S.; Murphy, S.E. Evidence for cytochrome P450 2A6 and 3A4 as major catalysts for N’-nitrosonornicotine alpha-hydroxylation by human liver microsomes. Carcinogenesis 1997, 18, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Klees, T.M.; Sheffels, P.; Dale, O.; Kharasch, E.D. Metabolism of alfentanil by cytochrome p4503a (cyp3a) enzymes. Drug Metab. Dispos. 2005, 33, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Zhou-Pan, X.R.; Gauthier, T.; Placidi, M.; Maurel, P.; Rahmani, R. Human liver microsomal cytochrome P450 3A isozymes mediated vindesine biotransformation. Metabolic drug interactions. Biochem. Pharmacol. 1993, 45, 853–861. [Google Scholar] [CrossRef]
- Preissner, S.; Kroll, K.; Dunkel, M.; Senger, C.; Goldsobel, G.; Kuzman, D.; Guenther, S.; Winnenburg, R.; Schroeder, M.; Preissner, R. SuperCYP: A comprehensive database on Cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 2010, 38, D237–D243. [Google Scholar] [CrossRef]
- Spratlin, J.; Sawyer, M.B. Pharmacogenetics of paclitaxel metabolism. Crit. Rev. Oncol. Hematol. 2007, 61, 222–229. [Google Scholar] [CrossRef]
- Royer, I.; Monsarrat, B.; Sonnier, M.; Wright, M.; Cresteil, T. Metabolism of docetaxel by human cytochromes P450: Interactions with paclitaxel and other antineoplastic drugs. Cancer Res. 1996, 56, 58–65. [Google Scholar]
- Harris, J.W.; Katki, A.; Anderson, L.W.; Chmurny, G.N.; Paukstelis, J.V.; Collins, J.M. Isolation, structural determination, and biological activity of 6 alpha-hydroxytaxol, the principal human metabolite of taxol. J. Med. Chem. 1994, 37, 706–709. [Google Scholar] [CrossRef]
- Baker, S.D.; Sparreboom, A.; Verweij, J. Clinical pharmacokinetics of docetaxel: Recent developments. Clin. Pharmacokinet. 2006, 45, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Kort, A.; Cheung, K.L.; Rosing, H.; Fukami, T.; Durmus, S.; Wagenaar, E.; Hendrikx, J.J.; Nakajima, M.; van Vlijmen, B.J.; et al. P-glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain Disposition and Oral Availability of the Novel Taxane Cabazitaxel (Jevtana) in Mice. Mol. Pharm. 2015, 12, 3714–3723. [Google Scholar] [CrossRef]
- Duckett, D.R.; Cameron, M.D. Metabolism considerations for kinase inhibitors in cancer treatment. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1175–1193. [Google Scholar] [CrossRef]
- Ling, J.; Johnson, K.A.; Miao, Z.; Rakhit, A.; Pantze, M.P.; Hamilton, M.; Lum, B.L.; Prakash, C. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab. Dispos. 2006, 34, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Pophali, P.A.; Patnaik, M.M. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. 2016, 22, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Magliocco, G.; Thomas, A.; Desmeules, J.; Daali, Y. Phenotyping of Human CYP450 Enzymes by Endobiotics: Current Knowledge and Methodological Approaches. Clin. Pharmacokinet. 2019, 58, 1373–1391. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Ahn, L.Y.; Choi, M.H.; Moon, J.Y.; Lee, J.; Jang, I.J.; Yu, K.S.; Cho, J.Y. Urinary 6β-Hydroxycortisol/Cortisol Ratio Most Highly Correlates With Midazolam Clearance Under Hepatic CYP3A Inhibition and Induction in Females: A Pharmacometabolomics Approach. AAPS J. 2016, 18, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Kim, B.; Rhee, S.J.; Lee, Y.; Park, J.S.; Lee, S.M.; Kim, S.M.; Lee, S.; Yu, K.S.; Jang, I.J.; et al. Assessment of induced CYP3A activity in pregnant women using 4β-hydroxycholesterol: Cholesterol ratio as an appropriate metabolic marker. Drug Metab. Pharmacokinet. 2018, 33, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Chiasson, J.L.; Gaudette, F.; Turgeon, J.; Michaud, V. Use of 4β-Hydroxycholesterol Plasma Concentrations as an Endogenous Biomarker of CYP3A Activity: Clinical Validation in Individuals With Type 2 Diabetes. Clin. Pharmacol. Ther. 2019, 106, 831–840. [Google Scholar] [CrossRef]
- Chen, Y.J.; Zhang, J.; Zhu, P.P.; Tan, X.W.; Lin, Q.H.; Wang, W.X.; Yin, S.S.; Gao, L.Z.; Su, M.M.; Liu, C.X.; et al. Stereoselective Oxidation Kinetics of Deoxycholate in Recombinant and Microsomal CYP3A Enzymes: Deoxycholate 19-Hydroxylation Is an In Vitro Marker of CYP3A7 Activity. Drug Metab. Dispos. 2019, 47, 574–581. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, S.H.; Yi, S.; Kim, A.H.; Kim, B.; Lee, S.; Yu, K.S.; Jang, I.J.; Cho, J.Y. Quantitative prediction of hepatic CYP3A activity using endogenous markers in healthy subjects after administration of CYP3A inhibitors or inducers. Drug Metab. Pharmacokinet. 2019, 34, 247–252. [Google Scholar] [CrossRef]
- Lee, J.; Kim, A.H.; Yi, S.; Lee, S.; Yoon, S.H.; Yu, K.S.; Jang, I.J.; Cho, J.Y. Distribution of Exogenous and Endogenous CYP3A Markers and Related Factors in Healthy Males and Females. AAPS J. 2017, 19, 1196–1204. [Google Scholar] [CrossRef]
- Bjorkhem-Bergman, L.; Backstrom, T.; Nylen, H.; Ronquist-Nii, Y.; Bredberg, E.; Andersson, T.B.; Bertilsson, L.; Diczfalusy, U. Comparison of endogenous 4beta-hydroxycholesterol with midazolam as markers for CYP3A4 induction by rifampicin. Drug Metab. Dispos. 2013, 41, 1488–1493. [Google Scholar] [CrossRef]
- Galteau, M.M.; Shamsa, F. Urinary 6β-hydroxycortisol: A validated test for evaluating drug induction or drug inhibition mediated through CYP3A in humans and in animals. Eur. J. Clin. Pharmacol. 2003, 59, 713–733. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, N.; Haefeli, W.E.; Mikus, G. CYP3A activity: Towards dose adaptation to the individual. Expert Opin. Drug Metab. Toxicol. 2016, 12, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Chetty, M.; Mattison, D.; Rostami-Hodjegan, A. Sex differences in the clearance of CYP3A4 substrates: Exploring possible reasons for the substrate dependency and lack of consensus. Curr. Drug Metab. 2012, 13, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, C.; Moore, A.; Hayes, P.; DeVane, C.L.; Pelonero, A. Influence of menstrual cycle and gender on alprazolam pharmacokinetics. Clin. Pharmacol. Ther. 1991, 50, 404–409. [Google Scholar] [CrossRef]
