
In Vitro Modeling of Diabetes Impact on Vascular Endothelium: Are Essentials Engaged to Tune Metabolism?


Abstract
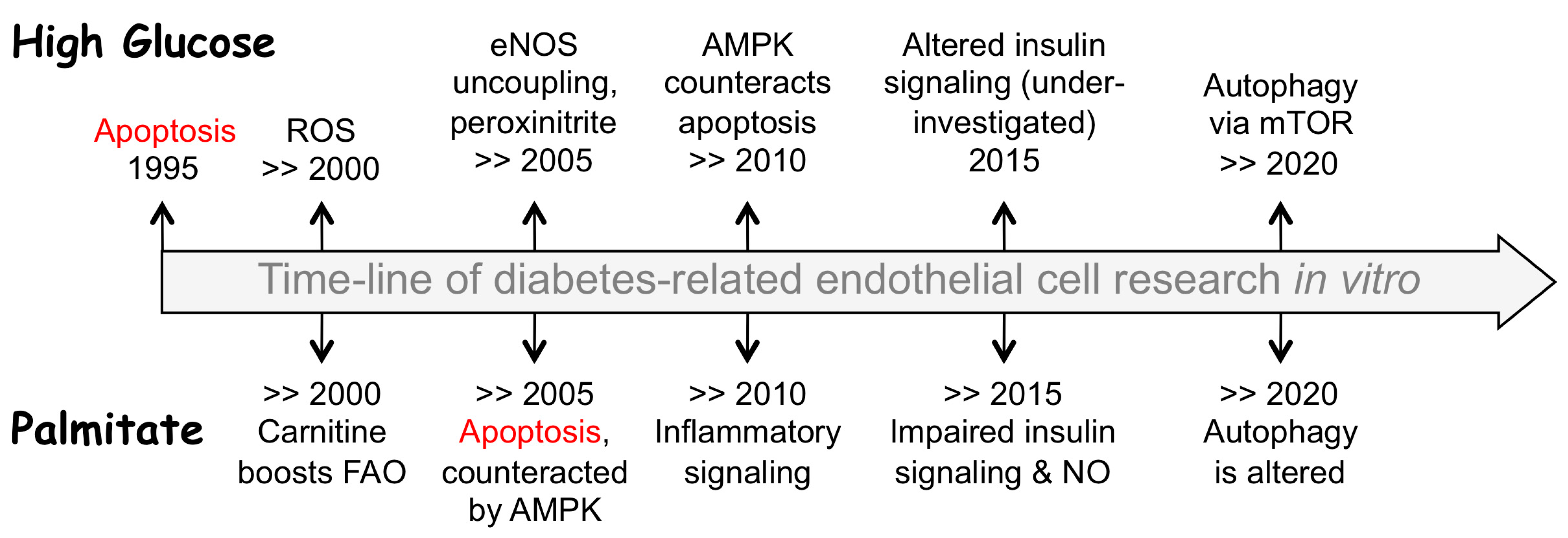
:1. Introduction
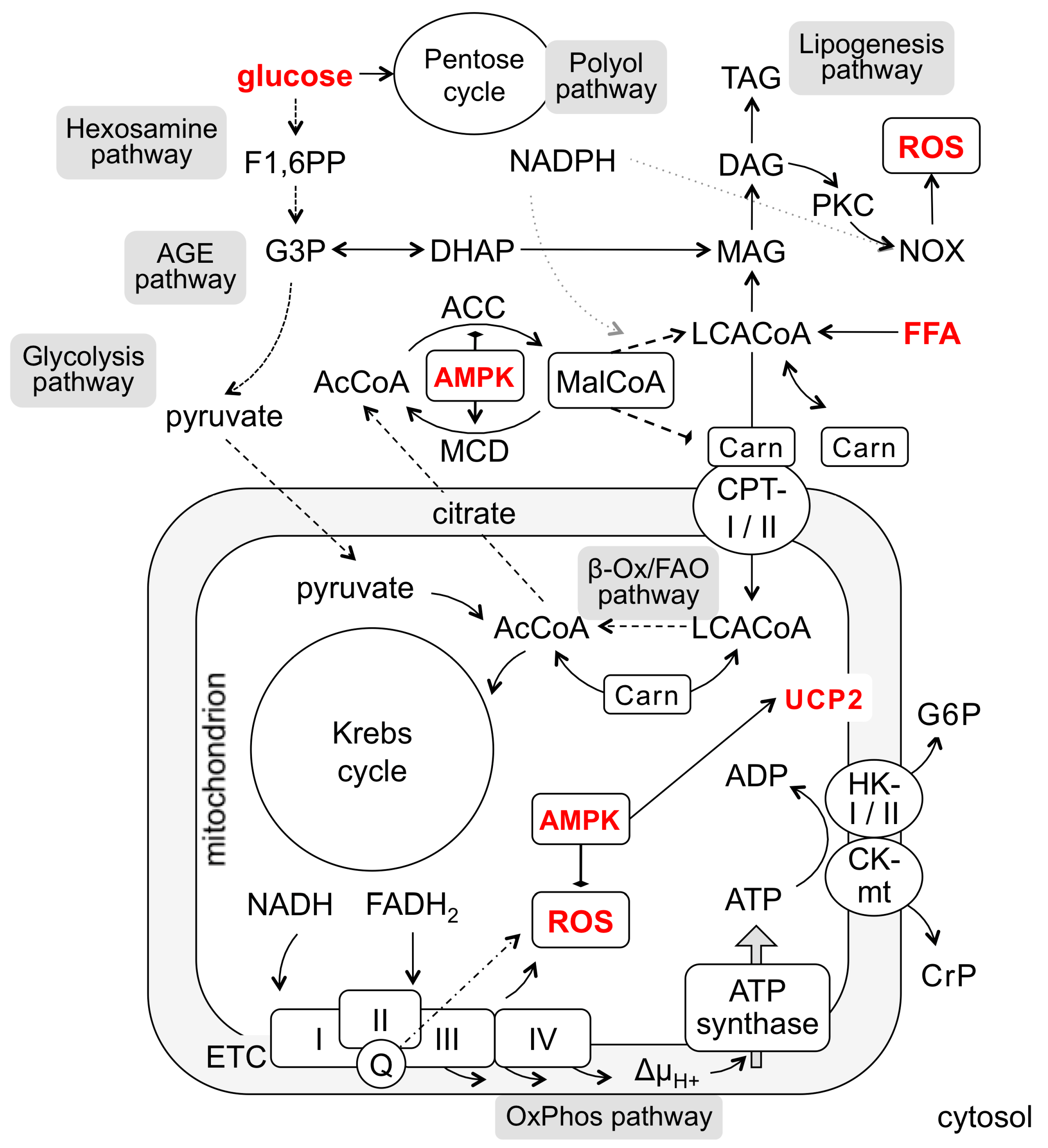
2. Hyperglycemia Sets Off Multiple Routes to Endothelial Dysfunction and Apoptosis
3. Hyperlipidemia Threats Endothelium through Increased FFA Levels
4. Carnitine Is Indispensable for FFA Utilization and Sequestration
5. AMPK Controls the Malonyl-CoA-Dependent Checkpoint of FFA Utilization
6. Mitochondrial Uncoupling as ROS Reduction Option: Potential Mechanisms
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| AcCoA | Acetyl-coenzyme A |
| ACSL | Acyl-CoA synthetase |
| ADP | Adenosine diphosphate |
| AGE | Advanced glycation end-products |
| AICAR | AMPK activator |
| Akt | Akt protein kinase |
| AMPK | AMP-activated protein kinase |
| ATCC | American tissue culture collection |
| ATP | Adenosine triphosphate |
| BAEC | Bovine aortic endothelial cells |
| Bax | BCL2 associated X, apoptosis regulator |
| BCL-2 | B-cell lymphoma 2, apoptosis regulator |
| BIM-1 | Bisindolylmaleimide 1, protein kinase C inhibitor |
| BSA | Bovine serum albumin |
| Carn | Carnitine |
| CCK-8 | Cell counting kit-8 |
| cGMP | Cyclic guanosine monophosphate |
| CK-mt | Mitochondrial isoform of creatine kinase |
| COX2 | Cyclooxygenase 2 |
| CPT-1 | Carnitine-palmitoyl transferase |
| CrP | Creatine phosphate |
| DAF2-DA | Diaminofluorescein diacetate, fluorescent nitric oxide probe |
| DAG | Diacylglycerol |
| DCF | H2DCFDA, a cell-permeant indicator for reactive oxygen species |
| DHAP | Dihydroxyacetone phosphate |
| DMEM | Cell growth medium formulation |
| DNA | Deoxyribonucleic acid |
| EA.hy926 | HUVEC-derived endothelial cell line |
| EBM | Endothelial basal medium |
| ECGS | Endothelial cell growth supplement |
| EGF | Epidermal growth factor |
| EGM | Endothelial growth medium |
| EGM-2mv | Microvascular Endothelial Cell Growth Medium-2 distributed by Lonza |
| eNOS | Endothelial NO synthase |
| EPC | Endothelial progenitor cells |
| Erk1/2 | Erk1/2 protein kinase |
| ER-stress | Endoplasmic reticulum stress |
| ET-1 | Endothelin-1 |
| ETC | Electron transport chain of mitochondria |
| F1,6PP | Fructose 1,6 biphosphate |
| F12 | Cell growth medium formulation |
| FADH2 | Flavin adenine dinucleotide reduced |
| FAO | Fatty acid oxidation |
| FBS | Fetal bovine serum |
| FFA | Free fatty acid |
| G3P | Glyceraldehyde 3-phosphate |
| G6P | Glucose-6-phosphate |
| GSH | Glutathione, reduced |
| GSK3β | Glycogen synthase kinase-3 beta |
| HAEC | Human aortic endothelial cells |
| HAT | Hypoxanthine-aminopterin-thymidine, cell growth medium supplement |
| HCAEC | Human coronary artery endothelial cells |
| HCEC | Human corneal endothelial cells |
| HG | High glucose |
| HK I/II | Hexokinase I/II isoform |
| HMEC-1 | Immortalized human dermal microvascular endothelial cells |
| HOMA-IR | Homeostatic model assessment of insulin resistance |
| HREC | Human retinal endothelial cells |
| HS-CoA | Free coenzyme A |
| HUAEC | Human umbilical artery endothelial cells |
| HUVEC | Human umbilical vein endothelial cells |
| ICAM-1 | Inter-cellular adhesion molecule 1 |
| IKKβ | Inhibitor of NF-kB kinase subunit beta |
| IL-6 | Interleukin-6 |
| iNOS | Inducible NOS |
| Ins | Insulin |
| IκBα | NF-κB inhibitor |
| JNK | JNK protein kinase |
| LCACoA | Long-chain acyl-coenzyme A |
| LD | lipid droplet |
| LNAME | L-Nω-nitroarginine methyl ester |
| LNMA | L-Nω-methylarginine, NOS inhibitor |
| LPL | Lipoprotein lipase |
| M199 | Cell growth medium formulation |
| MAG | Monoacylglycerol |
| MalCoA | Malonyl-coenzyme A |
| MCD | Malonyl-CoA decarboxylase |
| MCDB 131 | Cell growth medium formulation |
| MDA | Malondialdehyde |
| MEM | Cell growth medium formulation |
| MGO | Methylglyoxal |
| MHEC | Mouse heart endothelial cells |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MyD88 | Adaptor protein for Toll-like and interleukin-1 receptors |
| NAC | N-acetylcysteine |
| NAD+ | Nicotinamide adenine nucleotide oxidized |
| NADH | Nicotinamide adenine nucleotide reduced |
| NADPH | Nicotinamide adenine nucleotide phosphate reduced |
| NEFA | Non-esterified fatty acid |
| NF-kB | Nuclear factor kB, transcription factor |
| NGF | Nerve growth factor |
| NO | Nitric oxide |
| NOX | NADPH-oxidases |
| OxPhos | Oxidative phosphorylation |
| p21WAF-1/Cip1 | Cyclin-dependent kinase inhibitor 1 |
| p38 | p38 mitogen-activated protein kinases |
| PAI-1 | Plasminogen activator inhibitor-1 |
| P-Akt | Phosphorylated Akt |
| P-AMPK | Phosphorylated AMPK |
| PARP-1 | Poly(ADP-ribose) polymerase 1 |
| PCR | Polymerase chain reaction |
| P-eNOS | Phosphorylated eNOS |
| PGC-1α | Peroxisome proliferator–activated receptor gamma coactivator-1 alpha |
| PGE2 | Prostaglandin E2 |
