
Large-Scale Functional Assessment of Genes Involved in Rare Diseases with Intellectual Disabilities Unravels Unique Developmental and Behaviour Profiles in Mouse Models

 , , ,
, , ,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
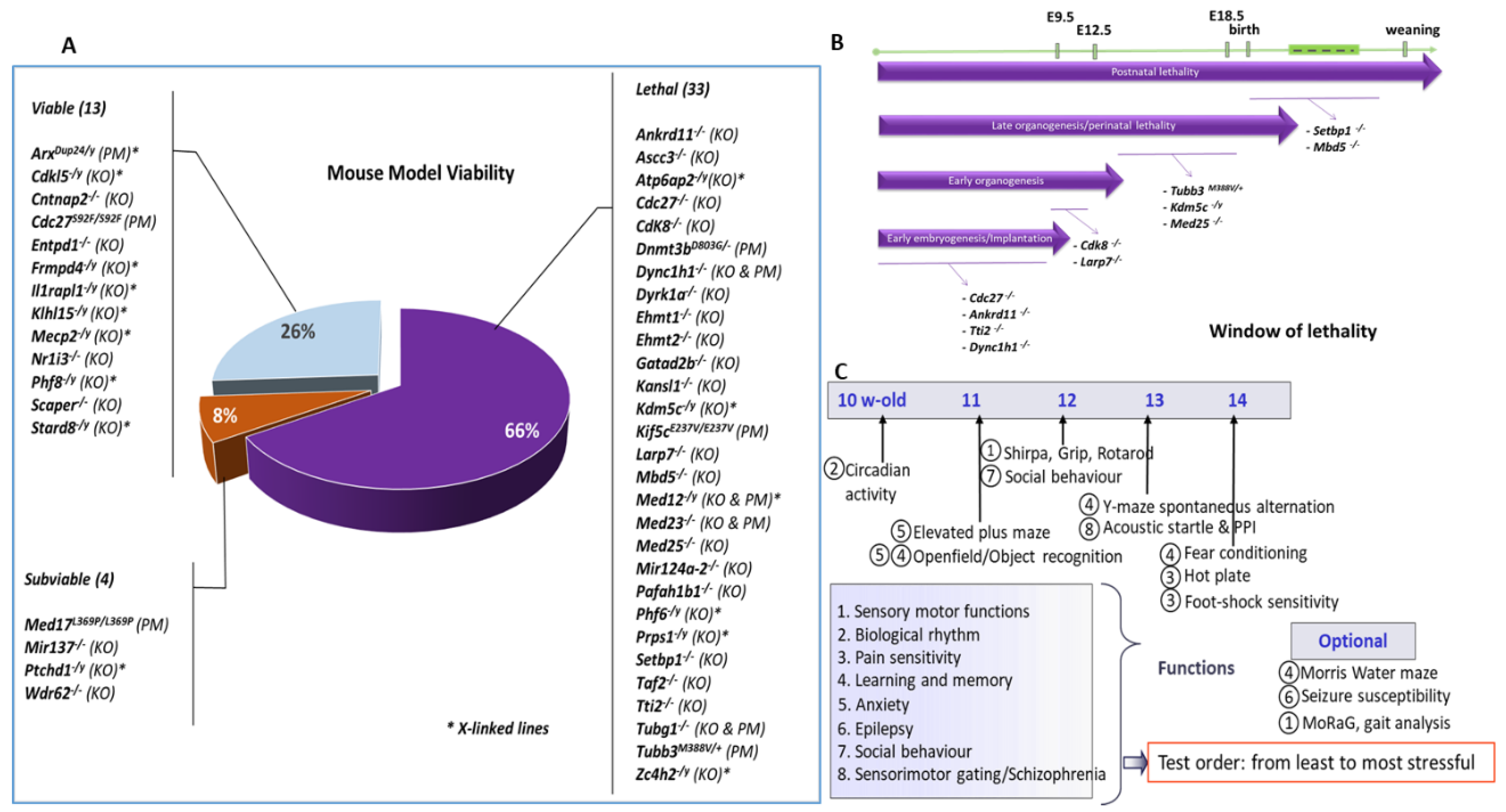
2.1. Generation of ID Mouse Models
2.2. Embryonic Pipeline
2.3. Behavioural Phenotyping Strategy
2.4. Statistical Analysis
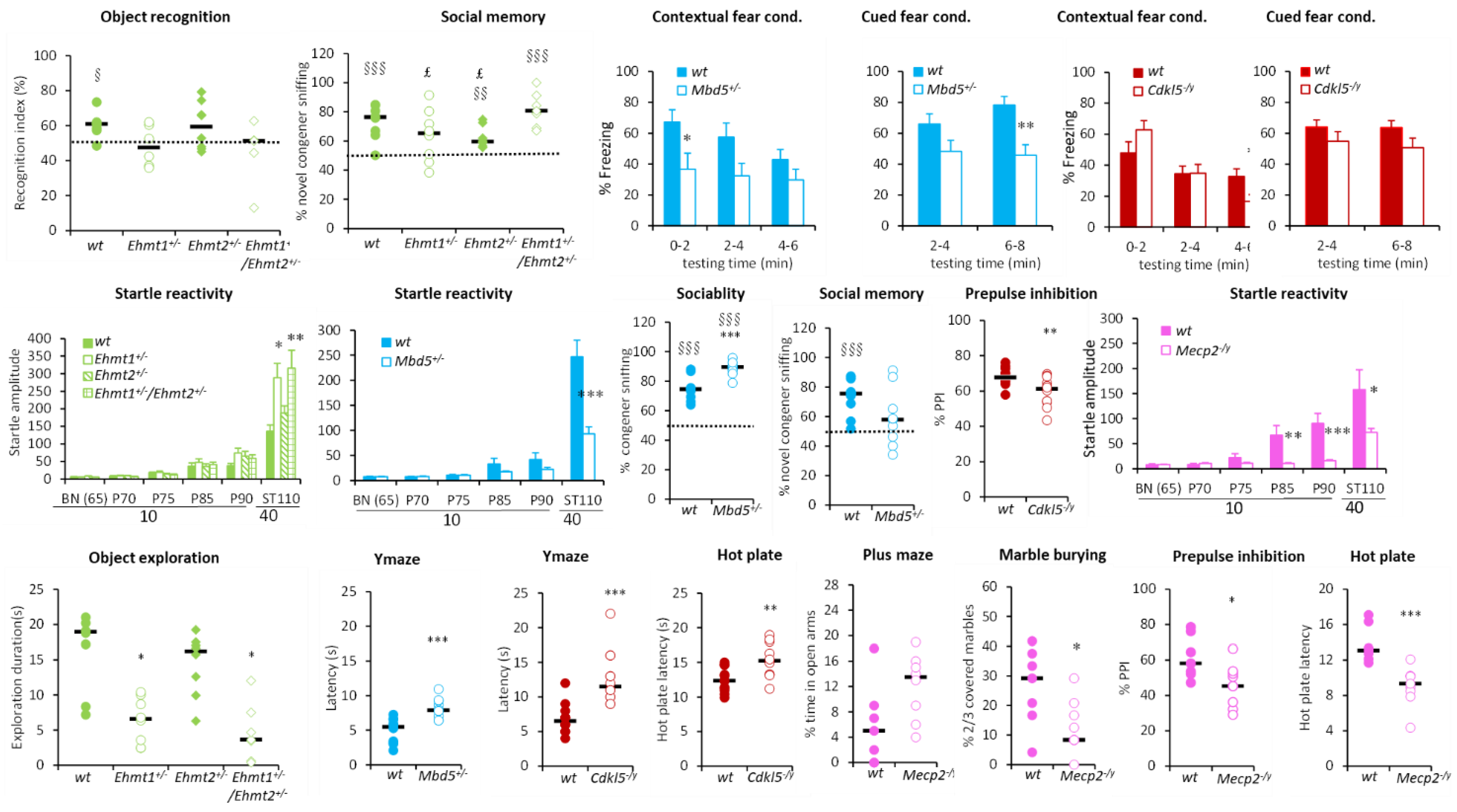
3. Results
3.1. Gene/Phenotype Relationship
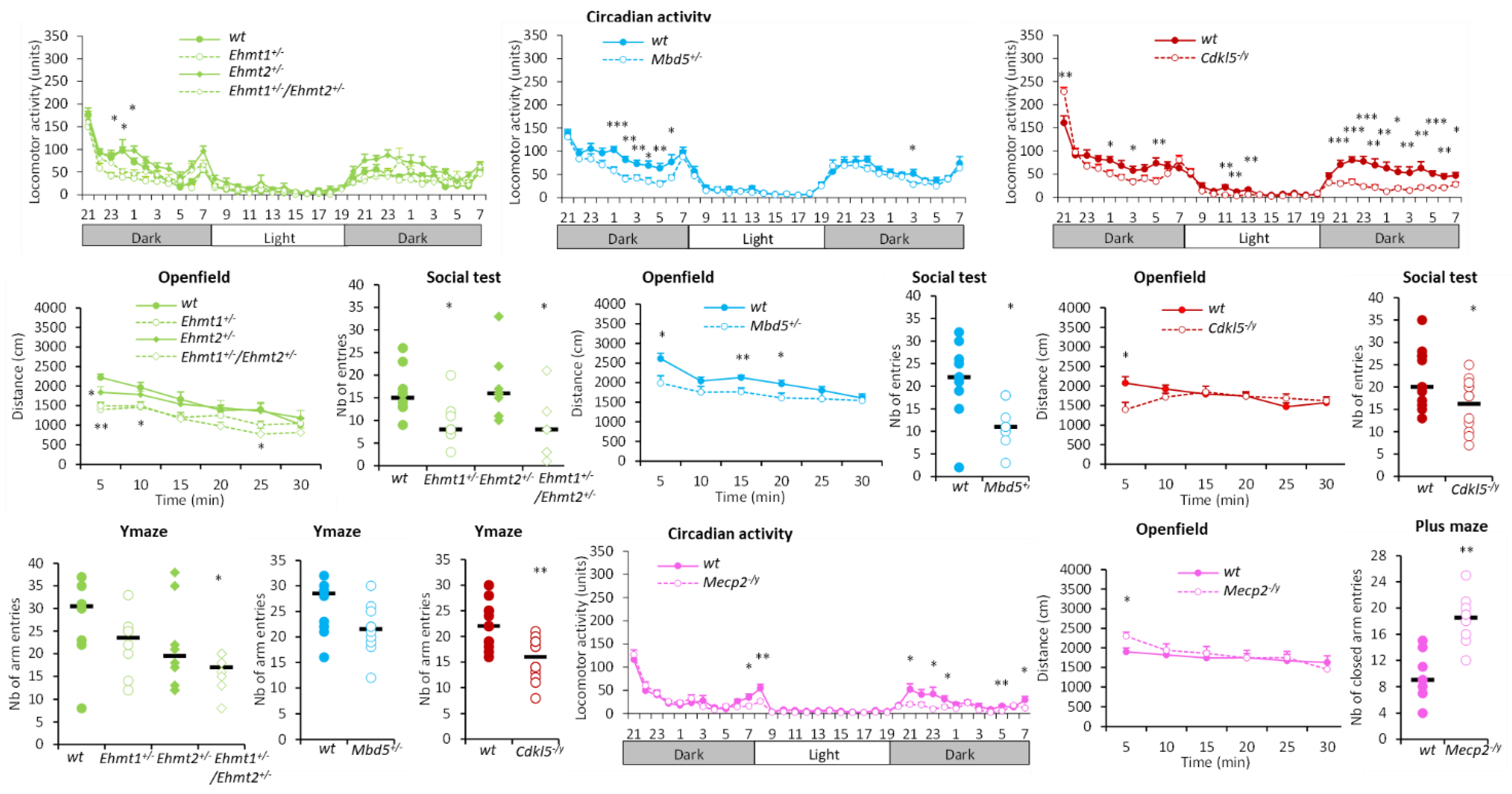
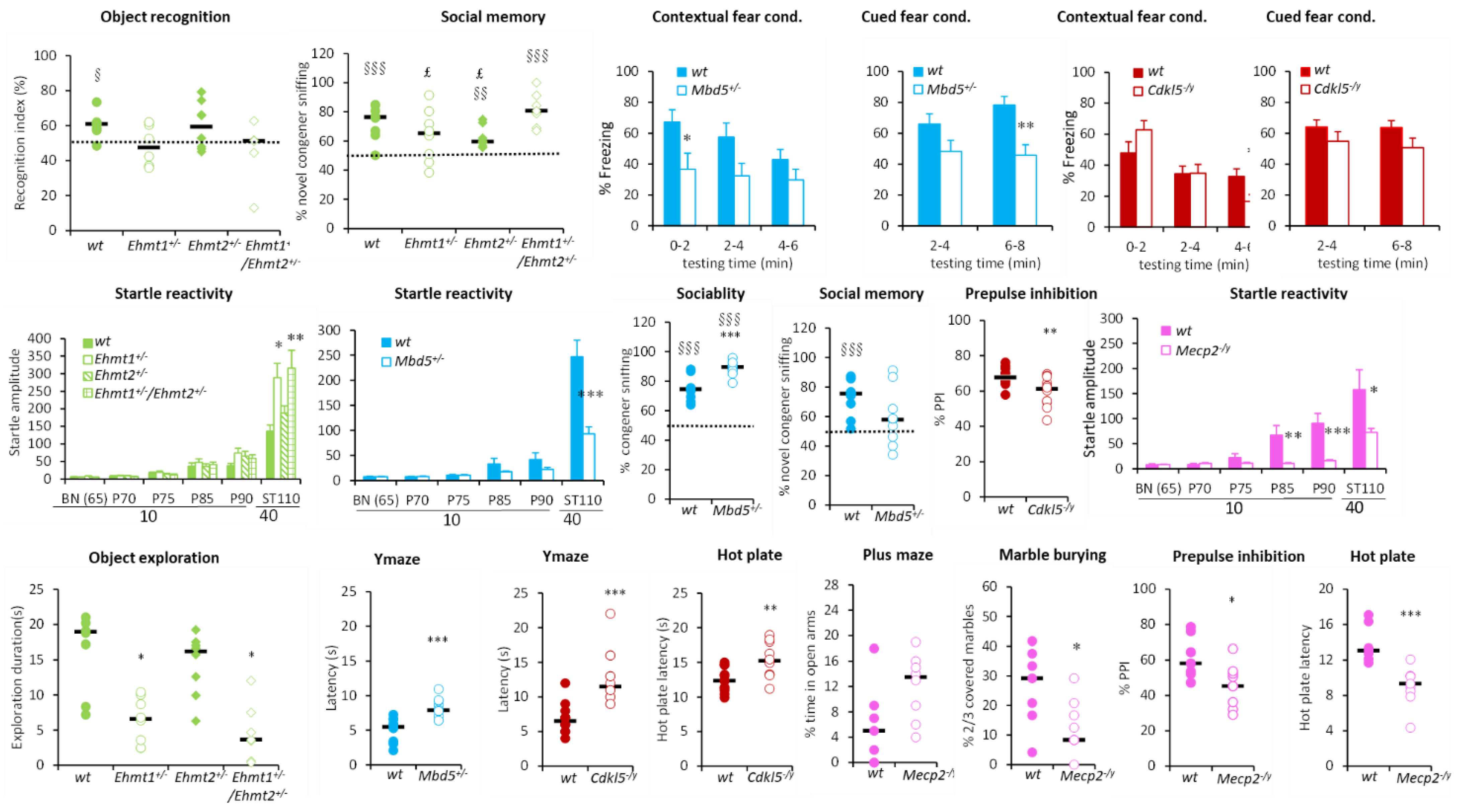
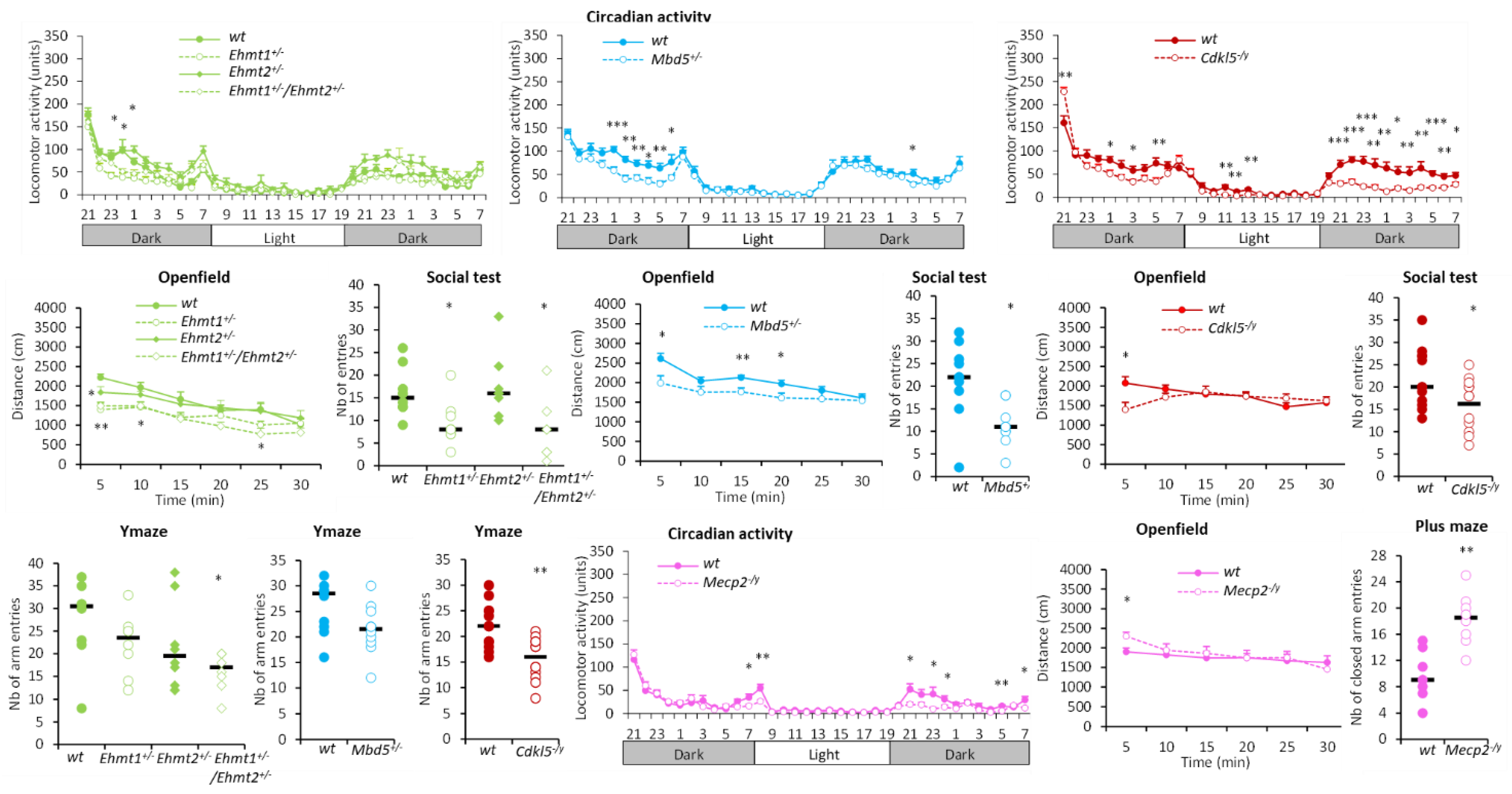
3.1.1. Hyperactivity Group
3.1.2. Hypoactivity Group
3.1.3. No Activity Phenotype Group
3.2. PCA and Cluster Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ilyas, M.; Mir, A.; Efthymiou, S.; Houlden, H. The genetics of intellectual disability: Advancing technology and gene editing. F1000Research 2020, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.; Kleefstra, T.; Kramer, J.M.; et al. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.R.; Matsumoto, K.; Kidd, A.M.; Goriely, A.; Taylor, I.B.; Fisher, R.B.; Hoogeboom, A.J.; Mathijssen, I.M.; Lourenco, M.T.; Morton, J.E.; et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: Frequent somatic mosaicism and explanation of the paucity of carrier males. Am. J. Hum. Genet. 2006, 78, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, I.; Weidner, C.; Ciccone, R.; Lapi, E.; McDonald-McGinn, D.; Kress, W.; Jakubiczka, S.; Collmann, H.; Zuffardi, O.; Zackai, E.; et al. Contiguous gene deletions involving EFNB1, OPHN1, PJA1 and EDA in patients with craniofrontonasal syndrome. Clin. Genet. 2007, 72, 506–516. [Google Scholar] [CrossRef]
- Billuart, P.; Bienvenu, T.; Ronce, N.; des Portes, V.; Vinet, M.C.; Zemni, R.; Roest Crollius, H.; Carrie, A.; Fauchereau, F.; Cherry, M.; et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 1998, 392, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Spiliopoulos, M.; Brancati, F.; Powell, E.; Duman, D.; Abrams, A.; Bademci, G.; Agolini, E.; Guo, S.; Konuk, B.; et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 2011, 89, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromme, P.; Mangelsdorf, M.E.; Shaw, M.A.; Lower, K.M.; Lewis, S.M.; Bruyere, H.; Lutcherath, V.; Gedeon, A.K.; Wallace, R.H.; Scheffer, I.E.; et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 2002, 30, 441–445. [Google Scholar] [CrossRef]
- Curie, A.; Nazir, T.; Brun, A.; Paulignan, Y.; Reboul, A.; Delange, K.; Cheylus, A.; Bertrand, S.; Rochefort, F.; Bussy, G.; et al. The c.429_452 duplication of the ARX gene: A unique developmental-model of limb kinetic apraxia. Orphanet J. Rare Dis. 2014, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Dubos, A.; Meziane, H.; Iacono, G.; Curie, A.; Riet, F.; Martin, C.; Loaec, N.; Birling, M.C.; Selloum, M.; Normand, E.; et al. A new mouse model of ARX dup24 recapitulates the patients’ behavioral and fine motor alterations. Hum. Mol. Genet. 2018, 27, 2138–2153. [Google Scholar] [CrossRef]
- Ramser, J.; Abidi, F.E.; Burckle, C.A.; Lenski, C.; Toriello, H.; Wen, G.; Lubs, H.A.; Engert, S.; Stevenson, R.E.; Meindl, A.; et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum. Mol. Genet. 2005, 14, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korvatska, O.; Strand, N.S.; Berndt, J.D.; Strovas, T.; Chen, D.H.; Leverenz, J.B.; Kiianitsa, K.; Mata, I.F.; Karakoc, E.; Greenup, J.L.; et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum. Mol. Genet. 2013, 22, 3259–3268. [Google Scholar] [CrossRef] [Green Version]