- Rivory, L.P.; Slaviero, K.A.; Clarke, S.J. Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br. J. Cancer 2002, 87, 277–280. [Google Scholar] [CrossRef]
- Chen, Y.C.; Gotzkowsky, S.K.; Nafziger, A.N.; Kulawy, R.W.; Rocci, M.L., Jr.; Bertino, J.S., Jr.; Kashuba, A.D. Poor correlation between 6β-hydroxycortisol:cortisol molar ratios and midazolam clearance as measure of hepatic CYP3A activity. Br. J. Clin. Pharmacol. 2006, 62, 187–195. [Google Scholar] [CrossRef]
- Wang, X.; Fu, X.; Van Ness, C.; Meng, Z.; Ma, X.; Huang, W. Bile Acid Receptors and Liver Cancer. Curr. Pathobiol. Rep. 2013, 1, 29–35. [Google Scholar] [CrossRef]
- Claudel, T.; Zollner, G.; Wagner, M.; Trauner, M. Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 867–878. [Google Scholar] [CrossRef]
- Evangelakos, I.; Heeren, J.; Verkade, E.; Kuipers, F. Role of bile acids in inflammatory liver diseases. Semin. Immunopathol. 2021, 43, 577–590. [Google Scholar] [CrossRef]
- Zollner, G.; Trauner, M. Mechanisms of cholestasis. Clin. Liver Dis. 2008, 12, 1–26. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, W.; Qatanani, M.; Evans, R.M.; Moore, D.D. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J. Biol. Chem. 2004, 279, 49517–49522. [Google Scholar] [CrossRef] [PubMed]
- Stedman, C.; Robertson, G.; Coulter, S.; Liddle, C. Feed-forward regulation of bile acid detoxification by CYP3A4: Studies in humanized transgenic mice. J. Biol. Chem. 2004, 279, 11336–11343. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.H.; Lammert, F. Nuclear xeno-sensors as receptors for cholestatic bile acids: The second line of defense. Hepatology 2002, 35, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Pozza, A.D.; Albertoni, L.; Lazzari, R.; Zigiotto, G.; Carrara, M.; Baldo, V.; Baldovin, T.; Floreani, A.; Martin, S. Pregnane X receptor and constitutive androstane receptor modulate differently CYP3A-mediated metabolism in early- and late-stage cholestasis. World J. Gastroenterol. 2017, 23, 7519–7530. [Google Scholar] [CrossRef]
- Liu, X.; Xue, R.; Yang, C.; Gu, J.; Chen, S.; Zhang, S. Cholestasis-induced bile acid elevates estrogen level via farnesoid X receptor-mediated suppression of the estrogen sulfotransferase SULT1E1. J. Biol. Chem. 2018, 293, 12759–12769. [Google Scholar] [CrossRef]
- Kandel, B.A.; Thomas, M.; Winter, S.; Damm, G.; Seehofer, D.; Burk, O.; Schwab, M.; Zanger, U.M. Genomewide comparison of the inducible transcriptomes of nuclear receptors CAR, PXR and PPAR alpha in primary human hepatocytes. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 1218–1227. [Google Scholar] [CrossRef]
- Pavek, P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front. Pharmacol. 2016, 7, 456. [Google Scholar] [CrossRef]
- Woolsey, S.J.; Mansell, S.E.; Kim, R.B.; Tirona, R.G.; Beaton, M.D. CYP3A Activity and Expression in Nonalcoholic Fatty Liver Disease. Drug Metab. Dispos. 2015, 43, 1484–1490. [Google Scholar] [CrossRef]
- Donato, M.T.; Lahoz, A.; Jimenez, N.; Perez, G.; Serralta, A.; Mir, J.; Castell, J.V.; Gomez-Lechon, M.J. Potential impact of steatosis on cytochrome P450 enzymes of human hepatocytes isolated from fatty liver grafts. Drug Metab. Dispos. 2006, 34, 1556–1562. [Google Scholar] [CrossRef]
- Yoshinari, K.; Takagi, S.; Yoshimasa, T.; Sugatani, J.; Miwa, M. Hepatic CYP3A expression is attenuated in obese mice fed a high-fat diet. Pharm. Res. 2006, 23, 1188–1200. [Google Scholar] [CrossRef]
- Maximos, S.; Chamoun, M.; Gravel, S.; Turgeon, J.; Michaud, V. Tissue Specific Modulation of cyp2c and cyp3a mRNA Levels and Activities by Diet-Induced Obesity in Mice: The Impact of Type 2 Diabetes on Drug Metabolizing Enzymes in Liver and Extra-Hepatic Tissues. Pharmaceutics 2017, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Ranger-Moore, J.; Jackson, J.P.; Ferguson, S.S.; Cherrington, N.J. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2009, 37, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, R.; de la Monte, S.M.; Ogasawara, K.; Adusumalli, S.; Barlock, B.B.; Akhlaghi, F. Nonalcoholic Fatty Liver Disease and Diabetes Are Associated with Decreased CYP3A4 Protein Expression and Activity in Human Liver. Mol. Pharm. 2018, 15, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Swathy, B.; Saradalekshmi, K.R.; Nair, I.V.; Nair, C.; Banerjee, M. Pharmacoepigenomic responses of antipsychotic drugs on pharmacogenes are likely to be modulated by miRNAs. Epigenomics 2017, 9, 811–821. [Google Scholar] [CrossRef]
- Yan, L.; Liu, J.; Zhao, Y.; Nie, Y.; Ma, X.; Kan, Q.; Zhang, L. Suppression of miR-628-3p and miR-641 is involved in rifampin-mediated CYP3A4 induction in HepaRG cells. Pharmacogenomics 2017, 18, 57–64. [Google Scholar] [CrossRef]
- Leti, F.; Malenica, I.; Doshi, M.; Courtright, A.; Van Keuren-Jensen, K.; Legendre, C.; Still, C.D.; Gerhard, G.S.; DiStefano, J.K. High-throughput sequencing reveals altered expression of hepatic microRNAs in nonalcoholic fatty liver disease-related fibrosis. Transl. Res. 2015, 166, 304–314. [Google Scholar] [CrossRef]
- Shulpekova, Y.; Zharkova, M.; Tkachenko, P.; Tikhonov, I.; Stepanov, A.; Synitsyna, A.; Izotov, A.; Butkova, T.; Shulpekova, N.; Lapina, N.; et al. The Role of Bile Acids in the Human Body and in the Development of Diseases. Molecules 2022, 27, 3401. [Google Scholar] [CrossRef]