| PGI2 | Prostaglandin I2 |
| PGIS | Prostaglandin I2 synthase |
| PI | Propidium iodide, fluorescent intercalating agent for DNA staining |
| PI3K | Phosphatidylinositol 3-kinase |
| P-IKK | Phosphorylated IκB kinase |
| P-IRS | Phosphorylated IRS |
| P-JNK | Phosphorylated JNK |
| PKC | Protein kinase C |
| PMA | Phorbol-12-myristate-13-acetate |
| P-mTOR | Phosphorylated mTOR |
| P-p65 | Phosphorylated p65 |
| PTEN | Dual specificity phosphatase, dephosphorylates phosphoinositides/proteins |
| P-Tyr | Phospho-tyrosine |
| Q | Coenzyme Q, ubiquinone |
| ROS | Reactive oxygen species |
| SAP | Secreted alkaline phosphatase |
| SIRT-1 | Sirtuin-1, deacetylase |
| SNP | Sodium nitroprusside |
| SOD | Superoxide dismutase |
| T2D | Type 2 diabetes mellitus |
| TAG | Triacylglycerol |
| TCA | Tricarboxylic acid cycle (Krebs cycle) |
| TLR4 | Toll-like receptor 4 |
| TNFα | Tumor necrosis factor alpha |
| TRPV1 | Transient receptor potential cation channel subfamily V member 1 |
| TUNEL | DNA fragmentation assay |
| UCP | Uncoupling protein |
| UCP-1 | Uncoupling protein 1 |
| UCP-2 | Uncoupling protein 2 |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| WB | Western blotting |
| 3-MA | 3-Methyladenine |
| 3-OMG | 3-O-Methylglucose |
| β-Ox | β-Oxidation of fatty acids |
| ΔμH+ | Membrane electrochemical potential |
Appendix A

{kind=link}
{kind=link}
| Cells and Study Design | Culture Conditions | Glucose Duration and Concentrations, Other Essentials | Assessed Parameters | Major Findings/Mechanism | Refs |
|---|---|---|---|---|---|
| Constant short-term HG treatment (1–3 days) | |||||
| HUVEC In vitro | M199, 20% FBS, ECGS | 5 vs. 30 mM; 24–48–72 h, (also includes the long-term treatment for 13 days) | Apoptosis (by DNA fragmentation, [3H]-thymidine incorporation) | The phenomenon first reported. Clusterin was not involved | [91] |
| HUVEC In vitro | M199, 20% FBS, ECGS, passage 4 | 5 vs. 30 mM, 48–72 h. Proinsulin or 3-O-methyl- glucose were also used | Apoptosis (by cell morphology, flow cytometry, DNA fragmentation) | HG, proinsulin, or 3-O-methylglucose induced apoptosis, which was inhibited by PKC activation and increased by PKC inhibition | [92] |
| HUVEC In vitro | M199, 20% FCS, ECGS, passage 3–5 | 5 vs. 19 or 33 mM, 24, 36, 48 h | Apoptosis, TUNEL assay, cells with hypodiploid DNA by flow cytometry, DNA fragmentation, ROS, intracellular Ca2+ | Taurine stabilized intracellular Ca2+, blocked HG-induced ROS production and apoptosis | [93] |
| HUVEC In vitro | M199, 20% FBS, ECGS, passage 3–5 | 5 vs. 33 mM, 2–48 h | Apoptosis, ROS (by DCF), eNOS (by WB), NF-kB, JNK1, caspase-3, PI3K pathway | HG increased eNOS expression that peaked at 6 h, then declined. ROS and apoptosis appeared later (24–48 h), were inhibited by ascorbate, vitamin C, and SNP, and increased by LNAME. HG-induced apoptosis was mediated by NF-kB, JNK1, caspase-3, and counteracted by PI3K > Akt > eNOS activation | [94,95,96] |
| BAEC, in vitro | DMEM, 10% FCS, passage 5–10 | 10–25 mM HG, 15 min–24 h, 1% FCS, subconfluent cells | ROS, apoptosis, cell death (trypan blue), NADPH, GSH, cAMP, glucose-6- phosphate dehydrogenase (G6PD) | Rapid (15 min) cAMP-dependent phosphorylation and inhibition of G6PD that decreased GSH and NADPH, increased ROS, and predisposed cells to apoptosis. | [97] |
| HAEC (Clonetics) In vitro | EGM, 2% FBS, ECGS (Clonetics) for growth, M199, 10% FBS for assays | 5 vs. 30 mM HG, 6–72 h | ROS (24 h), apoptosis (Annexin V, PI), GSH, mitochondrial membrane potential (ΔμH+, JC-1 fluorescent probe) | HG increased ROS, decreased ΔμH+ and GSH content, increased DNA fragmentation and apoptosis, all of them prevented by 10 mM N-acetylcysteine (NAC) | [98] |
| HUVEC, in vitro | M199, 20% FBS, ECGS | 5.5 vs. 30 mM HG, 8–12–48 h | Apoptosis, viability, COX2, PGE2, NF-kB, ROS, PI3K, caspase-3. | HG increased COX2, PGE2, ROS, NF-kB, caspase-3, and apoptosis. All these reduced by blocking PI3K-Akt | [99] |
| HUVEC, in vitro; diabetic mice, in vivo | DMEM, 10% FBS, passage 3–7 | 5.5 vs. 33 mM HG, 24 h | Viability (by CCK-8), apoptosis, caspase-3, ROS, autophagy (by LC3-I > LC3-II, mTOR, ULK1) | HG increased mitochondrial membrane potential, ROS, caspase-3, apoptosis, but inhibited autophagy via mTORC1, which was rescued by rapamycin or knocking down Raptor. | [57] |
| HUVEC, in vitro | ECM (ScienCell, 5% FBS, ECGS) | 25 mM HG, 72 h | Apoptosis, viability, ROS, SIRT-1/mTOR/AMPK signaling, Bax, Bcl-2, PARP, caspase-3 | HG induced apoptosis by decreased Bcl-2, increased Bax, caspase-3, PARP cleavage, ROS, lipid peroxidation, P-mTOR (S2448), and decreased P-AMPK (S-485). All prevented by fidarestat, the aldose reductase inhibitor, via Sirt1, AMPK, and mTOR | [60] |
