
In Silico Identification of Genes Associated with Breast Cancer Progression and Prognosis and Novel Therapeutic Targets

Abstract
:1. Introduction
2. Methods
2.1. LinkedOmics Analysis
2.2. UALCAN Analysis
2.3. Gene Expression Profiling Interactive Analysis 2 (GEPIA2)
2.4. DepMap Analysis
2.5. Statistical Analysis
3. Results
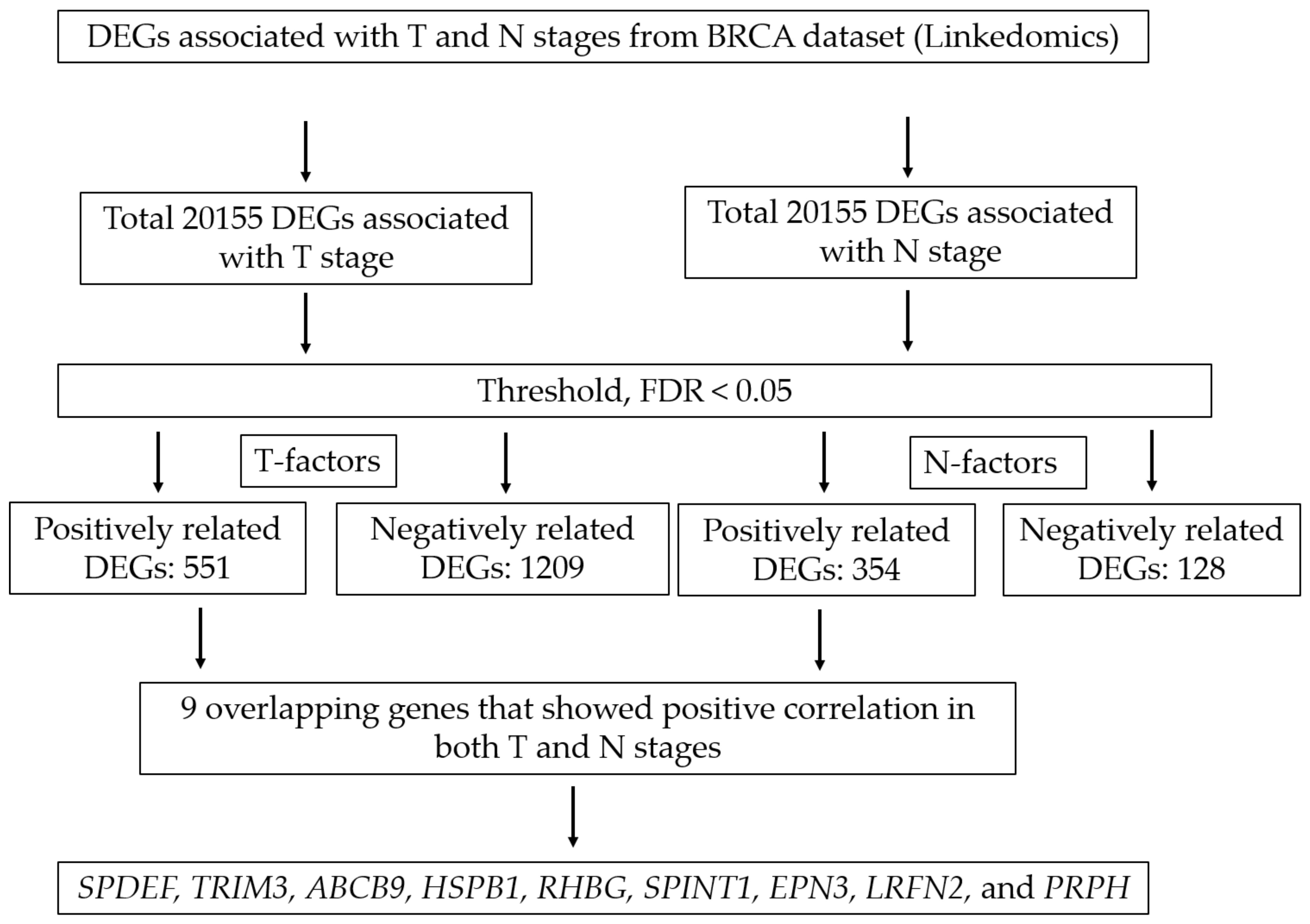
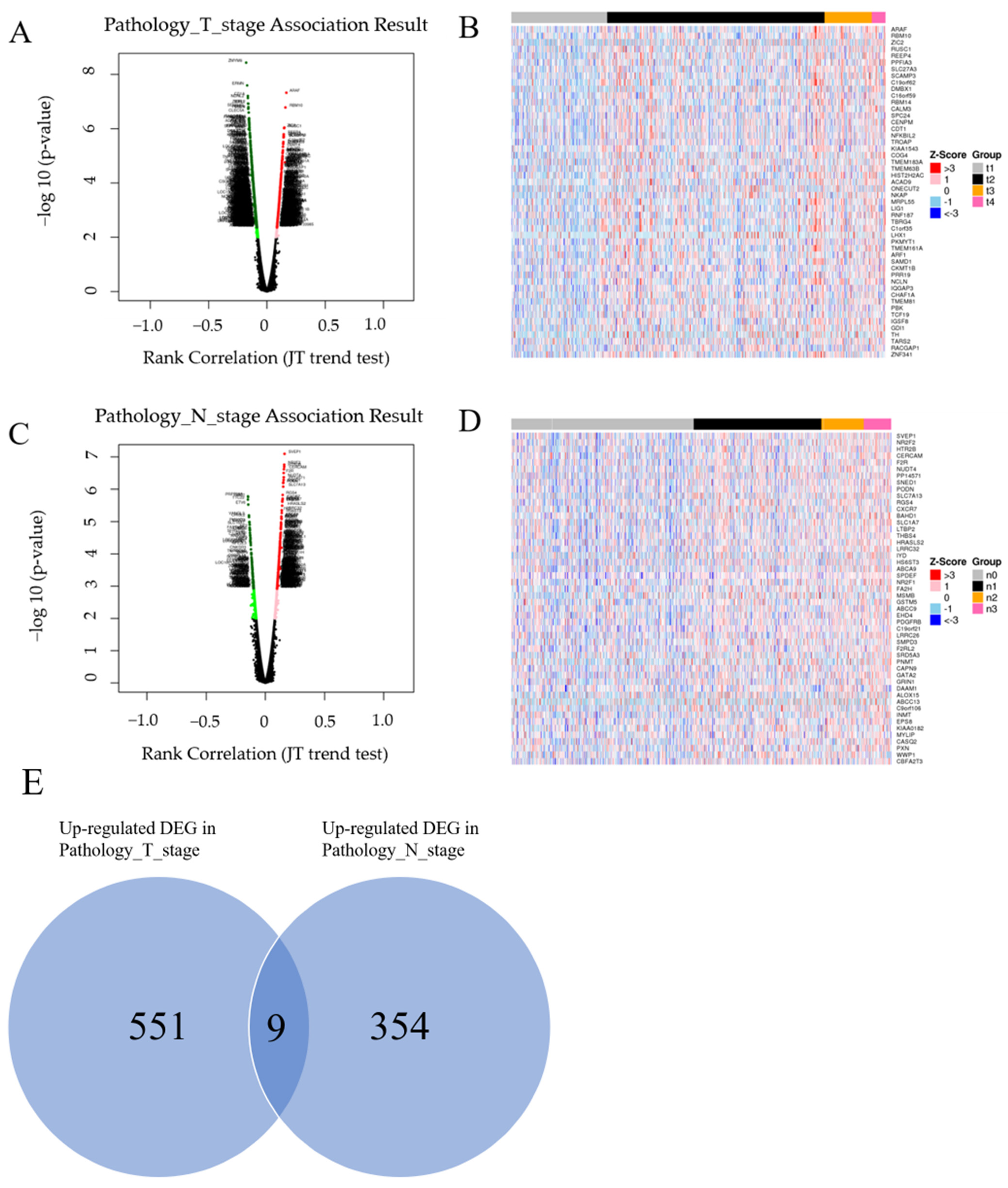
3.1. Screening Overlapping Upregulated DEGs
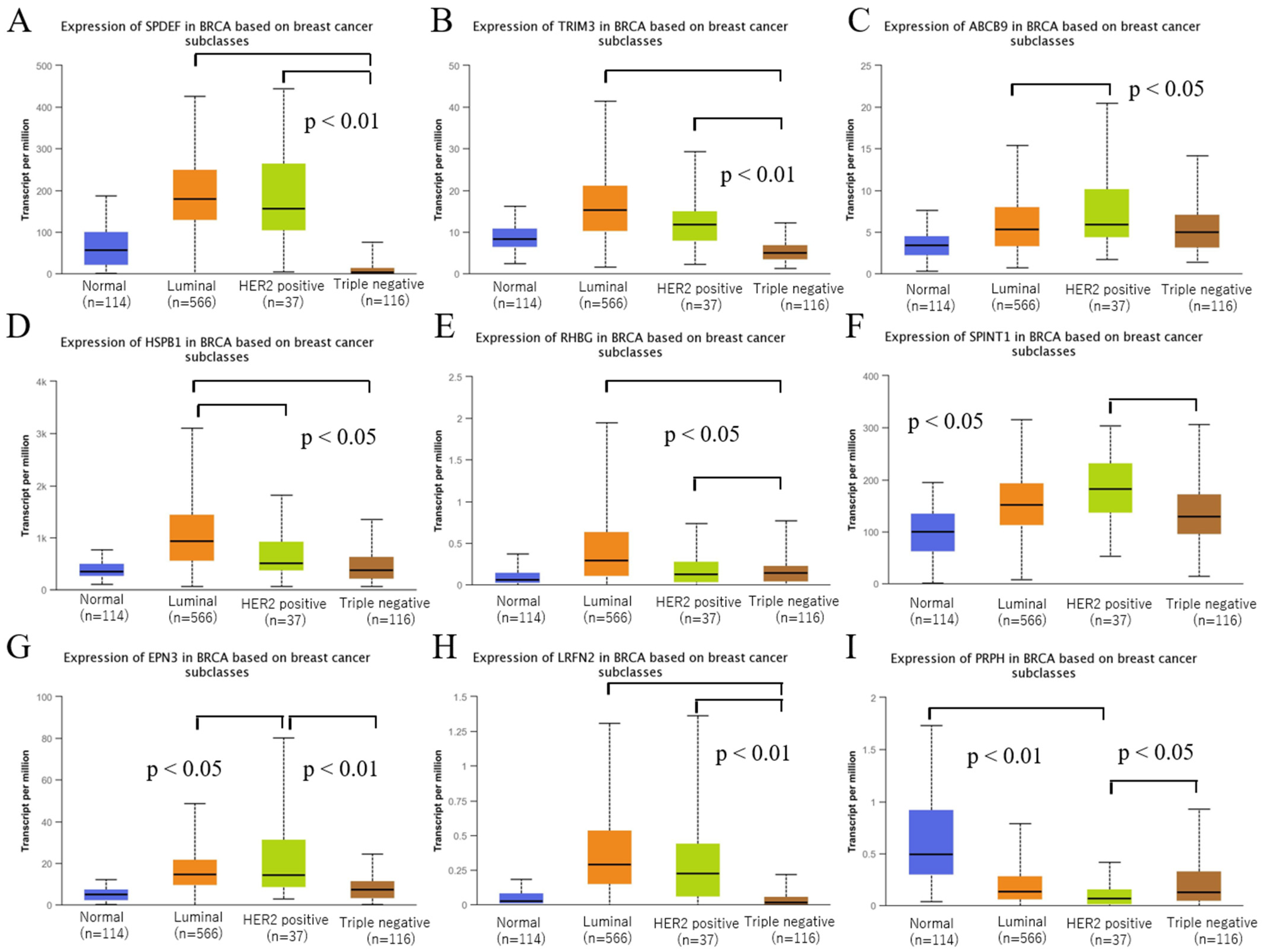
3.2. Validation of the Expression of Selected Genes in BC
3.3. Validation of the Expression of Selected Genes among Intrinsic Subtypes of BRCA
3.4. Survival Analysis for the Nine Genes
3.5. CRISPR-Cas9 and RNAi Screening Using DepMap
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Editorial Board. WHO classification of tumours. In Breast Tumours, 5th ed.; IARC: Lyon, France, 2019. [Google Scholar]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, J.; Rothman, J.; McDonald, K.A.; Nagahashi, M.; Takabe, K.; Wakai, T. Clinical target sequencing for precision medicine of breast cancer. Int. J. Clin. Oncol. 2019, 24, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Cserni, G.; Chmielik, E.; Cserni, B.; Tot, T. The new TNM-Based staging of breast cancer. Virchows Arch. 2018, 472, 697–703. [Google Scholar] [CrossRef]
- Hutchinson, S.A.; Websdale, A.; Cioccoloni, G.; Røberg-Larsen, H.; Lianto, P.; Kim, B.; Rose, A.; Soteriou, C.; Pramanik, A.; Wastall, L.M.; et al. Liver x receptor alpha drives chemoresistance in response to side-Chain hydroxycholesterols in triple negative breast cancer. Oncogene 2021, 40, 2872–2883. [Google Scholar] [CrossRef] [PubMed]