- Calpena, E.; Hervieu, A.; Kaserer, T.; Swagemakers, S.M.A.; Goos, J.A.C.; Popoola, O.; Ortiz-Ruiz, M.J.; Barbaro-Dieber, T.; Bownass, L.; Brilstra, E.H.; et al. De Novo Missense Substitutions in the Gene Encoding CDK8, a Regulator of the Mediator Complex, Cause a Syndromic Developmental Disorder. Am. J. Hum. Genet. 2019, 104, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalscheuer, V.M.; Tao, J.; Donnelly, A.; Hollway, G.; Schwinger, E.; Kubart, S.; Menzel, C.; Hoeltzenbein, M.; Tommerup, N.; Eyre, H.; et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 2003, 72, 1401–1411. [Google Scholar] [CrossRef]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, K.A.; Puffenberger, E.G.; Huentelman, M.J.; Gottlieb, S.; Dobrin, S.E.; Parod, J.M.; Stephan, D.A.; Morton, D.H. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 2006, 354, 1370–1377. [Google Scholar] [CrossRef]
- Zweier, C.; de Jong, E.K.; Zweier, M.; Orrico, A.; Ousager, L.B.; Collins, A.L.; Bijlsma, E.K.; Oortveld, M.A.; Ekici, A.B.; Reis, A.; et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 2009, 85, 655–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernes, S.C.; Newbury, D.F.; Abrahams, B.S.; Winchester, L.; Nicod, J.; Groszer, M.; Alarcon, M.; Oliver, P.L.; Davies, K.E.; Geschwind, D.H.; et al. A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 2008, 359, 2337–2345. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Pierre, B.S.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef]
- Harms, M.B.; Ori-McKenney, K.M.; Scoto, M.; Tuck, E.P.; Bell, S.; Ma, D.; Masi, S.; Allred, P.; Al-Lozi, M.; Reilly, M.M.; et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 2012, 78, 1714–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, R.S.; Kubart, S.; Hoeltzenbein, M.; Heye, B.; Vogel, I.; Hansen, C.P.; Menzel, C.; Ullmann, R.; Tommerup, N.; Ropers, H.H.; et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 2008, 82, 1165–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleefstra, T.; Smidt, M.; Banning, M.J.; Oudakker, A.R.; Van Esch, H.; de Brouwer, A.P.; Nillesen, W.; Sistermans, E.A.; Hamel, B.C.; de Bruijn, D.; et al. Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J. Med. Genet. 2005, 42, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Brunner, H.G.; Amiel, J.; Oudakker, A.R.; Nillesen, W.M.; Magee, A.; Genevieve, D.; Cormier-Daire, V.; van Esch, H.; Fryns, J.P.; et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006, 79, 370–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Haas, S.A.; Chelly, J.; Van Esch, H.; Raynaud, M.; de Brouwer, A.P.; Weinert, S.; Froyen, G.; Frints, S.G.; Laumonnier, F.; et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 2016, 21, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matosin, N.; Green, M.J.; Andrews, J.L.; Newell, K.A.; Fernandez-Enright, F. Possibility of a sex-specific role for a genetic variant in FRMPD4 in schizophrenia, but not cognitive function. Neuroreport 2016, 27, 33–38. [Google Scholar] [CrossRef]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Carrie, A.; Jun, L.; Bienvenu, T.; Vinet, M.C.; McDonell, N.; Couvert, P.; Zemni, R.; Cardona, A.; Van Buggenhout, G.; Frints, S.; et al. A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat. Genet. 1999, 23, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Piton, A.; Michaud, J.L.; Peng, H.; Aradhya, S.; Gauthier, J.; Mottron, L.; Champagne, N.; Lafreniere, R.G.; Hamdan, F.F.; S2D team; et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 2008, 17, 3965–3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koolen, D.A.; Kramer, J.M.; Neveling, K.; Nillesen, W.M.; Moore-Barton, H.L.; Elmslie, F.V.; Toutain, A.; Amiel, J.; Malan, V.; Tsai, A.C.; et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat. Genet. 2012, 44, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.R.; Amende, M.; Gurok, U.; Moser, B.; Gimmel, V.; Tzschach, A.; Janecke, A.R.; Tariverdian, G.; Chelly, J.; Fryns, J.P.; et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 2005, 76, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alazami, A.M.; Al-Owain, M.; Alzahrani, F.; Shuaib, T.; Al-Shamrani, H.; Al-Falki, Y.H.; Al-Qahtani, S.M.; Alsheddi, T.; Colak, D.; Alkuraya, F.S. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum. Mutat. 