| BAEC (primary cells) In vitro | DMEM, 10% FCS | 5 vs. 25 mM HG, 72 h, 2% FCS | ROS (by ESR), DAG, PKC. | HG increased ROS and PKC activity, DPI (2 h), but not LNMA/rotenone, negated ROS and the PKC increase. | [64] |
| HUVEC, in vitro | ECGM (Cell Applications) | 5.5 vs. 28 mM HG, 24 h, metabolic glucose, TCA intermediates | ROS (O2.−), ER-stress (by secreted alkaline phosphatase, SAP). | All intermediates induced ROS, but only glucose and glycolytic metabolites induced ER-stress. NOX and xanthine oxidases not involved | [100] |
| PIEC (porcine ileal artery) In vitro | DMEM, 10% FBS for growth, EBM for treatment | 5.5 vs. 30 mM HG, treatment, duration not indicated | ROS (dihydroethidium), NO (DAF2-DA), TRPV1, UCP2 expression. Signaling: PKA, AMPK, UCP2, p22phox, eNOS. | Capsaicin attenuated HG-induced endothelial dysfunction via TRPV1 activation, upregulation of UCP-2, reduced ROS, and increased NO | [101] |
| HUVEC, in vitro | EBM-2 (Lonza) | 5 vs. 25 mM HG, 24 h | Insulin signaling. | Insulin receptor decreased (both P-Tyr and expression), P-Akt increased, P-eNOS decreased. Erk1/2, p38 and JNK unchanged | [102] |
| Long-term HG treatment (5–14 days), including intermittent (oscillating) protocols | |||||
| HUVEC In vitro | M199, 20% heat- inactivated FBS, ECGS | Constant 5 vs. 20 mM HG vs. intermittent 5–20 mM glucose, 7–14 days | Viability, G0/G1 cell cycle arrest, apoptosis (by DNA fragmentation), Bcl-2/Bax expression | Bcl-2 was decreased by constant HG but unchanged by intermittent HG. Bax always increased. Apoptosis increased at >7 days and was greater in intermittent HG | [103] |
| Rat carotid artery, in vivo, glucose infusion | None | 5 vs. 30 mM HG, 48 h, constant or intermittent | Blood insulin, HOMA-IR, Bax, Bcl-2, caspase-3, ROS, p47phox NOX, GSH, MDA, IL-6, TNFa, ICAM-1. Apoptosis (by TUNEL assay) | Oscillating or constant HG induced insulin resistance. Oscillating more than constant HG reduced GSH, increased NOX, ROS, Bax/Bcl-2, caspase-3, MDA, inflammatory cytokines, and ICAM-1 expression. Oscillating but not constant HG induced apoptosis | [104] |
| HMEC-1 (dermal) HAEC (Cambrex) In vitro | MCDB 131, human serum, EGM (Cambrex), 2% FBS, ECGS | 5 vs. 20 mM HG, 7 days | Apoptosis (TUNEL, cell cycle), anoikis, necrosis, adhesion, dicarbonyls/AGEs, modifications by methylglyoxal (MGO) | Decreased collagen adhesion upon hyperglycemia-induced collagen IV modification by MGO, increased apoptosis of the detached cells (the attached not affected) | [105] |
| BAEC (Clonetics) or HAEC (ATCC) In vitro | F12, 10% FBS, ECGS | 5 vs. 25–30 or 44 mM HG, 3 or 7 days | eNOS dimerization and activity (by arginine to citrulline), peroxynitrite, O2.−, cGMP, PGI2 synthase (PGIS), apoptosis (by DNA fragmentation) | Peroxynitrite uncoupled eNOS resulting in O2.− production instead of NO. HG increased Ca2+-induced O2.−, peroxynitrite and PGIS nitration, inhibited cGMP and PGIS acivity, increased apoptosis, which depended on SOD and LNAME | [47,68] |
| MS-1 (the pancreatic islet cells, ATCC) In vitro | DMEM, 5% FBS | 5.5 vs. 30 mM HG, 7 days | Apoptosis (annexin V), iNOS expression, NO, peroxynitrite, O2.− (dihydroethidium), ROS (DCF), signaling (P-JNK) | HG increased iNOS expression, ROS, O2.−, nitrotyrosine, P-JNK, caspase-3 activity, Bax/Bcl-2 ratio and apoptosis, all prevented by JNK and iNOS inhibitors | [106] |
| HAEC (Clonetics) In vitro | Not clearly indicated | 5 vs. 22 mM HG, 5 days | eNOS (by PCR/WB), NO (by Griess assay), ROS (O2.−). | HG increased ROS (3-fold), eNOS (~2-fold) and NO (~40%) | [107] |
| BAEC, in vitro | MEM (Gibco), 0.4% FBS | 5 vs. 30 mM HG, 7 days | ROS, PKC, AGEs (MGO), sorbitol, NF-kB. | HG increased mitochondrial ROS, which induced PKC, AGEs, aldose reductase-mediated sorbitol, and NF-kB. All prevented by forced expression of UCP-1. | [70] |
| HUVEC, in vitro | M199, 20% heat- inactivated FBS, ECGS | Constant 5 vs. 20 mM HG vs. intermittent 5–20 mM glucose, 7–14 days | Signaling: PKC, NOX, nitrotyrosine, ROS, caspase-3, mitochondrial complex II. | HG > 7 days increased PKC activity, 8-OHdG, nitrotyrosine, NOX and caspase-3. All these were negated by PKC inhibitors and MnSOD mimetic. Thenoyltrifluoroacetone blocked nitrotyrosine while caspase-3 increased suggesting mitochondrial origin of ROS | [65,66] |
| EA.hy926, in vitro | DMEM, 10% FBS | 5.5 vs. 20 mM HG, 6 days | Aerobic metabolism, respiration, hexokinase-1, and UCP-2. | HG induced the Crabtree effect, no change in the Krebs cycle or respiratory chain. Upregulated UCP-2 and hexokinase-1 | [78] |
| SkMEC (primary cells from rat skeletal muscle) | EGM (Cell Applications Inc.) | 5.5 vs. 11 mM HG, 3 days | Cell viability (by trypan blue), mitochondrial respiration and coupling efficiency, H2O2 emission (by Amplex Ultra Red), citrate synthase activity. | Cell viability 89–98%, HG lowers mitochondrial respiration and increases H2O2/O2 emission, indicative of mitochondrial dysfunction | [108] |