- Gunyuz, Z.E.; Sahi-Ilhan, E.; Kucukkose, C.; Ipekgil, D.; Tok, G.; Mese, G.; Ozcivici, E.; Yalcin-Ozuysal, O. SEMA6D differentially regulates proliferation, migration, and invasion of breast cell lines. ACS Omega 2022, 7, 15769–15778. [Google Scholar] [CrossRef]
- Bhandari, A.; Xia, E.; Zhou, Y.; Guan, Y.; Xiang, J.; Kong, L.; Wang, Y.; Yang, F.; Wang, O.; Zhang, X. ITGA7 functions as a tumor suppressor and regulates migration and invasion in breast cancer. Cancer Manag. Res. 2018, 10, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Song, Z. Identification of core miRNAs and regulatory pathways in breast cancer by integrated bioinformatics analysis. Mol. Omics. 2021, 17, 277–287. [Google Scholar] [CrossRef]
- Zheng, S.; Zou, Y.; Liang, J.Y.; Xiao, W.; Yang, A.; Meng, T.; Lu, S.; Luo, Z.; Xie, X. Identification and validation of a combined hypoxia and immune index for triple-negative breast cancer. Mol. Oncol. 2020, 14, 2814–2833. [Google Scholar] [CrossRef]
- Craven, K.E.; Gökmen-Polar, Y.; Badve, S.S. CIBERSORT analysis of TCGA and METABRIC identifies subgroups with better outcomes in triple negative breast cancer. Sci. Rep. 2021, 11, 4691. [Google Scholar] [CrossRef]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a cancer dependency map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Larrue, C.; Scheidegger, N.K.; Seong, B.K.A.; Dharia, N.V.; Kuljanin, M.; Wechsler, C.S.; Kugener, G.; Robichaud, A.L.; Conway, A.S.; et al. An in vivo CRISPR screening platform for prioritizing therapeutic targets in AML. Cancer Discov. 2022, 12, 432–449. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.; Ding, J.; Qi, J.; Gu, Z. Characteristics and prognostic value of potential dependency genes in clear cell renal cell carcinoma based on a large-Scale CRISPR-Cas9 and RNAi screening database DepMap. Int. J. Med. Sci. 2021, 18, 2063–2075. [Google Scholar] [CrossRef]