2012, 33, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Wagenstaller, J.; Spranger, S.; Lorenz-Depiereux, B.; Kazmierczak, B.; Nathrath, M.; Wahl, D.; Heye, B.; Glaser, D.; Liebscher, V.; Meitinger, T.; et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 2007, 81, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.R.; Mullegama, S.V.; Rosenfeld, J.A.; Dagli, A.I.; Hatchwell, E.; Allen, W.P.; Williams, C.A.; Elsea, S.H. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur. J. Hum. Genet. 2010, 18, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkowski, M.E.; Mullegama, S.V.; Rosenfeld, J.A.; van Bon, B.W.; Shen, Y.; Repnikova, E.A.; Gastier-Foster, J.; Thrush, D.L.; Kathiresan, S.; Ruderfer, D.M.; et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011, 89, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Lam, C.W.; Yeung, W.L.; Ko, C.H.; Poon, P.M.; Tong, S.F.; Chan, K.Y.; Lo, I.F.; Chan, L.Y.; Hui, J.; Wong, V.; et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J. Med. Genet. 2000, 37, E41. [Google Scholar] [CrossRef]
- Risheg, H.; Graham, J.M., Jr.; Clark, R.D.; Rogers, R.C.; Opitz, J.M.; Moeschler, J.B.; Peiffer, A.P.; May, M.; Joseph, S.M.; Jones, J.R.; et al. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat. Genet. 2007, 39, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.E.; Tarpey, P.S.; Lubs, H.A.; Verloes, A.; May, M.M.; Risheg, H.; Friez, M.J.; Futreal, P.A.; Edkins, S.; Teague, J.; et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J. Med. Genet. 2007, 44, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulto-van Silfhout, A.T.; de Vries, B.B.; van Bon, B.W.; Hoischen, A.; Ruiterkamp-Versteeg, M.; Gilissen, C.; Gao, F.; van Zwam, M.; Harteveld, C.L.; van Essen, A.J.; et al. Mutations in MED12 cause X-linked Ohdo syndrome. Am. J. Hum. Genet. 2013, 92, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, R.; Straussberg, R.; Mandel, H.; Fattal-Valevski, A.; Ben-Zeev, B.; Naamati, A.; Shaag, A.; Zenvirt, S.; Konen, O.; Mimouni-Bloch, A.; et al. Infantile cerebral and cerebellar atrophy is associated with a mutation in the MED17 subunit of the transcription preinitiation mediator complex. Am. J. Hum. Genet. 2010, 87, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Boissel, S.; Zarhrate, M.; Rio, M.; Munnich, A.; Egly, J.M.; Colleaux, L. MED23 mutation links intellectual disability to dysregulation of immediate early gene expression. Science 2011, 333, 1161–1163. [Google Scholar] [CrossRef]
- Leal, A.; Huehne, K.; Bauer, F.; Sticht, H.; Berger, P.; Suter, U.; Morera, B.; Del Valle, G.; Lupski, J.R.; Ekici, A.; et al. Identification of the variant Ala335Val of MED25 as responsible for CMT2B2: Molecular data, functional studies of the SH3 recognition motif and correlation between wild-type MED25 and PMP22 RNA levels in CMT1A animal models. Neurogenetics 2009, 10, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basel-Vanagaite, L.; Smirin-Yosef, P.; Essakow, J.L.; Tzur, S.; Lagovsky, I.; Maya, I.; Pasmanik-Chor, M.; Yeheskel, A.; Konen, O.; Orenstein, N.; et al. Homozygous MED25 mutation implicated in eye-intellectual disability syndrome. Hum. Genet. 2015, 134, 577–587. [Google Scholar] [CrossRef]
- Willemsen, M.H.; Valles, A.; Kirkels, L.A.; Mastebroek, M.; Olde Loohuis, N.; Kos, A.; Wissink-Lindhout, W.M.; de Brouwer, A.P.; Nillesen, W.M.; Pfundt, R.; et al. Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. J. Med. Genet. 2011, 48, 810–818. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Kleefstra, T.; Kramer, J.M.; Neveling, K.; Willemsen, M.H.; Koemans, T.S.; Vissers, L.E.; Wissink-Lindhout, W.; Fenckova, M.; van den Akker, W.M.; Kasri, N.N.; et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 2012, 91, 73–82. [Google Scholar] [CrossRef]
- Chong, S.S.; Pack, S.D.; Roschke, A.V.; Tanigami, A.; Carrozzo, R.; Smith, A.C.; Dobyns, W.B.; Ledbetter, D.H. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum. Mol. Genet. 1997, 6, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, M.; Ono, J.; Okada, S.; Masuno, M.; Nakamura, Y.; Kurahashi, H. Alteration of the LIS1 gene in Japanese patients with isolated lissencephaly sequence or Miller-Dieker syndrome. Hum. Genet. 1998, 103, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Lower, K.M.; Turner, G.; Kerr, B.A.; Mathews, K.D.; Shaw, M.A.; Gedeon, A.K.; Schelley, S.; Hoyme, H.E.; White, S.M.; Delatycki, M.B.; et al. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat. Genet. 2002, 32, 661–665. [Google Scholar] [CrossRef]