| Cells and Study Design | Culture Conditions | FFA Type, Duration and Concentrations, Other Essentials | Assessed Parameters | Major Findings/Mechanism | Refs |
|---|---|---|---|---|---|
| Short-term FFA treatment (up to 2 days) | |||||
| Human skin microvascular EC | EGM (Clontech), ECGS | 0.1 mM palmitate, 4–24 h | Apoptosis, ROS, DNA fragmentation, [3H]-thymidine incorporation | Palmitate increased ROS, DNA fragmentation, [3H]-thymidine incorporation. NAC rescued all these almost completely | [109] |
| HUVEC | M199, 20% delipidated FBS, 0.4% albumin; human plasma +/− intralipid | 0.1–0.3 mM, 0.05 vs. 0.37 mM palmitate; 24 or 48 h; also oleate, stearate, linoleate, γ-linoleate, arachidonate | Apoptosis (time-, dose-, and double-bond dependent), G0/G1 cell cycle arrest | Upregulation of pro-apoptotic Bak and Cdk inhibitor p21WAF-1/Cip1. Reduced expression of apoptotic inhibitor Bcl-2, NF-κB inhibitor IκBα, ET-1, and eNOS | [110] |
| HAEC (from Bio Whittaker) | EBM, 2% FBS, growth factors (Bio Whittaker) | 0.3 mM linoleate, 16 h | ROS (by DCF), ET-1, NO, eNOS, NF-kB, caspase-3, apoptosis (by DNA fragmentation) | Linoleate increased ROS, ET-1, reduced NO, activated NF-kB and induced apoptosis; all rescued by UCP-2 adenoviral transduction | [77] |
| HCAECs (coronary artery) | EGM-2MV (Clonetics/Bio Whittaker) | 1 mM, palmitate, also oleate, stearate, palmitoleate, linoleate, 24 h, 2.5 or 5% BSA | Apoptosis, NF-kB, caspase-3, cytokine expression | Caspase-3, acyl-CoA formation (triacsin-C), no effect of etomoxir. FFA (but not NEFA) activated NF-kB. Blocking NF-kB or IKK or NEFA added on the top of FFA prevented FFA-induced apoptosis | [111] |
| HUVEC, HAEC, HREC, EPC | M199, 20% delipidated FBS, 0.4% albumin | 0.1–0.3 mM palmitate; 24 h; also oleate, stearate, linoleate, γ-linoleate, arachidonate | Apoptosis, membrane rigidity, incorporation of FFA into cell membranes | PUFA-induced apoptosis was mediated by c-myc in cells other than HUVEC but required caspase activation in all cells | [112] |
| EA.hy926 | DMEM, 10% FBS | 0.5 mM palmitate, oleate, 1–28 h; 6:1 to albumin | Necroptosis rather than apoptosis, autophagy | PA (but not oleate) induced autophagy (by LC3-I > LC3-II), which was rescued by autophagy inhibitors (3-MA, bafilomycin A1, wortmannin) | [58] |
| HUVEC | DMEM, 10% FBS | 0.1, 0.2, 0.3 mM; 24 or 48 h; FFA is not specified | Viability, apoptosis | FFA increased inflammatory cytokine expression and NF-kB signaling (MyD88, NF-kB, IKKb), all prevented by TLR4 interference | [61] |
| HUVEC | DMEM, 10% FBS, passage 20–25 | 0.3 mM palmitate, 24 h | Viability, autophagy (by LCI > LCII, loss of p62), cytosolic Ca2+, NO, tube formation, wound healing | FFA induced autophagy, increased NOX4, P-PKCa and ROS, decreased NO, wound healing, tube formation and cell viability—all prevented by 3-MA | [56] |
| EA.hy926 | DMEM, 10% FBS, 1% HAT | 0.36 mM oleate, 4 h | ROS, Ca2+, eNOS. | Oleate increased ROS, decreased Ca2+ and eNOS activity in response to histamine or ATP | [113] |
| BAEC (primary) | DMEM, 10% FBS for growth, EBM for treatment | 0.1 mM palmitate, oleate, linoleate, 3 h in EBM (Clontech), 30 mM BSA | Insulin signaling, IKK (by WB), NO (by DAF). | All FFAs inhibited insulin signaling and NO production, and increased IKKb. Dominant-negative IKK reversed the effects of palmitate | [114] |
| HAEC (primary, aortic) | EGM, 10% FBS for growth, EBM for treatment | 0.1–1 mM palmitate, oleate, linoleate, 24 h, 2% BSA | Ins signaling, eNOS (by Arg > citrulline), JNK/p38, PTEN (ChIP assay). | Palmitate and linoleate (but not oleate) inhibited Ins cascade and eNOS activity, increased JNK and p38, upregulated PTEN. | [115] |
| HUVEC | DMEM, 10% FBS | 0.1 mM palmitate, 24 h | NF-kB, insulin signaling (by WB), NO (by DAF). | FFA increased IL-6 and TNF-α production, P-IKKb, P-p65, P-IRS (S307), and decreased P-IRS (Tyr), P-Akt, P-eNOS, NO in response to insulin | [116] |
| HUVEC | RPMI-1640, 10% FBS | 0.1 mM palmitate, 0.5, 2 and 12 h | NO (by DAF), glucose uptake, insulin signaling (by WB). | FFA induced ROS, IL-6, TNF-a (all inhibited by piceatannol, resveratrol analog. FFA reduced P-IRS (Tyr), P-eNOS, NO, glucose uptake | [62] |
| HUVEC | Medium 200, ECGS, ? % FBS | 0.25, 0.5, 0.75 mM palmitate, 6–18 h | Autophagy, ER-stress, insulin signaling (by WB), glucose uptake. | FFA decreased P-Akt, Glut4 exposure and glucose uptake, increased LC3-II, caspase activity, ER-stress, P-IRS (S307), P-JNK | [117] |
| MHEC (mouse heart), EA.hy926 | DMEM, 20% FBS, ECGS DMEM, 10% FBS | 0.1–0.4 mM palmitate, 1–8 h | Autophagy (by LCI > LCII), ROS, lipid droplets (by Bodipy), adhesion of leukocytes. | PA induced ROS and LD formation, but impaired autophagy. Blocking CPT1 (by etomoxir) promoted, but blocking ACSL (by triacsin-C) inhibited autophagy. Metformin reduced LD and rescued autophagy, the effects blunted by silencing of AMPK | [59] |