- Merhi, A.; De Mees, C.; Abdo, R.; Alberola, J.V.; Marini, A.M. Wnt/β-catenin signaling regulates the expression of the ammonium permease gene RHBG in human cancer cells. PLoS ONE 2015, 10, e0128683. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, L.; Wang, J.; Ge, B.; Wang, Q.; Wang, T.; Liu, C.; Wei, B.; Wang, Q.; Gao, Y. LRFN2 binding to NMDAR inhibits the progress of ESCC via regulating the Wnt/β-Catenin and NF-κB signaling pathway. Cancer Sci. 2022, 113, 3566–3578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liem, R.K.H. α-internexin and peripherin: Expression, assembly, functions, and roles in disease. Methods Enzymol. 2016, 568, 477–507. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Hayama, Y.; Kato, S.; Shimomura, A.; Shimomura, T.; Irie, K.; Kaneko, R.; Yanagawa, Y.; Kobayashi, K.; Ochiya, T. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci. 2019, 22, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Morimura, N.; Yasuda, H.; Yamaguchi, K.; Katayama, K.I.; Hatayama, M.; Tomioka, N.H.; Odagawa, M.; Kamiya, A.; Iwayama, Y.; Maekawa, M.; et al. Autism-like behaviours and enhanced memory formation and synaptic plasticity in Lrfn2/SALM1-deficient mice. Nat. Commun. 2017, 8, 15800. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.; Kim, S.; Chung, H.S.; Kim, K.; Han, K.; Kim, H.; Jun, H.; Kaang, B.K.; Kim, E. SALM synaptic cell adhesion-like molecules regulate the differentiation of excitatory synapses. Neuron 2006, 50, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Morimura, N.; Inoue, T.; Katayama, K.; Aruga, J. Comparative analysis of structure, expression and PSD95-binding capacity of Lrfn, a novel family of neuronal transmembrane proteins. Gene 2006, 380, 72–83. [Google Scholar] [CrossRef]