- Laumonnier, F.; Holbert, S.; Ronce, N.; Faravelli, F.; Lenzner, S.; Schwartz, C.E.; Lespinasse, J.; Van Esch, H.; Lacombe, D.; Goizet, C.; et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J. Med. Genet. 2005, 42, 780–786. [Google Scholar] [CrossRef] [Green Version]
- de Brouwer, A.P.; Williams, K.L.; Duley, J.A.; van Kuilenburg, A.B.; Nabuurs, S.B.; Egmont-Petersen, M.; Lugtenberg, D.; Zoetekouw, L.; Banning, M.J.; Roeffen, M.; et al. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007, 81, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Sohn, K.M.; Shy, M.E.; Krajewski, K.M.; Hwang, M.; Park, J.H.; Jang, S.Y.; Won, H.H.; Choi, B.O.; Hong, S.H.; et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5). Am. J. Hum. Genet. 2007, 81, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Han, D.; Li, J.; Han, B.; Ouyang, X.; Cheng, J.; Li, X.; Jin, Z.; Wang, Y.; Bitner-Glindzicz, M.; et al. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am. J. Hum. Genet. 2010, 86, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Noor, A.; Whibley, A.; Marshall, C.R.; Gianakopoulos, P.J.; Piton, A.; Carson, A.R.; Orlic-Milacic, M.; Lionel, A.C.; Sato, D.; Pinto, D.; et al. Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2010, 2, 49ra68. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, A.; Noor, A.; Degagne, B.; Baker, K.; Bok, L.A.; Brady, A.F.; Chitayat, D.; Chung, B.H.; Cytrynbaum, C.; Dyment, D.; et al. Phenotypic spectrum associated with PTCHD1 deletions and truncating mutations includes intellectual disability and autism spectrum disorder. Clin. Genet. 2015, 88, 224–233. [Google Scholar] [CrossRef]
- Tatour, Y.; Sanchez-Navarro, I.; Chervinsky, E.; Hakonarson, H.; Gawi, H.; Tahsin-Swafiri, S.; Leibu, R.; Lopez-Molina, M.I.; Fernandez-Sanz, G.; Ayuso, C.; et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J. Med. Genet. 2017, 54, 698–704. [Google Scholar] [CrossRef]
- Hoischen, A.; van Bon, B.W.; Gilissen, C.; Arts, P.; van Lier, B.; Steehouwer, M.; de Vries, P.; de Reuver, R.; Wieskamp, N.; Mortier, G.; et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010, 42, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Filges, I.; Shimojima, K.; Okamoto, N.; Rothlisberger, B.; Weber, P.; Huber, A.R.; Nishizawa, T.; Datta, A.N.; Miny, P.; Yamamoto, T. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J. Med. Genet. 2011, 48, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, G.; Scordo, M.R.; Pescucci, C.; Nannetti, G.; Biagini, E.; Scandurra, V.; Gerundino, F.; Magi, A.; Benelli, M.; Torricelli, F. 372 kb microdeletion in 18q12.3 causing SETBP1 haploinsufficiency associated with mild mental retardation and expressive speech impairment. Eur. J. Med. Genet. 2012, 55, 216–221. [Google Scholar] [CrossRef]
- Hellman-Aharony, S.; Smirin-Yosef, P.; Halevy, A.; Pasmanik-Chor, M.; Yeheskel, A.; Har-Zahav, A.; Maya, I.; Straussberg, R.; Dahary, D.; Haviv, A.; et al. Microcephaly thin corpus callosum intellectual disability syndrome caused by mutated TAF2. Pediatr. Neurol. 2013, 49, 411–416.e411. [Google Scholar] [CrossRef] [PubMed]
- Langouet, M.; Saadi, A.; Rieunier, G.; Moutton, S.; Siquier-Pernet, K.; Fernet, M.; Nitschke, P.; Munnich, A.; Stern, M.H.; Chaouch, M.; et al. Mutation in TTI2 reveals a role for triple T complex in human brain development. Hum. Mutat. 2013, 34, 1472–1476. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Poirier, K.; Saillour, Y.; Bahi-Buisson, N.; Jaglin, X.H.; Fallet-Bianco, C.; Nabbout, R.; Castelnau-Ptakhine, L.; Roubertie, A.; Attie-Bitach, T.; Desguerre, I.; et al. Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 2010, 19, 4462–4473. [Google Scholar] [CrossRef]