| Long-term FFA treatment (more than 2 days) | |||||
| HUVEC | M199 or DMEM, 20% FBS, ECGS | 2 mM oleate/palmitate mix (2:1), 2% albumin, 3 days | Apoptosis, insulin signaling | Caspase-9 activation, increased bax-a and reduced bcl-2 expression. Decreased P-Akt (S473), P-eNOS. Apoptosis prevented by insulin (10 nM) in PI3K-dependent manner | [118] |
| HUVEC | M199, 20% FBS | 0.75 mM oleate/palmitate mix (2:1), 72 h; 0.5, 0.75, 1 mM up to 4 days | Apoptosis, proliferation | FFA inhibited proliferation and induces apoptosis. The responses were partly rescued by GSK3β knockdown | [119] |
| HUVEC | EGM-2mv (Lonza), 50 μM carnitine, passage 3 | 0.5–1.5 mM palmitate, up to 14 days | Cell viability (by light microscopy), monolayer permeability (by ECIS-z), insulin signaling (by WB), ROS (by DCF), NO (by DAF), MDA (by WB) | Palmitate dose-dependently induced cell death associated with oxidative/dicarbonyl stress, increased permeability, ROS, NO, MDA. AICAR and carnitine were overwhelmingly protective | [28] |
| BAEC (primary) | DMEM, 10% FCS | 0.2 mM palmitate, 24–72 h, 1% FCS, 3 days | ROS (by ESR), DAG and PKC. | ROS, DAG, PKC activity increased, DPI (for 2 h) negated the ROS and PKC upregulation responses | [64] |
| SkMEC (primary cells from rat skeletal muscle) | EGM (Cell Applications Inc.) | 1.7 or 2.2 mM palmitate (albumin concentration not indicated), 3 days | Cell viability (trypan blue), mitochondrial respiration and coupling efficiency, H2O2 emission (by Amplex Ultra Red), citrate synthase activity. | Cell viability 89–98%, palmitate lowers mitochondrial respiration and increases H2O2/O2 emission, indicative of mitochondrial dysfunction | [108] |
References
- Del Turco, S.; Gaggini, M.; Daniele, G.; Basta, G.; Folli, F.; Sicari, R.; Gastaldelli, A. Insulin resistance and endothelial dysfunction: A mutual relationship in cardiometabolic risk. Curr. Pharm. Des. 2013, 19, 2420–2431. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.A. Endothelial dysfunction in insulin resistance and type 2 diabetes. J. Intern. Med. 2007, 262, 173–183. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. [Google Scholar] [CrossRef] [PubMed]
- La Sala, L.; Prattichizzo, F.; Ceriello, A. The link between diabetes and atherosclerosis. Eur. J. Prev. Cardiol. 2019, 26, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M. Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes 1999, 48, 937–942. [Google Scholar] [CrossRef]
- Little, P.J.; Askew, C.D.; Xu, S.; Kamato, D. Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship. Biomedicines 2021, 9, 699. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Domínguez-Pérez, M.; Mercado, I.; Villarreal-Molina, M.T.; Jacobo-Albavera, L. Use of Human Umbilical Vein Endothelial Cells (HUVEC) as a Model to Study Cardiovascular Disease: A Review. Appl. Sci. 2020, 10, 938. [Google Scholar] [CrossRef] [Green Version]
- Ham, R.G. Nutritional requirements of primary cultures. a neglected problem of modern biology. In Vitro 1974, 10, 119–129. [Google Scholar] [CrossRef]
- Knedler, A.; Ham, R.G. Optimized medium for clonal growth of human microvascular endothelial cells with minimal serum. In Vitro Cell Dev. Biol. 1987, 23, 481–491. [Google Scholar] [CrossRef]
- Leopold, B.; Strutz, J.; Weiß, E.; Gindlhuber, J.; Birner-Gruenberger, R.; Hackl, H.; Appel, H.M.; Cvitic, S.; Hiden, U. Outgrowth, proliferation, viability, angiogenesis and phenotype of primary human endothelial cells in different purchasable endothelial culture media: Feed wisely. Histochem. Cell Biol. 2019, 152, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, E.P.; Cheng, S.; Larson, M.G.; Walford, G.A.; Lewis, G.D.; McCabe, E.; Yang, E.; Farrell, L.; Fox, C.S.; O’Donnell, C.J.; et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J. Clin. Investig. 2011, 121, 1402–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef] [Green Version]
- Giugliano, D.; Ceriello, A.; Paolisso, G. Oxidative stress and diabetic vascular complications. Diabetes Care 1996, 19, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Wojtczak, L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic. Biol. Med. 2008, 45, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Diez-Roux, G.; Lang, R.A. Macrophages induce apoptosis in normal cells in vivo. Development 1997, 124, 3633–3638. [Google Scholar] [CrossRef]
- Winn, R.K.; Harlan, J.M. The role of endothelial cell apoptosis in inflammatory and immune diseases. J. Thromb. Haemost. 2005, 3, 1815–1824. [Google Scholar] [CrossRef]
- Best, P.J.; Hasdai, D.; Sangiorgi, G.; Schwartz, R.S.; Holmes, D.R., Jr.; Simari, R.D.; Lerman, A. Apoptosis. Basic concepts and implications in coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef]