- Shulpekova, Y.; Shirokova, E.; Zharkova, M.; Tkachenko, P.; Tikhonov, I.; Stepanov, A.; Sinitsyna, A.; Izotov, A.; Butkova, T.; Shulpekova, N.; et al. A Recent Ten-Year Perspective: Bile Acid Metabolism and Signaling. Molecules 2022, 27, 1983. [Google Scholar] [CrossRef]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. Int. J. Mol. Sci. 2022, 23, 4305. [Google Scholar] [CrossRef]
- Beuers, U.; Spengler, U.; Zwiebel, F.M.; Pauletzki, J.; Fischer, S.; Paumgartner, G. Effect of ursodeoxycholic acid on the kinetics of the major hydrophobic bile acids in health and in chronic cholestatic liver disease. Hepatology 1992, 15, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Hohenester, S.; de Buy Wenniger, L.J.; Kremer, A.E.; Jansen, P.L.; Elferink, R.P. The biliary HCO(3)(-) umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Fleming, I. Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediat. 2007, 82, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Vascular cytochrome p450 enzymes: Physiology and pathophysiology. Trends Cardiovasc. Med. 2008, 18, 20–25. [Google Scholar] [CrossRef]
- Fleming, I. The cytochrome P450 pathway in angiogenesis and endothelial cell biology. Cancer Metastasis Rev. 2011, 30, 541–555. [Google Scholar] [CrossRef]
- Fleming, I. The factor in EDHF: Cytochrome P450 derived lipid mediators and vascular signaling. Vasc. Pharmacol. 2016, 86, 31–40. [Google Scholar] [CrossRef]
- Imig, J.D. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am. J. Physiol. Ren. Physiol. 2005, 289, F496–F503. [Google Scholar] [CrossRef]
- Capdevila, J.; Wang, W. Role of cytochrome P450 epoxygenase in regulating renal membrane transport and hypertension. Curr. Opin. Nephrol. Hypertens. 2013, 22, 163–169. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Wang, W.; Falck, J.R. Arachidonic acid monooxygenase: Genetic and biochemical approaches to physiological/pathophysiological relevance. Prostaglandins Other Lipid Mediat. 2015, 120, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, X.A.; Wang, D.W. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv. Drug Deliv. Rev. 2011, 63, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Edin, M.L.; Lee, C.R.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnes, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Investig. 2012, 122, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-α-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695. [Google Scholar] [CrossRef]
- Pozzi, A.; Capdevila, J.H. PPARα Ligands as Antitumorigenic and Antiangiogenic Agents. PPAR Res. 2008, 2008, 906542. [Google Scholar] [CrossRef]
- Yang, S.; Wei, S.; Pozzi, A.; Capdevila, J.H. The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch. Biochem. Biophys. 2009, 489, 82–91. [Google Scholar] [CrossRef]
- Mitra, R.; Guo, Z.; Milani, M.; Mesaros, C.; Rodriguez, M.; Nguyen, J.; Luo, X.; Clarke, D.; Lamba, J.; Schuetz, E.; et al. CYP3A4 mediates growth of estrogen receptor-positive breast cancer cells in part by inducing nuclear translocation of phospho-Stat3 through biosynthesis of (+/-)-14,15-epoxyeicosatrienoic acid (EET). J. Biol. Chem. 2011, 286, 17543–17559. [Google Scholar] [CrossRef]
- Munzenmaier, D.H.; Harder, D.R. Cerebral microvascular endothelial cell tube formation: Role of astrocytic epoxyeicosatrienoic acid release. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1163–H1167. [Google Scholar] [CrossRef]
- Zhang, C.; Harder, D.R. Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic Acid. Stroke 2002, 33, 2957–2964. [Google Scholar] [CrossRef]
- Medhora, M.; Daniels, J.; Mundey, K.; Fisslthaler, B.; Busse, R.; Jacobs, E.R.; Harder, D.R. Epoxygenase-driven angiogenesis in human lung microvascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H215–H224. [Google Scholar] [CrossRef]
- Cheranov, S.Y.; Karpurapu, M.; Wang, D.; Zhang, B.; Venema, R.C.; Rao, G.N. An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood 2008, 111, 5581–5591. [Google Scholar] [CrossRef] [PubMed]
- Seubert, J.M.; Zeldin, D.C.; Nithipatikom, K.; Gross, G.J. Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat. 2007, 82, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Floriano-Sanchez, E.; Rodriguez, N.C.; Bandala, C.; Coballase-Urrutia, E.; Lopez-Cruz, J. CYP3A4 expression in breast cancer and its association with risk factors in Mexican women. Asian Pac. J. Cancer Prev. 2014, 15, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Popescu, V.; Yang, S.; Mei, S.; Shi, M.; Puolitaival, S.M.; Caprioli, R.M.; Capdevila, J.H. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J. Biol. Chem. 2010, 285, 12840–12850. [Google Scholar] [CrossRef]
- Guo, Z.; Sevrioukova, I.F.; Denisov, I.G.; Zhang, X.; Chiu, T.L.; Thomas, D.G.; Hanse, E.A.; Cuellar, R.A.D.; Grinkova, Y.V.; Langenfeld, V.W.; et al. Heme Binding Biguanides Target Cytochrome P450-Dependent Cancer Cell Mitochondria. Cell. Chem. Biol. 2017, 24, 1259–1275.e6. [Google Scholar] [CrossRef]
- Zhang, B.; Cao, H.; Rao, G.N. Fibroblast growth factor-2 is a downstream mediator of phosphatidylinositol 3-kinase-Akt signaling in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. J. Biol. Chem. 2006, 281, 905–914. [Google Scholar] [CrossRef]