- Mattes, M.D.; Bhatia, J.K.; Metzger, D.; Ashamalla, H.; Katsoulakis, E. Breast cancer subtype as a predictor of lymph node metastasis according to the SEER registry. J. Breast Cancer 2015, 18, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Feldman, R.J.; Sementchenko, V.I.; Gayed, M.; Fraig, M.M.; Watson, D.K. Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res. 2003, 63, 4626–4631. [Google Scholar]
- Turner, D.P.; Moussa, O.; Sauane, M.; Fisher, P.B.; Watson, D.K. Prostate-derived ETS factor is a mediator of metastatic potential through the inhibition of migration and invasion in breast cancer. Cancer Res. 2007, 67, 1618–1625. [Google Scholar] [CrossRef] [Green Version]
- Ye, T.; Li, J.; Feng, J.; Guo, J.; Wan, X.; Xie, D.; Liu, J. The subtype-specific molecular function of SPDEF in breast cancer and insights into prognostic significance. J. Cell. Mol. Med. 2021, 25, 7307–7320. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Wang, B.; Tan, X.; Wu, L.; Li, X.; Li, Z.; Cai, Y.; Fan, R.; Yang, X.; Zhang, C.; et al. TRIM3 facilitates estrogen signaling and modulates breast cancer cell progression. Cell Commun. Signal. 2022, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Dang Cong, T.; Nguyen Thanh, T.; Nguyen Phan, Q.A.; Hoang Thi, A.P.; Nguyen Tran, B.S.; Nguyen Vu, Q.H. Correlation between HER2 expression and clinicopathological features of breast cancer: A cross- sectional study in Vietnam. Asian Pac. J. Cancer Prev. 2020, 21, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Shokouh, T.Z.; Ezatollah, A.; Barand, P. Interrelationships between Ki67, HER2/neu, p53, ER, and PR status and their associations with tumor grade and lymph node involvement in breast carcinoma subtypes: Retrospective-observational analytical study. Medicine 2015, 94, e1359. [Google Scholar] [CrossRef]