- Gauthier, M.S.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.J.; Gauthier, M.S.; Hess, D.T.; Apovian, C.M.; Cacicedo, J.M.; Gokce, N.; Farb, M.; Valentine, R.J.; Ruderman, N.B. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J. Lipid Res. 2012, 53, 792–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ido, Y.; Carling, D.; Ruderman, N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: Inhibition by the AMP-activated protein kinase activation. Diabetes 2002, 51, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Hu, V.W.; Black, G.E.; Torres-Duarte, A.; Abramson, F.P. 3H-thymidine is a defective tool with which to measure rates of DNA synthesis. FASEB J. 2002, 16, 1456–1457. [Google Scholar] [CrossRef]
- Orlov, S.N.; Pchejetski, D.V.; Sarkissian, S.D.; Adarichev, V.; Taurin, S.; Pshezhetsky, A.V.; Tremblay, J.; Maximov, G.V.; deBlois, D.; Bennett, M.R.; et al. [3H]-thymidine labelling of DNA triggers apoptosis potentiated by E1A-adenoviral protein. Apoptosis 2003, 8, 199–208. [Google Scholar] [CrossRef]
- Watanabe, M.; Hitomi, M.; van der Wee, K.; Rothenberg, F.; Fisher, S.A.; Zucker, R.; Svoboda, K.K.; Goldsmith, E.C.; Heiskanen, K.M.; Nieminen, A.L. The pros and cons of apoptosis assays for use in the study of cells, tissues, and organs. Microsc. Microanal. 2002, 8, 375–391. [Google Scholar] [CrossRef] [Green Version]
- Samsonov, M.V.; Podkuychenko, N.V.; Khapchaev, A.Y.; Efremov, E.E.; Yanushevskaya, E.V.; Vlasik, T.N.; Lankin, V.Z.; Stafeev, I.S.; Skulachev, M.V.; Shestakova, M.V.; et al. AICAR Protects Vascular Endothelial Cells from Oxidative Injury Induced by the Long-Term Palmitate Excess. Int. J. Mol. Sci. 2021, 23, 211. [Google Scholar] [CrossRef]
- Arner, P.; Ryden, M. Fatty Acids, Obesity and Insulin Resistance. Obes. Facts 2015, 8, 147–155. [Google Scholar] [CrossRef]
- Hales, C.N.; Walker, J.B.; Garland, P.B.; Randle, P.J. Fasting Plasma Concentrations of Insulin, Non-Esterified Fatty Acids, Glycerol, and Glucose in the Early Detection of Diabetes Mellitus. Lancet 1965, 285, 65–67. [Google Scholar] [CrossRef]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.M.; Park, Y.S.; Walewicz, D.; Russell-Lopez, C.; Windsor, S.; Isley, W.L.; Coppack, S.W.; Harris, W.S. Systemic and forearm triglyceride metabolism: Fate of lipoprotein lipase-generated glycerol and free fatty acids. Diabetes 2004, 53, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.M.; Nelson, R.H. Contribution of triglyceride-rich lipoproteins to plasma free fatty acids. Horm. Metab. Res. 2007, 39, 726–729. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Cunha, D.A.; Ladriere, L.; Igoillo-Esteve, M.; Bugliani, M.; Marchetti, P.; Cnop, M. In vitro use of free fatty acids bound to albumin: A comparison of protocols. Biotechniques 2015, 58, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, S. Plasma albumin as a fatty acid carrier. Adv. Mol. Cell Biol. 2004, 33, 29–46. [Google Scholar] [CrossRef]
- Stafeev, I.S.; Vorotnikov, A.V.; Ratner, E.I.; Menshikov, M.Y.; Parfyonova, Y.V. Latent Inflammation and Insulin Resistance in Adipose Tissue. Int. J. Endocrinol. 2017, 2017, 5076732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmann, W.C.; Dubelaar, M.L. Aspects of fatty acid metabolism in vascular endothelial cells. Biochimie 1988, 70, 681–686. [Google Scholar] [CrossRef]
- Hulsmann, W.C.; Dubelaar, M.L. Carnitine requirement of vascular endothelial and smooth muscle cells in imminent ischemia. Mol. Cell Biochem. 1992, 116, 125–129. [Google Scholar] [CrossRef]
- Dagher, Z.; Ruderman, N.; Tornheim, K.; Ido, Y. The effect of AMP-activated protein kinase and its activator AICAR on the metabolism of human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 1999, 265, 112–115. [Google Scholar] [CrossRef]
- Dagher, Z.; Ruderman, N.; Tornheim, K.; Ido, Y. Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ. Res. 2001, 88, 1276–1282. [Google Scholar] [CrossRef]
- Cacicedo, J.M.; Yagihashi, N.; Keaney, J.F., Jr.; Ruderman, N.B.; Ido, Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 324, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Cacicedo, J.M.; Itani, S.; Yagihashi, N.; Saha, A.K.; Ye, J.M.; Chen, K.; Zou, M.; Carling, D.; Boden, G.; et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): Possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem. Soc. Trans. 2003, 31, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Noland, R.C.; Kovalik, J.P.; Seiler, S.E.; Davies, M.N.; DeBalsi, K.L.; Ilkayeva, O.R.; Stevens, R.D.; Kheterpal, I.; Zhang, J.; et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell. Metab. 2012, 15, 764–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D. Banting lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.H.; Shi, C.; Cohen, R.A. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor–mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes 2002, 51, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Gong, J.; Peterson, A.L.; Lu, X.; Zhang, P.; Dennery, P.A. Fatty Acid Oxidation Protects against Hyperoxia-induced Endothelial Cell Apoptosis and Lung Injury in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2019, 60, 667–677. [Google Scholar] [CrossRef]