- Jiang, J.G.; Ning, Y.G.; Chen, C.; Ma, D.; Liu, Z.J.; Yang, S.; Zhou, J.; Xiao, X.; Zhang, X.A.; Edin, M.L.; et al. Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer Res. 2007, 67, 6665–6674. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, D.; Dou, X.; Niu, N.; Huang, W.; Bai, J.; Zhang, G. Elevated 14,15- epoxyeicosatrienoic acid by increasing of cytochrome P450 2C8, 2C9 and 2J2 and decreasing of soluble epoxide hydrolase associated with aggressiveness of human breast cancer. BMC Cancer 2014, 14, 841. [Google Scholar] [CrossRef]
- Luo, J.; Yao, J.F.; Deng, X.F.; Zheng, X.D.; Jia, M.; Wang, Y.Q.; Huang, Y.; Zhu, J.H. 14, 15-EET induces breast cancer cell EMT and cisplatin resistance by up-regulating integrin alphavbeta3 and activating FAK/PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2018, 37, 23. [Google Scholar] [CrossRef]
- Guo, Z.; Johnson, V.; Barrera, J.; Porras, M.; Hinojosa, D.; Hernandez, I.; McGarrah, P.; Potter, D.A. Targeting cytochrome P450-dependent cancer cell mitochondria: Cancer associated CYPs and where to find them. Cancer Metastasis Rev. 2018, 37, 409–423. [Google Scholar] [CrossRef]
- Thuy Phuong, N.T.; Kim, J.W.; Kim, J.A.; Jeon, J.S.; Lee, J.Y.; Xu, W.J.; Yang, J.W.; Kim, S.K.; Kang, K.W. Role of the CYP3A4-mediated 11,12-epoxyeicosatrienoic acid pathway in the development of tamoxifen-resistant breast cancer. Oncotarget 2017, 8, 71054–71069. [Google Scholar] [CrossRef] [PubMed]
- Oguro, A.; Sakamoto, K.; Funae, Y.; Imaoka, S. Overexpression of CYP3A4, but not of CYP2D6, promotes hypoxic response and cell growth of Hep3B cells. Drug Metab. Pharmacokinet. 2011, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, H.; Dong, H.; Liao, J.; Hammock, B.D.; Yang, G.Y. Soluble epoxide hydrolase deficiency inhibits dextran sulfate sodium-induced colitis and carcinogenesis in mice. Anticancer Res. 2013, 33, 5261–5271. [Google Scholar]
- Nanda, P.; Ghosh, A. Genome Scale-Differential Flux Analysis reveals deregulation of lung cell metabolism on SARS-CoV-2 infection. PLoS Comput. Biol. 2021, 17, e1008860. [Google Scholar] [CrossRef] [PubMed]
- Hoff, U.; Bubalo, G.; Fechner, M.; Blum, M.; Zhu, Y.; Pohlmann, A.; Hentschel, J.; Arakelyan, K.; Seeliger, E.; Flemming, B.; et al. A synthetic epoxyeicosatrienoic acid analogue prevents the initiation of ischemic acute kidney injury. Acta Physiol. 2019, 227, e13297. [Google Scholar] [CrossRef]
- Hellmold, H.; Rylander, T.; Magnusson, M.; Reihner, E.; Warner, M.; Gustafsson, J.A. Characterization of cytochrome P450 enzymes in human breast tissue from reduction mammaplasties. J. Clin. Endocrinol. Metab. 1998, 83, 886–895. [Google Scholar] [CrossRef]
- Murray, G.I.; Weaver, R.J.; Paterson, P.J.; Ewen, S.W.; Melvin, W.T.; Burke, M.D. Expression of xenobiotic metabolizing enzymes in breast cancer. J. Pathol. 1993, 169, 347–353. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Ando, A.; Takamura, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Prediction of response to docetaxel by CYP3A4 mRNA expression in breast cancer tissues. Int. J. Cancer 2002, 97, 129–132. [Google Scholar] [CrossRef]
- Finnstrom, N.; Bjelfman, C.; Soderstrom, T.G.; Smith, G.; Egevad, L.; Norlen, B.J.; Wolf, C.R.; Rane, A. Detection of cytochrome P450 mRNA transcripts in prostate samples by RT-PCR. Eur. J. Clin. Investig. 2001, 31, 880–886. [Google Scholar] [CrossRef]
- Murray, G.I.; Taylor, V.E.; McKay, J.A.; Weaver, R.J.; Ewen, S.W.; Melvin, W.T.; Burke, M.D. The immunohistochemical localization of drug-metabolizing enzymes in prostate cancer. J. Pathol. 1995, 177, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, J.; Mantyla, M.; Kangas, L.; Wirta, P.; Hakkola, J.; Paakki, P.; Evisalmi, S.; Pelkonen, O.; Raunio, H. Expression of cytochrome P450 genes encoding enzymes active in the metabolism of tamoxifen in human uterine endometrium. Pharmacol. Toxicol. 1998, 82, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.A.; Vadlamuri, V.; Ghosh, S.; Glover, D.D. Expression and cyclic variability of CYP3A4 and CYP3A7 isoforms in human endometrium and cervix during the menstrual cycle. Drug Metab. Dispos. 2003, 31, 1–6. [Google Scholar] [CrossRef]
- DeLoia, J.A.; Zamboni, W.C.; Jones, J.M.; Strychor, S.; Kelley, J.L.; Gallion, H.H. Expression and activity of taxane-metabolizing enzymes in ovarian tumors. Gynecol. Oncol. 2008, 108, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Baumann, F.; Knupfer, H.; Brauckhoff, M.; Horn, L.C.; Schonfelder, M.; Kohler, U.; Preiss, R. CYP3A4, CYP2C9 and CYP2B6 expression and ifosfamide turnover in breast cancer tissue microsomes. Br. J. Cancer 2004, 90, 911–916. [Google Scholar] [CrossRef]
- Haas, S.; Pierl, C.; Harth, V.; Pesch, B.; Rabstein, S.; Bruning, T.; Ko, Y.; Hamann, U.; Justenhoven, C.; Brauch, H.; et al. Expression of xenobiotic and steroid hormone metabolizing enzymes in human breast carcinomas. Int. J. Cancer 2006, 119, 1785–1791. [Google Scholar] [CrossRef]
- Murray, G.I.; Patimalla, S.; Stewart, K.N.; Miller, I.D.; Heys, S.D. Profiling the expression of cytochrome P450 in breast cancer. Histopathology 2010, 57, 202–211. [Google Scholar] [CrossRef]
- Downie, D.; McFadyen, M.C.; Rooney, P.H.; Cruickshank, M.E.; Parkin, D.E.; Miller, I.D.; Telfer, C.; Melvin, W.T.; Murray, G.I. Profiling cytochrome P450 expression in ovarian cancer: Identification of prognostic markers. Clin. Cancer Res. 2005, 11, 7369–7375. [Google Scholar] [CrossRef]
- Fujimura, T.; Takahashi, S.; Urano, T.; Kumagai, J.; Murata, T.; Takayama, K.; Ogushi, T.; Horie-Inoue, K.; Ouchi, Y.; Kitamura, T.; et al. Expression of cytochrome P450 3A4 and its clinical significance in human prostate cancer. Urology 2009, 74, 391–397. [Google Scholar] [CrossRef]