- Wei, L.; Liu, T.T.; Wang, H.H.; Hong, H.M.; Yu, A.L.; Feng, H.P.; Chang, W.W. Hsp27 participates in the maintenance of breast cancer stem cells through regulation of epithelial-mesenchymal transition and nuclear factor-κB. Breast Cancer Res. 2011, 13, R101. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, X.; Wang, H.; Wang, Y.; Chen, P.; Wang, L. Heat shock Protein 27 enhances SUMOylation of heat shock protein B8 to accelerate the progression of breast cancer. Am. J. Pathol. 2020, 190, 2464–2477. [Google Scholar] [CrossRef]
- Wu, Q.; Yin, G.; Luo, J.; Zhang, Y.; Ai, T.; Tian, J.; Jin, Y.; Lei, J.; Liu, S. Comprehensive analysis of the expression and prognostic value of SPINT1/2 in breast carcinoma. Front. Endocrinol. 2021, 12, 665666. [Google Scholar] [CrossRef]
- Gong, J.P.; Yang, L.; Tang, J.W.; Sun, P.; Hu, Q.; Qin, J.W.; Xu, X.M.; Sun, B.C.; Tang, J.H. Overexpression of microRNA-24 increases the sensitivity to paclitaxel in drug-resistant breast carcinoma cell lines via targeting ABCB9. Oncol. Lett. 2016, 12, 3905–3911. [Google Scholar] [CrossRef] [Green Version]
- Pandya, K.; Wyatt, D.; Gallagher, B.; Shah, D.; Baker, A.; Bloodworth, J.; Zlobin, A.; Pannuti, A.; Green, A.; Ellis, I.O.; et al. PKCα attenuates Jagged-1-Mediated Notch signaling in ErbB-2-Positive breast cancer to reverse trastuzumab resistance. Clin. Cancer Res. 2016, 22, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Elizalde, P.V.; Cordo Russo, R.I.; Chervo, M.F.; Schillaci, R. ErbB-2 nuclear function in breast cancer growth, metastasis and resistance to therapy. Endocr. Relat. Cancer 2016, 23, T243–T257. [Google Scholar] [CrossRef] [Green Version]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Symbol | Gene Name | p-Value (T Stage) | p-Value (N Stage) |