- Faergeman, N.J.; Knudsen, J. Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem. J. 1997, 323, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Karlic, H.; Lohninger, A. Supplementation of L-carnitine in athletes: Does it make sense? Nutrition 2004, 20, 709–715. [Google Scholar] [CrossRef]
- McGarry, J.D.; Mannaerts, G.P.; Foster, D.W. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Investig. 1977, 60, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zammit, V.A. The malonyl-CoA-long-chain acyl-CoA axis in the maintenance of mammalian cell function. Biochem. J. 1999, 343, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Saha, A.K. Metabolic syndrome: Adenosine monophosphate-activated protein kinase and malonyl coenzyme A. Obesity 2006, 14, 25S–33S. [Google Scholar] [CrossRef] [Green Version]
- Saggerson, D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 2008, 28, 253–272. [Google Scholar] [CrossRef]
- Park, H.; Kaushik, V.K.; Constant, S.; Prentki, M.; Przybytkowski, E.; Ruderman, N.B.; Saha, A.K. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J. Biol. Chem. 2002, 277, 32571–32577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Liu, H.; Xiang, H.; Zhou, J.; Zeng, Z.; Chen, R.; Zhao, S.; Xiao, J.; Shu, Z.; Chen, S.; et al. Palmitic acid-induced autophagy increases reactive oxygen species via the Ca(2+)/PKCalpha/NOX4 pathway and impairs endothelial function in human umbilical vein endothelial cells. Exp. Ther. Med. 2019, 17, 2425–2432. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Han, D.; Sun, Z.; Ma, S.; Gao, L.; Chen, J.; Li, X.; Li, X.; Fan, M.; Li, C.; et al. Endothelial deletion of mTORC1 protects against hindlimb ischemia in diabetic mice via activation of autophagy, attenuation of oxidative stress and alleviation of inflammation. Free Radic. Biol. Med. 2017, 108, 725–740. [Google Scholar] [CrossRef]
- Khan, M.J.; Rizwan Alam, M.; Waldeck-Weiermair, M.; Karsten, F.; Groschner, L.; Riederer, M.; Hallstrom, S.; Rockenfeller, P.; Konya, V.; Heinemann, A.; et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J. Biol. Chem. 2012, 287, 21110–21120. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Ren, G.; Kim, T.; Bhatnagar, S.; Yang, Q.; Bahk, Y.Y.; Kim, J.A. Metformin reduces saturated fatty acid-induced lipid accumulation and inflammatory response by restoration of autophagic flux in endothelial cells. Sci. Rep. 2020, 10, 13523. [Google Scholar] [CrossRef]
- Pal, P.B.; Sonowal, H.; Shukla, K.; Srivastava, S.K.; Ramana, K.V. Aldose reductase regulates hyperglycemia-induced HUVEC death via SIRT1/AMPK-alpha1/mTOR pathway. J. Mol. Endocrinol. 2019, 63, 11–25. [Google Scholar] [CrossRef]
- Chen, L.; Yu, C.X.; Song, B.; Cai, W.; Liu, C.; Guan, Q.B. Free fatty acids mediates human umbilical vein endothelial cells inflammation through toll-like receptor-4. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.O.; Son, Y.; Lee, J.H.; Cheong, Y.K.; Park, S.H.; Chung, H.T.; Pae, H.O. Resveratrol analog piceatannol restores the palmitic acid-induced impairment of insulin signaling and production of endothelial nitric oxide via activation of anti-inflammatory and antioxidative heme oxygenase-1 in human endothelial cells. Mol. Med. Rep. 2015, 12, 937–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, S.L.; Moncada, S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Piconi, L.; Quagliaro, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes/Metab. Res. Rev. 2006, 22, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD(P)H-oxidase activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintero, M.; Colombo, S.L.; Godfrey, A.; Moncada, S. Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 5379–5384. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Green, K.; Brand, M.D.; Murphy, M.P. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004, 53, S110–S118. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.; Zhao, Y. Uncoupling protein 2 and metabolic diseases. Mitochondrion 2017, 34, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox. Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [Green Version]