- Bilguvar, K.; Ozturk, A.K.; Louvi, A.; Kwan, K.Y.; Choi, M.; Tatli, B.; Yalnizoglu, D.; Tuysuz, B.; Caglayan, A.O.; Gokben, S.; et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 2010, 467, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, A.K.; Khurshid, M.; Desir, J.; Carvalho, O.P.; Cox, J.J.; Thornton, G.; Kausar, R.; Ansar, M.; Ahmad, W.; Verloes, A.; et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat. Genet. 2010, 42, 1010–1014. [Google Scholar] [CrossRef]
- Hirata, H.; Nanda, I.; van Riesen, A.; McMichael, G.; Hu, H.; Hambrock, M.; Papon, M.A.; Fischer, U.; Marouillat, S.; Ding, C.; et al. ZC4H2 mutations are associated with arthrogryposis multiplex congenita and intellectual disability through impairment of central and peripheral synaptic plasticity. Am. J. Hum. Genet. 2013, 92, 681–695. [Google Scholar] [CrossRef]
- Frints, S.G.M.; Hennig, F.; Colombo, R.; Jacquemont, S.; Terhal, P.; Zimmerman, H.H.; Hunt, D.; Mendelsohn, B.A.; Kordass, U.; Webster, R.; et al. Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita. Hum. Mutat. 2019, 40, 2270–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balemans, M.C.; Kasri, N.N.; Kopanitsa, M.V.; Afinowi, N.O.; Ramakers, G.; Peters, T.A.; Beynon, A.J.; Janssen, S.M.; van Summeren, R.C.; Eeftens, J.M.; et al. Hippocampal dysfunction in the Euchromatin histone methyltransferase 1 heterozygous knockout mouse model for Kleefstra syndrome. Hum. Mol. Genet. 2013, 22, 852–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambino, F.; Kneib, M.; Pavlowsky, A.; Skala, H.; Heitz, S.; Vitale, N.; Poulain, B.; Khelfaoui, M.; Chelly, J.; Billuart, P.; et al. IL1RAPL1 controls inhibitory networks during cerebellar development in mice. Eur. J. Neurosci. 2009, 30, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.; Young, J.; Yuva-Paylor, L.; Spencer, C.; Antalffy, B.; Noebels, J.; Armstrong, D.; Paylor, R.; Zoghbi, H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacono, G.; Dubos, A.; Meziane, H.; Benevento, M.; Habibi, E.; Mandoli, A.; Riet, F.; Selloum, M.; Feil, R.; Zhou, H.; et al. Increased H3K9 methylation and impaired expression of Protocadherins are associated with the cognitive dysfunctions of the Kleefstra syndrome. Nucleic Acids Res. 2018, 46, 4950–4965. [Google Scholar] [CrossRef]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef] [Green Version]
- Mantamadiotis, T.; Lemberger, T.; Bleckmann, S.C.; Kern, H.; Kretz, O.; Martin Villalba, A.; Tronche, F.; Kellendonk, C.; Gau, D.; Kapfhammer, J.; et al. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 2002, 31, 47–54. [Google Scholar] [CrossRef]
- Monory, K.; Massa, F.; Egertova, M.; Eder, M.; Blaudzun, H.; Westenbroek, R.; Kelsch, W.; Jacob, W.; Marsch, R.; Ekker, M.; et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 2006, 51, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.A.; Natsume, R.; Sakimura, K.; Nishiyama, A. NG2 cells are uniformly distributed and NG2 is not required for barrel formation in the somatosensory cortex. Mol. Cell Neurosci. 2011, 46, 689–698. [Google Scholar] [CrossRef]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, N.A.; Meehan, T.F.; Morgan, H.; Mason, J.C.; Blake, A.; Kurbatova, N.; Smedley, D.; Jacobsen, J.; Mott, R.F.; Iyer, V.; et al. Applying the ARRIVE Guidelines to an In Vivo Database. PLoS Biol. 2015, 13, e1002151. [Google Scholar] [CrossRef]
- Riet, F.; Mittelhaeuser, C.; Lux, A.; Bour, R.; Selloum, M.; Sorg, T.; Herault, Y.; Meziane, H. Behavioral Testing Design for Evaluation of Cognitive Disabilities. Curr. Protoc. 2022, 2, e382. [Google Scholar] [CrossRef] [PubMed]
- Cacheiro, P.; Munoz-Fuentes, V.; Murray, S.A.; Dickinson, M.E.; Bucan, M.; Nutter, L.M.J.; Peterson, K.A.; Haselimashhadi, H.; Flenniken, A.M.; Morgan, H.; et al. Human and mouse essentiality screens as a resource for disease gene discovery. Nat. Commun. 2020, 11, 655. [Google Scholar] [CrossRef] [Green Version]