|---|---|---|---|---|
| 25,803 | SPDEF | SAM pointed domain-containing ETS transcription factor | 6.28 × 10−4 | 7.07 × 10−6 |
| 10,612 | TRIM3 | Tripartite motif containing 3 | 3.96 × 10−3 | 3.05 × 10−5 |
| 23,457 | ABCB9 | ATP binding cassette subfamily B member 9 | 7.00 × 10−5 | 3.35 × 10−5 |
| 3315 | HSPB1 | Heat shock protein family B (small) member 1 | 4.35 × 10−3 | 3.91 × 10−5 |
| 57,127 | RHBG | Rh family B glycoprotein | 6.17 × 10−4 | 9.17 × 10−5 |
| 6692 | SPINT1 | Serine peptidase inhibitor, Kunitz type 1 | 2.39 × 10−3 | 1.82 × 10−4 |
| 55,040 | EPN3 | Epsin 3 | 1.56 × 10−3 | 2.72 × 10−4 |
| 57,497 | LRFN2 | Leucine rich repeat and fibronectin type III domain-containing 2 | 4.02 × 10−3 | 4.11 × 10−4 |
| 5630 | PRPH | Peripherin | 5.85 × 10−4 | 8.92 × 10−4 |
| Gene Symbol | Cell Line | Subtype | Gene Effect Score |
|---|---|---|---|
| SPDEF | EVSAT | Unknown | −1.24 |
| MDAMB361 | HER2 | −1.23 | |
| OCUBM | Unknown | −1.21 | |
| MDAMB453 | HER2 | −1.08 | |
| MDAMB415 | HER2 | −0.76 | |
| HCC1419 | HER2 | −0.76 | |
| AU565 | HER2 | −0.74 | |
| EFM19 | Luminal | −0.71 | |
| CAMA1 | Luminal | −0.68 | |
| SKBR3 | HER2 | −0.66 | |
| MFM223 | Luminal | −0.65 | |
| HCC1428 | Luminal | −0.63 | |
| ZR751 | Luminal | −0.53 | |
| KPL1 | Luminal | −0.52 | |
| SPINT1 | VP228 | Unknown | −1.00 |
| HCC1954 | TN | −0.84 | |
| HCC1937 | TN | −0.58 | |
| SUM149PT | TN | −0.53 | |
| SUM159PT | TN | −0.52 | |
| HCC1187 | TN | −0.51 |
| Gene Symbol | Cell Line | Subtype | Gene Effect Score | Cell Line | Subtype | Gene Effect Score |
|---|---|---|---|---|---|---|