- Leskela, S.; Honrado, E.; Montero-Conde, C.; Landa, I.; Cascon, A.; Leton, R.; Talavera, P.; Cozar, J.M.; Concha, A.; Robledo, M.; et al. Cytochrome P450 3A5 is highly expressed in normal prostate cells but absent in prostate cancer. Endocr. Relat. Cancer 2007, 14, 645–654. [Google Scholar] [CrossRef]
- Kapucuoglu, N.; Coban, T.; Raunio, H.; Pelkonen, O.; Edwards, R.J.; Boobis, A.R.; Iscan, M. Expression of CYP3A4 in human breast tumour and non-tumour tissues. Cancer Lett. 2003, 202, 17–23. [Google Scholar] [CrossRef] [PubMed]
- El-Rayes, B.F.; Ali, S.; Heilbrun, L.K.; Lababidi, S.; Bouwman, D.; Visscher, D.; Philip, P.A. Cytochrome p450 and glutathione transferase expression in human breast cancer. Clin. Cancer Res. 2003, 9, 1705–1709. [Google Scholar] [PubMed]
- Maksymchuk, O.V.; Kashuba, V.I. Altered expression of cytochrome P450 enzymes involved in metabolism of androgens and vitamin D in the prostate as a risk factor for prostate cancer. Pharmacol. Rep. 2020, 72, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, A.M.; Hakkola, J.; Vaarala, M.H.; Kauppila, S.; Hirvikoski, P.; Vuoristo, J.T.; Edwards, R.J.; Paavonen, T.K. Characterization of androgen-regulated expression of CYP3A5 in human prostate. Carcinogenesis 2007, 28, 916–921. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Y.; Wang, M.T.; Zeng, S.; Nie, D. Human pregnane X receptor and resistance to chemotherapy in prostate cancer. Cancer Res. 2007, 67, 10361–10367. [Google Scholar] [CrossRef]
- Qiao, E.Q.; Yang, H.J. Effect of pregnane X receptor expression on drug resistance in breast cancer. Oncol. Lett. 2014, 7, 1191–1196. [Google Scholar] [CrossRef]
- Ushakov, D.S.; Dorozhkova, A.S.; Babayants, E.V.; Ovchinnikov, V.Y.; Kushlinskii, D.N.; Adamyan, L.V.; Gulyaeva, L.F.; Kushlinskii, N.E. Expression of microRNA Potentially Regulated by AhR and CAR in Malignant Tumors of the Endometrium. Bull. Exp. Biol. Med. 2018, 165, 688–691. [Google Scholar] [CrossRef]
- Masuyama, H.; Nakamura, K.; Nobumoto, E.; Hiramatsu, Y. Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells. Int. J. Oncol. 2016, 49, 1211–1220. [Google Scholar] [CrossRef]
- Masuyama, H.; Hiramatsu, Y.; Kodama, J.; Kudo, T. Expression and potential roles of pregnane X receptor in endometrial cancer. J. Clin. Endocrinol. Metab. 2003, 88, 4446–4454. [Google Scholar] [CrossRef]
- Masuyama, H.; Suwaki, N.; Tateishi, Y.; Nakatsukasa, H.; Segawa, T.; Hiramatsu, Y. The pregnane X receptor regulates gene expression in a ligand- and promoter-selective fashion. Mol. Endocrinol. 2005, 19, 1170–1180. [Google Scholar] [CrossRef]
- Masuyama, H.; Nakatsukasa, H.; Takamoto, N.; Hiramatsu, Y. Down-regulation of pregnane X receptor contributes to cell growth inhibition and apoptosis by anticancer agents in endometrial cancer cells. Mol. Pharmacol. 2007, 72, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, W.; Li, D.; Yin, X.; Zhang, X.; Olsen, N.; Zheng, S.G. Vitamin D and Chronic Diseases. Aging Dis. 2017, 8, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Badenhoop, K. Vitamin D and type 1 diabetes mellitus: State of the art. Trends Endocrinol. Metab. 2005, 16, 261–266. [Google Scholar] [CrossRef]
- Nikolac Gabaj, N.; Unic, A.; Miler, M.; Pavicic, T.; Culej, J.; Bolanca, I.; Herman Mahecic, D.; Milevoj Kopcinovic, L.; Vrtaric, A. In sickness and in health: Pivotal role of vitamin D. Biochem. Med. 2020, 30, 020501. [Google Scholar] [CrossRef]
- Jeon, S.M.; Shin, E.A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Mondul, A.M.; Weinstein, S.J.; Layne, T.M.; Albanes, D. Vitamin D and Cancer Risk and Mortality: State of the Science, Gaps, and Challenges. Epidemiol. Rev. 2017, 39, 28–48. [Google Scholar] [CrossRef]
- Amrein, K.; Scherkl, M.; Hoffmann, M.; Neuwersch-Sommeregger, S.; Kostenberger, M.; Tmava Berisha, A.; Martucci, G.; Pilz, S.; Malle, O. Vitamin D deficiency 2.0: An update on the current status worldwide. Eur. J. Clin. Nutr. 2020, 74, 1498–1513. [Google Scholar] [CrossRef]
- Wang, S. Epidemiology of vitamin D in health and disease. Nutr. Res. Rev. 2009, 22, 188–203. [Google Scholar] [CrossRef]
- Karakaya Molla, G.; Unal Uzun, O.; Koc, N.; Ozen Yesil, B.; Bayhan, G.I. Evaluation of nutritional status in pediatric patients diagnosed with Covid-19 infection. Clin. Nutr. ESPEN 2021, 44, 424–428. [Google Scholar] [CrossRef]
- Khazai, N.; Judd, S.E.; Tangpricha, V. Calcium and vitamin D: Skeletal and extraskeletal health. Curr. Rheumatol. Rep. 2008, 10, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Fleet, J.C. The role of vitamin D in the endocrinology controlling calcium homeostasis. Mol. Cell. Endocrinol. 2017, 453, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Norton, R. Vitamin D and respiratory health. Clin. Exp. Immunol. 2009, 158, 20–25. [Google Scholar] [CrossRef]
- Kumar, V.; Kancharla, S.; Jena, M.K. In silico virtual screening-based study of nutraceuticals predicts the therapeutic potentials of folic acid and its derivatives against COVID-19. Virusdisease 2021, 32, 29–37. [Google Scholar] [CrossRef]
- Grant, W.B. Epidemiology of disease risks in relation to vitamin D insufficiency. Prog. Biophys. Mol. Biol. 2006, 92, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Dinicolantonio, J.J.; Milani, R.V.; O’Keefe, J.H. Vitamin D and cardiovascular health. Circulation 2013, 128, 2404–2406. [Google Scholar] [CrossRef]
- Ferder, M.; Inserra, F.; Manucha, W.; Ferder, L. The world pandemic of vitamin D deficiency could possibly be explained by cellular inflammatory response activity induced by the renin-angiotensin system. Am. J. Physiol. Cell Physiol. 2013, 304, C1027–C1039. [Google Scholar] [CrossRef]
- Al-Timimi, D.J.; Ali, A.F. Serum 25(OH) D in Diabetes Mellitus Type 2: Relation to Glycaemic Control. J. Clin. Diagn. Res. 2013, 7, 2686–2688. [Google Scholar] [CrossRef]