- Dubos, A.; Castells-Nobau, A.; Meziane, H.; Oortveld, M.A.; Houbaert, X.; Iacono, G.; Martin, C.; Mittelhaeuser, C.; Lalanne, V.; Kramer, J.M.; et al. Conditional depletion of intellectual disability and Parkinsonism candidate gene ATP6AP2 in fly and mouse induces cognitive impairment and neurodegeneration. Hum. Mol. Genet. 2015, 24, 6736–6755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ung, D.C.; Iacono, G.; Meziane, H.; Blanchard, E.; Papon, M.A.; Selten, M.; van Rhijn, J.R.; Montjean, R.; Rucci, J.; Martin, S.; et al. Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Mol. Psychiatry 2018, 23, 1356–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzel, A.; Giedion, A. A syndrome of severe midface retraction, multiple skull anomalies, clubfeet, and cardiac and renal malformations in sibs. Am. J. Med. Genet. 1978, 1, 361–375. [Google Scholar] [CrossRef]
- Jansen, N.A.; Braden, R.O.; Srivastava, S.; Otness, E.F.; Lesca, G.; Rossi, M.; Nizon, M.; Bernier, R.A.; Quelin, C.; van Haeringen, A.; et al. Clinical delineation of SETBP1 haploinsufficiency disorder. Eur. J. Hum. Genet. 2021, 29, 1198–1205. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Cederquist, G.Y.; Gupta, M.L., Jr.; Engle, E.C. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin Genet. Dev. 2011, 21, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.R.; Kleefstra, T. The chromosome 9q subtelomere deletion syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2007, 145C, 383–392. [Google Scholar] [CrossRef]
- Balemans, M.C.; Huibers, M.M.; Eikelenboom, N.W.; Kuipers, A.J.; van Summeren, R.C.; Pijpers, M.M.; Tachibana, M.; Shinkai, Y.; van Bokhoven, H.; Van der Zee, C.E. Reduced exploration, increased anxiety, and altered social behavior: Autistic-like features of euchromatin histone methyltransferase 1 heterozygous knockout mice. Behav. Brain Res. 2010, 208, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Sampath, S.C.; Intrator, A.; Min, A.; Gertler, T.S.; Surmeier, D.J.; Tarakhovsky, A.; Greengard, P. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 2009, 64, 678–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoubridge, C.; Fullston, T.; Gecz, J. ARX spectrum disorders: Making inroads into the molecular pathology. Hum. Mutat. 2010, 31, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Tekin, M.; Kavaz, A.; Berberoglu, M.; Fitoz, S.; Ekim, M.; Ocal, G.; Akar, N. The KBG syndrome: Confirmation of autosomal dominant inheritance and further delineation of the phenotype. Am. J. Med. Genet. Part A 2004, 130A, 284–287. [Google Scholar] [CrossRef]
- Gupta, H.V.; Vengoechea, J.; Sahaya, K.; Virmani, T. A splice site mutation in ATP6AP2 causes X-linked intellectual disability, epilepsy, and parkinsonism. Park. Relat. Disord. 2015, 21, 1473–1475. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Muller vom Hagen, J.; Haack, T.B.; Wilhelm, C.; Lindig, T.; Beck-Wodl, S.; Nabuurs, S.B.; van Kuilenburg, A.B.; de Brouwer, A.P.; Schols, L. X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: Evidence from a family with a novel PRPS1 mutation. Orphanet J. Rare Dis. 2014, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Roessler, B.J.; Nosal, J.M.; Smith, P.R.; Heidler, S.A.; Palella, T.D.; Switzer, R.L.; Becker, M.A. Human X-linked phosphoribosylpyrophosphate synthetase superactivity is associated with distinct point mutations in the PRPS1 gene. J. Biol. Chem. 1993, 268, 26476–26481. [Google Scholar] [CrossRef]
- Becker, M.A.; Smith, P.R.; Taylor, W.; Mustafi, R.; Switzer, R.L. The genetic and functional basis of purine nucleotide feedback-resistant phosphoribosylpyrophosphate synthetase superactivity. J. Clin. Investig. 1995, 96, 2133–2141. [Google Scholar] [CrossRef]
- Brancati, F.; D’Avanzo, M.G.; Digilio, M.C.; Sarkozy, A.; Biondi, M.; De Brasi, D.; Mingarelli, R.; Dallapiccola, B. KBG syndrome in a cohort of Italian patients. Am. J. Med. Genet. A 2004, 131, 144–149. [Google Scholar] [CrossRef]
- Bowl, M.R.; Simon, M.M.; Ingham, N.J.; Greenaway, S.; Santos, L.; Cater, H.; Taylor, S.; Mason, J.; Kurbatova, N.; Pearson, S.; et al. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nat. Commun. 2017, 8, 886. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chr | Human Variant(s) | Syndrome(s) | References | Mouse Models |