| SPDEF | EVSAT | Unknown | −1.74 | SKBR3 | HER2 | −1.35 |
| MDAMB330 | Unknown | −1.07 | MDAMB453 | HER2 | −1.05 | |
| ZR75B | Unknown | −1.05 | MDAMB361 | HER2 | −1.03 | |
| 600MPE | Unknown | −0.94 | HCC228 | HER2 | −0.94 | |
| HCC1419 | HER2 | −0.92 | SKBR5 | Unknown | −0.88 | |
| SUM225CWN | TN | −0.88 | OCUMBM | Unknown | −0.82 | |
| UACC812 | Luminal | −0.78 | ZR7530 | HER2 | −0.77 | |
| CAMA1 | Luminal | −0.76 | CAL148 | TN | −0.74 | |
| UACC893 | HER2 | −0.66 | BT483 | Luminal | −0.65 | |
| BT474 | HER2 | −0.58 | SUM190PT | TN | −0.57 | |
| LY2 | Unknown | −0.54 | ZR751 | Luminal | −0.53 | |
| HCC1428 | Luminal | −0.51 | EFM19 | Luminal | −0.51 | |
| TRIM3 | EFM192A | HER2 | −0.63 | 600MPE | Unknown | −0.55 |
| ABCB9 | EVSAT | Unknown | −0.81 | |||
| RHBG | CAL851 | TN | −0.63 | MACLS2 | Unknown | −0.52 |
| PRPH | BT474 | HER2 | −1.13 | SUM52PE | Luminal | −0.88 |
| HCC3153 | Unknown | −0.62 | CAL51 | TN | −0.58 | |
| SUM225CWN | TN | −0.53 | UACC893 | HER2 | −0.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uchida, S.; Sugino, T. In Silico Identification of Genes Associated with Breast Cancer Progression and Prognosis and Novel Therapeutic Targets. Biomedicines 2022, 10, 2995. https://doi.org/10.3390/biomedicines10112995
Uchida S, Sugino T. In Silico Identification of Genes Associated with Breast Cancer Progression and Prognosis and Novel Therapeutic Targets. Biomedicines. 2022; 10(11):2995. https://doi.org/10.3390/biomedicines10112995
Chicago/Turabian StyleUchida, Shiro, and Takashi Sugino. 2022. "In Silico Identification of Genes Associated with Breast Cancer Progression and Prognosis and Novel Therapeutic Targets" Biomedicines 10, no. 11: 2995. https://doi.org/10.3390/biomedicines10112995