- Roizen, J.D.; Li, D.; O’Lear, L.; Javaid, M.K.; Shaw, N.J.; Ebeling, P.R.; Nguyen, H.H.; Rodda, C.P.; Thummel, K.E.; Thacher, T.D.; et al. CYP3A4 mutation causes vitamin D-dependent rickets type 3. J. Clin. Investig. 2018, 128, 1913–1918. [Google Scholar] [CrossRef]
- Harinarayan, C.V. Vitamin D and diabetes mellitus. Hormones 2014, 13, 163–181. [Google Scholar] [CrossRef]
- Lim, S.; Kim, M.J.; Choi, S.H.; Shin, C.S.; Park, K.S.; Jang, H.C.; Billings, L.K.; Meigs, J.B. Association of vitamin D deficiency with incidence of type 2 diabetes in high-risk Asian subjects. Am. J. Clin. Nutr. 2013, 97, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Vimaleswaran, K.S.; Berry, D.J.; Lu, C.; Tikkanen, E.; Pilz, S.; Hiraki, L.T.; Cooper, J.D.; Dastani, Z.; Li, R.; Houston, D.K.; et al. Causal relationship between obesity and vitamin D status: Bi-directional Mendelian randomization analysis of multiple cohorts. PLoS Med. 2013, 10, e1001383. [Google Scholar] [CrossRef] [PubMed]
- Moreira TS, H.M. The role of vitamin D deficiency in the pathogenesis of type 2 diabetes mellitus. E-SPEN Eur. E-J. Clin. Nutr. Metab. 2010, 5, e155–e165. [Google Scholar] [CrossRef]
- Sapkota, B.R.; Hopkins, R.; Bjonnes, A.; Ralhan, S.; Wander, G.S.; Mehra, N.K.; Singh, J.R.; Blackett, P.R.; Saxena, R.; Sanghera, D.K. Genome-wide association study of 25(OH) Vitamin D concentrations in Punjabi Sikhs: Results of the Asian Indian diabetic heart study. J. Steroid Biochem. Mol. Biol. 2016, 158, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Hu, M.; Duan, R.; Liu, C.; Guo, H.; Zhang, M.; Yu, Y.; Wang, X.; Liu, L.; Liu, X. Increased levels of fatty acids contributed to induction of hepatic CYP3A4 activity induced by diabetes—In vitro evidence from HepG2 cell and Fa2N-4 cell lines. J. Pharmacol. Sci. 2014, 124, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Chiasson, J.L.; Turgeon, J.; Grangeon, A.; Michaud, V. Modulation of CYP450 Activities in Patients With Type 2 Diabetes. Clin. Pharmacol. Ther. 2019, 106, 1280–1289. [Google Scholar] [CrossRef]
- Dostalek, M.; Court, M.H.; Yan, B.; Akhlaghi, F. Significantly reduced cytochrome P450 3A4 expression and activity in liver from humans with diabetes mellitus. Br. J. Pharmacol. 2011, 163, 937–947. [Google Scholar] [CrossRef]
- Giammanco, M.; Di Majo, D.; La Guardia, M.; Aiello, S.; Crescimannno, M.; Flandina, C.; Tumminello, F.M.; Leto, G. Vitamin D in cancer chemoprevention. Pharm. Biol. 2015, 53, 1399–1434. [Google Scholar] [CrossRef]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, Q.; Yang, X.; Qian, S.Y.; Guo, B. Vitamin D Enhances the Efficacy of Irinotecan through miR-627-Mediated Inhibition of Intratumoral Drug Metabolism. Mol. Cancer Ther. 2016, 15, 2086–2095. [Google Scholar] [CrossRef]
- Morgan, E.T. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin. Pharmacol. Ther. 2009, 85, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.T. Drug Metabolism in Diseases; Xie, W., Ed.; Academic Press: Boston, MA, USA, 2017; pp. 21–58. [Google Scholar]
- Wu, K.C.; Lin, C.J. The regulation of drug-metabolizing enzymes and membrane transporters by inflammation: Evidences in inflammatory diseases and age-related disorders. J. Food Drug Anal. 2019, 27, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, C.; Rollason, V.; Desmeules, J.A.; Samer, C.F. Influence of Inflammation on Cytochromes P450 Activity in Adults: A Systematic Review of the Literature. Front. Pharmacol. 2021, 12, 733935. [Google Scholar] [CrossRef]
- Rendic, S.; Guengerich, F.P. Update information on drug metabolism systems--2009, part II: Summary of information on the effects of diseases and environmental factors on human cytochrome P450 (CYP) enzymes and transporters. Curr. Drug Metab. 2010, 11, 4–84. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.; Hermann, M. Immunological response as a source to variability in drug metabolism and transport. Front. Pharmacol. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Yanev, S.G. Immune system drug metabolism interactions: Toxicological insight. Adipobiology 2014, 6, 31–36. [Google Scholar] [CrossRef][Green Version]
- Renton, K.W. Regulation of drug metabolism and disposition during inflammation and infection. Expert Opin. Drug Metab. Toxicol. 2005, 1, 629–640. [Google Scholar] [CrossRef]
- Molanaei, H.; Stenvinkel, P.; Qureshi, A.R.; Carrero, J.J.; Heimburger, O.; Lindholm, B.; Diczfalusy, U.; Odar-Cederlof, I.; Bertilsson, L. Metabolism of alprazolam (a marker of CYP3A4) in hemodialysis patients with persistent inflammation. Eur. J. Clin. Pharmacol. 2012, 68, 571–577. [Google Scholar] [CrossRef]
- White, C.M.; Sicignano, D.J.; Smith, K. Impact of Interferons and Biological Drug Inhibitors of IL-2 and IL-6 on Small-Molecule Drug Metabolism Through the Cytochrome P450 System. Ann. Pharmacother. 2022, 56, 170–180. [Google Scholar] [CrossRef]
- Wonganan, P.; Jonsson-Schmunk, K.; Callahan, S.M.; Choi, J.H.; Croyle, M.A. Evaluation of the HC-04 cell line as an in vitro model for mechanistic assessment of changes in hepatic cytochrome P450 3A during adenovirus infection. Drug Metab. Dispos. 2014, 42, 1191–1201. [Google Scholar] [CrossRef]