|---|---|---|---|---|---|
| Ankrd11 | 8 | LoF mutations and deletion (heterozygous) | KBG Syndrome | [7] | Ankrd11Camk2a |
| Arx | X | LoF mutations, deletion, duplication/expansion (hemizygous) 24bp duplication most frequent mutation | Early infantile epileptic encephalopathy 1, Hydranencephaly with abnormal genitalia, X-linked lissencephaly 2, X-linked Mental retardation, Partington syndrome, Proud syndrome | [8,9,10] | ArxDup24/y |
| Ascc3 | 10 | - | - | - | Ascc3Camk2a |
| Atp6ap2 | X | Splice site and missense (hemizygous) | ID +/− Parkinsonism with spasticity | [11,12] | Atp6ap2Camk2a/y |
| Cdc27 | 11 | - | - | - | Cdc27−/−; Cdc27S92F |
| Cdk8 | 5 | Missense substitutions | - | [13] | Cdk8−/−; Cdk8Camk2a/y |
| Cdkl5 | X | Translocations, microdeletions, missense, LoF mutations & mosaic exonic deletions (hemizygous in males & heterozygous in females) | CDKL5 Deficiency disorder (CDD) & Early Infantile Epileptic Encephalopathy 2 (EIEE2) | [14,15] | Cdkl5−/y |
| Cntnap2 | 6 | LoF mutations & SNPs (homozygous or compound heterozygous) | Cortical Dysplasia-Focal Epilepsy Syndrome (CDFES), Pitt-Hopkins like syndrome 1 & Autism & Specific Language Impairment | [16,17,18] | Cntnap2−/− |
| Dnmt3b | 2 | LoF mutations (homozygous or compound heterozygous) | Immunodeficiency-centromeric instability-facial anomalies syndrome 1 | [19,20] | Dnmt3bD803G/− |
| Dync1h1 | 12 | LoF mutations (heterozygous) | Autosomal dominant axonal Charcot-Marie-Tooth type 20 disease (CMT20), Autosomal dominant mental retardation 13 (MRD13), Autosomal dominant lower extremity-predominant Spinal muscular atrophy 1 (SMALED1) | [21,22,23] | Dync1h1−/−; Dync1h1K3334N |
| Dyrk1a | 16 | Translocations, LoF mutations & deletions (heterozygous) | Autosomal Dominant Mental Retardation 7 (MRD7) | [24] | Dyrk1a−/−; Dyrk1aCamk2a; Dyrk1aDlx5−6/+ |
| Ehmt1 | 2 | Translocations, microdeletion & LoF mutations (heterozygous) | Kleefstra syndrome | [25,26] | Ehmt1+/− |
| Ehmt2 | 17 | - | - | - | Ehmt2+/− |
| Entpd1 | 19 | LoF mutations (homozygous) | Autosomal Recessive Spastic Paraplegia 64 (SPG64) | [27] | Entpd1−/− |
| Frmpd4 | X | LoF mutations, missense and exon deletion (hemizygous) | X-linked Intellectual disability & Schizophrenia | [28,29] | Frmpd4−/y |
| Gatad2b | 3 | LoF mutations & deletions (heterozygous) | Intellectual disability | [30] | Gatad2b−/− |
| Il1rap1l1 | X | LoF mutations (hemizygous) | X-linked intellectual disability | [31,32] | Il1rpl1−/y |
| Kansl1 | 11 | LoF mutations & deletions (heterozygous) | Koolen-De Vries syndrome | [33] | Kansl1−/− |
| Kdm5c | X | LoF mutations (hemizygous) | Claes-Jensen type X-linked syndromic mental retardation | [34] | Kdm5c−/y; Kdm5cCamk2a/y; Kdm5cDlx5−6/y |
| Kif5c | 2 | LoF mutations (heterozygous) | Complex Cortical Dysplasia with Other Brain Malformations 2 (CDCBM2) | [22] | Kif5c+/−; Kif5cE237V |
| Klhl15 | X | LoF mutations & deletions (hemizygous) | Intellectual disability | [28] | Klhl15−/y |
| Larp7 | 3 | LoF mutations & duplications (homozygous) | Alazami syndrome | [3,35] | Larp7−/y |
| Mbd5 | 2 | LoF mutations, translocation, duplications & deletions (heterozygous) | Autosomal Dominant Mental Retardation 1 (MRD1) & 2q23.1 duplication and deletion syndromes | [36,37,38] | Mbd5−/− |
| Mecp2 | X | LoF mutations (heterozygous in females) | Rett syndrome, Atypical Rett Syndrome or Angelman-like Phenotype & Autism | [39,40] | Mecp2−/y |
| Med12 | X | Missense mutations (hemizygous) p.(R961W) most frequent mutation | Lujan-Fryns syndrome, X-linked Ohdo syndrome & Opitz-Kaveggia syndrome | [41,42,43] | Med12−/y; Med12R961X |
| Med17 | 9 | p.(L371P) missense mutation (homozygous) | Postnatal progressive microcephaly with seizures and brain atrophy | [44] | Med17L369P |
| Med23 | 10 | p.(R617Q) missense mutation (homozygous) | Autosmal recessive intellectual disability 18 | [45] | Med23−/−; Med23R617Q |