- Morcos, P.N.; Moreira, S.A.; Brennan, B.J.; Blotner, S.; Shulman, N.S.; Smith, P.F. Influence of chronic hepatitis C infection on cytochrome P450 3A4 activity using midazolam as an in vivo probe substrate. Eur. J. Clin. Pharmacol. 2013, 69, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Morcos, P.N.; Chang, L.; Kulkarni, R.; Giraudon, M.; Shulman, N.; Brennan, B.J.; Smith, P.F.; Tran, J.Q. A randomised study of the effect of danoprevir/ritonavir or ritonavir on substrates of cytochrome P450 (CYP) 3A and 2C9 in chronic hepatitis C patients using a drug cocktail. Eur. J. Clin. Pharmacol. 2013, 69, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.E.; Brown, K.C.; Werner, R.E.; Gotzkowsky, K.; Gaedigk, A.; Blake, M.; Hein, D.W.; van der Horst, C.; Kashuba, A.D. Variability in drug metabolizing enzyme activity in HIV-infected patients. Eur. J. Clin. Pharmacol. 2010, 66, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Jetter, A.; Fatkenheuer, G.; Frank, D.; Klaassen, T.; Seeringer, A.; Doroshyenko, O.; Kirchheiner, J.; Hein, W.; Schomig, E.; Fuhr, U.; et al. Do activities of cytochrome P450 (CYP)3A, CYP2D6 and P-glycoprotein differ between healthy volunteers and HIV-infected patients? Antivir. Ther. 2010, 15, 975–983. [Google Scholar] [CrossRef]
- Wang, G.; Xiao, B.; Deng, J.; Gong, L.; Li, Y.; Li, J.; Zhong, Y. The Role of Cytochrome P450 Enzymes in COVID-19 Pathogenesis and Therapy. Front. Pharmacol. 2022, 13, 791922. [Google Scholar] [CrossRef]
- Lenoir, C.; Terrier, J.; Gloor, Y.; Curtin, F.; Rollason, V.; Desmeules, J.A.; Daali, Y.; Reny, J.L.; Samer, C.F. Impact of SARS-CoV-2 Infection (COVID-19) on Cytochromes P450 Activity Assessed by the Geneva Cocktail. Clin. Pharmacol. Ther. 2021, 110, 1358–1367. [Google Scholar] [CrossRef]
- Deb, S.; Arrighi, S. Potential Effects of COVID-19 on Cytochrome P450-Mediated Drug Metabolism and Disposition in Infected Patients. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 185–203. [Google Scholar] [CrossRef]
- Gregoire, M.; Le Turnier, P.; Gaborit, B.J.; Veyrac, G.; Lecomte, R.; Boutoille, D.; Canet, E.; Imbert, B.M.; Bellouard, R.; Raffi, F. Lopinavir pharmacokinetics in COVID-19 patients. J. Antimicrob. Chemother. 2020, 75, 2702–2704. [Google Scholar] [CrossRef]
- Schoergenhofer, C.; Jilma, B.; Stimpfl, T.; Karolyi, M.; Zoufaly, A. Pharmacokinetics of Lopinavir and Ritonavir in Patients Hospitalized With Coronavirus Disease 2019 (COVID-19). Ann. Intern. Med. 2020, 173, 670–672. [Google Scholar] [CrossRef]
- Testa, S.; Prandoni, P.; Paoletti, O.; Morandini, R.; Tala, M.; Dellanoce, C.; Giorgi-Pierfranceschi, M.; Betti, M.; Danzi, G.B.; Pan, A.; et al. Direct oral anticoagulant plasma levels’ striking increase in severe COVID-19 respiratory syndrome patients treated with antiviral agents: The Cremona experience. J. Thromb. Haemost. 2020, 18, 1320–1323. [Google Scholar] [CrossRef]
- Cojutti, P.G.; Londero, A.; Della Siega, P.; Givone, F.; Fabris, M.; Biasizzo, J.; Tascini, C.; Pea, F. Comparative Population Pharmacokinetics of Darunavir in SARS-CoV-2 Patients vs. HIV Patients: The Role of Interleukin-6. Clin. Pharmacokinet. 2020, 59, 1251–1260. [Google Scholar] [CrossRef]
- Marzolini, C.; Stader, F.; Stoeckle, M.; Franzeck, F.; Egli, A.; Bassetti, S.; Hollinger, A.; Osthoff, M.; Weisser, M.; Gebhard, C.E.; et al. Effect of Systemic Inflammatory Response to SARS-CoV-2 on Lopinavir and Hydroxychloroquine Plasma Concentrations. Antimicrob. Agents Chemother. 2020, 64, e01177-20. [Google Scholar] [CrossRef] [PubMed]
- Arvinte, C.; Singh, M.; Marik, P.E. Serum Levels of Vitamin C and Vitamin D in a Cohort of Critically Ill COVID-19 Patients of a North American Community Hospital Intensive Care Unit in May 2020: A Pilot Study. Med. Drug Discov. 2020, 8, 100064. [Google Scholar] [CrossRef] [PubMed]
- Dostalek, M.; Akhlaghi, F.; Puzanovova, M. Effect of diabetes mellitus on pharmacokinetic and pharmacodynamic properties of drugs. Clin. Pharmacokinet. 2012, 51, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Darakjian, L.; Deodhar, M.; Turgeon, J.; Michaud, V. Chronic Inflammatory Status Observed in Patients with Type 2 Diabetes Induces Modulation of Cytochrome P450 Expression and Activity. Int. J. Mol. Sci. 2021, 22, 4967. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, B.M.; Syversen, S.W.; Vistnes, M.; Lie, E.; Mehus, L.L.; Molden, E. Associations between Cytokine Levels and CYP3A4 Phenotype in Patients with Rheumatoid Arthritis. Drug Metab. Dispos. 2018, 46, 1384–1389. [Google Scholar] [CrossRef]
- Wollmann, B.M.; Syversen, S.W.; Lie, E.; Gjestad, C.; Mehus, L.L.; Olsen, I.C.; Molden, E. 4beta-Hydroxycholesterol Level in Patients With Rheumatoid Arthritis Before vs. After Initiation of bDMARDs and Correlation With Inflammatory State. Clin. Transl. Sci. 2017, 10, 42–49. [Google Scholar] [CrossRef]
- Lee, E.B.; Daskalakis, N.; Xu, C.; Paccaly, A.; Miller, B.; Fleischmann, R.; Bodrug, I.; Kivitz, A. Disease-Drug Interaction of Sarilumab and Simvastatin in Patients with Rheumatoid Arthritis. Clin. Pharmacokinet. 2017, 56, 607–615. [Google Scholar] [CrossRef]
- Schmitt, C.; Kuhn, B.; Zhang, X.; Kivitz, A.J.; Grange, S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin. Pharmacol. Ther. 2011, 89, 735–740. [Google Scholar] [CrossRef]
- Zhuang, Y.; de Vries, D.E.; Xu, Z.; Marciniak, S.J., Jr.; Chen, D.; Leon, F.; Davis, H.M.; Zhou, H. Evaluation of disease-mediated therapeutic protein-drug interactions between an anti-interleukin-6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J. Clin. Pharmacol. 2015, 55, 1386–1394. [Google Scholar] [CrossRef]
- Langmann, T.; Moehle, C.; Mauerer, R.; Scharl, M.; Liebisch, G.; Zahn, A.; Stremmel, W.; Schmitz, G. Loss of detoxification in inflammatory bowel disease: Dysregulation of pregnane X receptor target genes. Gastroenterology 2004, 127, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Kenny, J.R.; Dickmann, L.; Maciuca, R.; Looney, C.; Tang, M.T. Assessment of Disease-Related Therapeutic Protein Drug-Drug Interaction for Etrolizumab in Patients With Moderately to Severely Active Ulcerative Colitis. J. Clin. Pharmacol. 2016, 56, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Lirio, R.A.; Schneider, J.; Aubrecht, J.; Kadali, H.; Baratta, M.; Gulati, P.; Suri, A.; Lin, T.; Vasudevan, R.; et al. Assessment of Vedolizumab Disease-Drug-Drug Interaction Potential in Patients With Inflammatory Bowel Diseases. Clin. Pharmacol. Drug Dev. 2021, 10, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Urquhart, B.L.; Ponich, T.; Chande, N.; Gregor, J.C.; Beaton, M.; Kim, R.B. Crohn’s Disease Is Associated with Decreased CYP3A4 and P-Glycoprotein Protein Expression. Mol. Pharm. 2019, 16, 4059–4064. [Google Scholar] [CrossRef] [PubMed]
- Shakhnovich, V.; Vyhlidal, C.; Friesen, C.; Hildreth, A.; Singh, V.; Daniel, J.; Kearns, G.L.; Leeder, J.S. Decreased Pregnane X Receptor Expression in Children with Active Crohn’s Disease. Drug Metab. Dispos. 2016, 44, 1066–1069. [Google Scholar] [CrossRef]
- Vyhlidal, C.A.; Chapron, B.D.; Ahmed, A.; Singh, V.; Casini, R.; Shakhnovich, V. Effect of Crohn’s Disease on Villous Length and CYP3A4 Expression in the Pediatric Small Intestine. Clin. Transl. Sci. 2021, 14, 729–736. [Google Scholar] [CrossRef]
- Wilson, A.; Tirona, R.G.; Kim, R.B. CYP3A4 Activity is Markedly Lower in Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2017, 23, 804–813. [Google Scholar] [CrossRef]
- Bragde, H.; Jansson, U.; Jarlsfelt, I.; Soderman, J. Gene expression profiling of duodenal biopsies discriminates celiac disease mucosa from normal mucosa. Pediatr. Res. 2011, 69, 530–537. [Google Scholar] [CrossRef]
- Harvey, R.D.; Morgan, E.T. Cancer, inflammation, and therapy: Effects on cytochrome p450-mediated drug metabolism and implications for novel immunotherapeutic agents. Clin. Pharmacol. Ther. 2014, 96, 449–457. [Google Scholar] [CrossRef]
- Trousil, S.; Lee, P.; Edwards, R.J.; Maslen, L.; Lozan-Kuehne, J.P.; Ramaswami, R.; Aboagye, E.O.; Clarke, S.; Liddle, C.; Sharma, R. Altered cytochrome 2E1 and 3A P450-dependent drug metabolism in advanced ovarian cancer correlates to tumour-associated inflammation. Br. J. Pharmacol. 2019, 176, 3712–3722. [Google Scholar] [CrossRef]
- Kacevska, M.; Robertson, G.R.; Clarke, S.J.; Liddle, C. Inflammation and CYP3A4-mediated drug metabolism in advanced cancer: Impact and implications for chemotherapeutic drug dosing. Expert Opin. Drug Metab. Toxicol. 2008, 4, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Charles, K.A.; Rivory, L.P.; Brown, S.L.; Liddle, C.; Clarke, S.J.; Robertson, G.R. Transcriptional repression of hepatic cytochrome P450 3A4 gene in the presence of cancer. Clin. Cancer Res. 2006, 12, 7492–7497. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.F. S. Bile Acid Activated Receptors: Integrating Immune and Metabolic Regulation in Non-Alcoholic Fatty Liver Disease. Liver Res. 2021, 5, 119–141. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Molina-Ortiz, D.; Camacho-Carranza, R.; Gonzalez-Zamora, J.F.; Shalkow-Kalincovstein, J.; Cardenas-Cardos, R.; Nosti-Palacios, R.; Vences-Mejia, A. Differential expression of cytochrome P450 enzymes in normal and tumor tissues from childhood rhabdomyosarcoma. PLoS ONE 2014, 9, e93261. [Google Scholar] [CrossRef] [PubMed]
- Torres-Zarate, C.; Vences-Mejia, A.; Espinosa-Aguirre, J.J.; Diaz-Diaz, E.; Palacios-Acosta, J.M.; Cardenas-Cardos, R.; Hernandez-Arrazola, D.; Shalkow-Klincovstein, J.; Jurado, R.R.; Santes-Palacios, R.; et al. Expression of Cytochrome P450 Enzymes in Pediatric Non-Rhabdomyosarcoma Soft Tissue Sarcomas: Possible Role in Carcinogenesis and Treatment Response. Int. J. Toxicol. 2022, 41, 234–242. [Google Scholar] [CrossRef]
- Zia, H.; Murray, G.I.; Vyhlidal, C.A.; Leeder, J.S.; Anwar, A.E.; Bui, M.M.; Ahmed, A.A. CYP3A isoforms in Ewing’s sarcoma tumours: An immunohistochemical study with clinical correlation. Int. J. Exp. Pathol. 2015, 96, 81–86. [Google Scholar] [CrossRef]
- Tsunedomi, R.; Iizuka, N.; Hamamoto, Y.; Uchimura, S.; Miyamoto, T.; Tamesa, T.; Okada, T.; Takemoto, N.; Takashima, M.; Sakamoto, K.; et al. Patterns of expression of cytochrome P450 genes in progression of hepatitis C virus-associated hepatocellular carcinoma. Int. J. Oncol. 2005, 27, 661–667. [Google Scholar]
- Jiang, F.; Chen, L.; Yang, Y.C.; Wang, X.M.; Wang, R.Y.; Li, L.; Wen, W.; Chang, Y.X.; Chen, C.Y.; Tang, J.; et al. CYP3A5 Functions as a Tumor Suppressor in Hepatocellular Carcinoma by Regulating mTORC2/Akt Signaling. Cancer Res. 2015, 75, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Wang, X.; Zhu, G.; Han, C.; Su, H.; Liao, X.; Yang, C.; Qin, W.; Huang, K.; Peng, T. The prognostic value of differentially expressed CYP3A subfamily members for hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Wang, L.; Liang, Y.; Dong, G.; Xia, W.; Hu, J.; Xu, L.; Jiang, F. CYP3A5 suppresses metastasis of lung adenocarcinoma through ATOH8/Smad1 axis. Am. J. Cancer Res. 2020, 10, 3194–3211. [Google Scholar] [PubMed]
- Nokin, M.J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 11. [Google Scholar] [CrossRef]
- Mitra, R.; Goodman, O.B., Jr. CYP3A5 regulates prostate cancer cell growth by facilitating nuclear translocation of AR. Prostate 2015, 75, 527–538. [Google Scholar] [CrossRef]
- Gorjala, P.; Kittles, R.A.; Goodman, O.B., Jr.; Mitra, R. Role of CYP3A5 in Modulating Androgen Receptor Signaling and Its Relevance to African American Men with Prostate Cancer. Cancers 2020, 12, 989. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).