| Med25 | 7 | Missense mutations (homozygous) | Autosmal recessive Charcot-Marie-Tooth type 2B2 disease & Basel-Vanagait-Smirin-Yosef syndrome (BVSYS) | [46,47] | Med25−/− |
| mIR124a-2 | 3 | - | - | - | Mir124−/− |
| mIR137 | 3 | Microdeletions (heterozygous) | Intellectual disability, autism & schizophrenia | [48,49] | Mir137−/− |
| Nr1i3 | 1 | LoF mutations (heterozygous) | Core features of Kleefstra syndrome | [50] | Nr1i3−/− |
| Pafah1b1 | 11 | LoF mutations, deletion & translocation (heterozygous) | Lissencephaly 1, Miller-Dieker lissencephaly syndrome & Subcortical laminar heterotopia | [51,52] | Pafah1b1−/− |
| Phf6 | X | LoF Mutations (heterozygous in females & hemizygous in males) | Borjeson-Forssman-Lehmann syndrome (BFLS) | [53] | Phf6−/y |
| Phf8 | X | LoF mutations & deletion (hemizygous) | Siderius X-linked Mental retardation syndrome (MRXSSD) | [54] | Phf8−/y |
| Prps1 | X | LoF & GoF mutations (hemizygous) | LoF: Arts syndrome, X-linked recessive Charcot-Marie-Tooth disease 5 & X-linked non syndromic hearing loss (NSHL) vs. GoF: PRPS-related Gout syndrome & Phosphoribosylpyrophosphate Synthetase Superactivity | [55,56,57] | Prps1−/y; Prps1Camk2a/y; Prps1Cspg4/y |
| Ptchd1 | X | LoF mutations & deletion (hemizygous) | X-linked Autism Susceptibility (AUTSX4) | [58,59] | Ptchd1−/y |
| Scaper | 9 | LoF mutation (homozygous & compound heterozgous) | Retinitis pigmentosa with intellectual disability | [60] | Scaper−/− |
| Setbp1 | 18 | GoF mutations (Schinzel-Giedion Syndrome) & LoF mutations (autosmal dominant mental retardation 29) (heterozygous) | Schinzel-Giedion Syndrome vs. Autosomal dominant Mental Retardation syndrome 29 (MRD29) | [61,62,63] | Setbp1−/− |
| Stard8 | X | - | - | - | Stard8−/y |
| Taf2 | 15 | Missense mutations (homozygous) | Autosomal recessive Mental retardation 40 (MRT40) | [3,64] | Taf2−/− |
| Tti2/C8orf41 | 8 | Missense mutations (homozygous) | Autosomal recessive Mental retardation 39 (MRT39) | [3,65] | Tti2−/− |
| Tubb3 | 8 | LoF mutations (heterozygous) | Complex Cortical Dysplasia with Other Brain Malformations (CDCBM1) & Congenital Fibrosis of Extraocular Muscles 3a (CFEOM3A) | [66,67] | Tubb3M388V |
| Tubg1 | 11 | Missense mutations (heterozygous) | Complex Cortical dysplasia with other brain malformations 4 (CDCBM4) | [22] | Tubg1−/−; Tubg1Y92C |
| Wdr62 | 7 | LoF mutations (homozygous) | Autosomal recessive primary Microcephaly 2 with or without cortical malformations | [68,69] | Wdr62−/− |
| Zc4h2 | X | Missense & in frame insertion (hemizygous in males), LoF mutations, splice site, missense & (partial) gene deletions (heterozygous in females) | ZC4H2-Associated Rare Disorders (ZARD), previously known as Wieacker-Wolff syndrome (WRWF) | [70,71] | Zc4h2−/y |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meziane, H.; Birling, M.-C.; Wendling, O.; Leblanc, S.; Dubos, A.; Selloum, M.; Pavlovic, G.; Sorg, T.; Kalscheuer, V.M.; Billuart, P.; et al. Large-Scale Functional Assessment of Genes Involved in Rare Diseases with Intellectual Disabilities Unravels Unique Developmental and Behaviour Profiles in Mouse Models. Biomedicines 2022, 10, 3148. https://doi.org/10.3390/biomedicines10123148
Meziane H, Birling M-C, Wendling O, Leblanc S, Dubos A, Selloum M, Pavlovic G, Sorg T, Kalscheuer VM, Billuart P, et al. Large-Scale Functional Assessment of Genes Involved in Rare Diseases with Intellectual Disabilities Unravels Unique Developmental and Behaviour Profiles in Mouse Models. Biomedicines. 2022; 10(12):3148. https://doi.org/10.3390/biomedicines10123148
Chicago/Turabian StyleMeziane, Hamid, Marie-Christine Birling, Olivia Wendling, Sophie Leblanc, Aline Dubos, Mohammed Selloum, Guillaume Pavlovic, Tania Sorg, Vera M. Kalscheuer, Pierre Billuart, and et al. 2022. "Large-Scale Functional Assessment of Genes Involved in Rare Diseases with Intellectual Disabilities Unravels Unique Developmental and Behaviour Profiles in Mouse Models" Biomedicines 10, no. 12: 3148. https://doi.org/10.3390/biomedicines10123148