- Pecqueur, C.; Alves-Guerra, C.; Ricquier, D.; Bouillaud, F. UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life 2009, 61, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Crichton, P.G.; Lee, Y.; Kunji, E.R. The molecular features of uncoupling protein 1 support a conventional mitochondrial carrier-like mechanism. Biochimie 2017, 134, 35–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.U.; Lee, I.K.; Han, J.; Song, D.K.; Kim, Y.M.; Song, H.S.; Kim, H.S.; Lee, W.J.; Koh, E.H.; Song, K.H.; et al. Effects of recombinant adenovirus-mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ. Res. 2005, 96, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Koziel, A.; Woyda-Ploszczyca, A.; Kicinska, A.; Jarmuszkiewicz, W. The influence of high glucose on the aerobic metabolism of endothelial EA.hy926 cells. Pflügers Arch. Eur. J. Physiol. 2012, 464, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Dopheide, J.; Schuhmacher, S.; Thomas, S.R.; Chen, K.; Daiber, A.; Wenzel, P.; Munzel, T.; Keaney, J.F., Jr. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 2008, 118, 1347–1357. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, J.; Wu, J.; Viollet, B.; Zou, M.H. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 2008, 57, 3222–3230. [Google Scholar] [CrossRef]
- Zou, M.H.; Wu, Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin. Exp. Pharmacol. Physiol. 2008, 35, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef] [PubMed]
- da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.E.; Machado, L.B.; Santiago, A.P.; da-Silva, W.S.; De Felice, F.G.; Holub, O.; Oliveira, M.F.; Galina, A. Mitochondrial creatine kinase activity prevents reactive oxygen species generation: Antioxidant role of mitochondrial kinase-dependent ADP re-cycling activity. J. Biol. Chem. 2006, 281, 37361–37371. [Google Scholar] [CrossRef] [Green Version]
- Vyssokikh, M.Y.; Holtze, S.; Averina, O.A.; Lyamzaev, K.G.; Panteleeva, A.A.; Marey, M.V.; Zinovkin, R.A.; Severin, F.F.; Skulachev, M.V.; Fasel, N.; et al. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc. Natl. Acad. Sci. USA 2020, 117, 6491–6501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, H.; Hickner, R.C.; Ormsbee, M.J. The Potential Role of Creatine in Vascular Health. Nutrients 2021, 13, 857. [Google Scholar] [CrossRef]
- Decking, U.K.; Alves, C.; Wallimann, T.; Wyss, M.; Schrader, J. Functional aspects of creatine kinase isoenzymes in endothelial cells. Am. J. Physiol. Cell. Physiol. 2001, 281, C320–C328. [Google Scholar] [CrossRef] [Green Version]
- Nomura, A.; Zhang, M.; Sakamoto, T.; Ishii, Y.; Morishima, Y.; Mochizuki, M.; Kimura, T.; Uchida, Y.; Sekizawa, K. Anti-inflammatory activity of creatine supplementation in endothelial cells in vitro. Br. J. Pharmacol. 2003, 139, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, E.; Guescini, M.; Calcabrini, C.; Vallorani, L.; Diaz, A.R.; Fimognari, C.; Canonico, B.; Luchetti, F.; Papa, S.; Battistelli, M.; et al. Creatine Prevents the Structural and Functional Damage to Mitochondria in Myogenic, Oxidatively Stressed C2C12 Cells and Restores Their Differentiation Capacity. Oxid. Med. Cell. Longev. 2016, 2016, 5152029. [Google Scholar] [CrossRef]
- Sestili, P.; Martinelli, C.; Bravi, G.; Piccoli, G.; Curci, R.; Battistelli, M.; Falcieri, E.; Agostini, D.; Gioacchini, A.M.; Stocchi, V. Creatine supplementation affords cytoprotection in oxidatively injured cultured mammalian cells via direct antioxidant activity. Free Radic. Biol. Med. 2006, 40, 837–849. [Google Scholar] [CrossRef]
- Baumgartner-Parzer, S.M.; Wagner, L.; Pettermann, M.; Grillari, J.; Gessl, A.; Waldhausl, W. High-glucose—Triggered apoptosis in cultured endothelial cells. Diabetes 1995, 44, 1323–1327. [Google Scholar] [CrossRef]
- Du, X.L.; Sui, G.Z.; Stockklauser-Farber, K.; Weiss, J.; Zink, S.; Schwippert, B.; Wu, Q.X.; Tschope, D.; Rosen, P. Induction of apoptosis by high proinsulin and glucose in cultured human umbilical vein endothelial cells is mediated by reactive oxygen species. Diabetologia 1998, 41, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.D.; Wang, J.H.; Fennessy, F.; Redmond, H.P.; Bouchier-Hayes, D. Taurine prevents high-glucose-induced human vascular endothelial cell apoptosis. Am. J. Physiol. Cell Physiol. 1999, 277, C1229-1238. [Google Scholar] [CrossRef]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-kappaB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Ho, F.M.; Liu, S.H.; Liau, C.S.; Huang, P.J.; Lin-Shiau, S.Y. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH(2)-terminal kinase and caspase-3. Circulation 2000, 101, 2618–2624. [Google Scholar] [CrossRef] [Green Version]
- Ho, F.M.; Liu, S.H.; Liau, C.S.; Huang, P.J.; Shiah, S.G.; Lin-Shiau, S.Y. Nitric oxide prevents apoptosis of human endothelial cells from high glucose exposure during early stage. J. Cell. Biochem. 1999, 75, 258–263. [Google Scholar] [CrossRef]
- Zhang, Z.; Apse, K.; Pang, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J. Biol. Chem. 2000, 275, 40042–40047. [Google Scholar] [CrossRef] [Green Version]
- Recchioni, R.; Marcheselli, F.; Moroni, F.; Pieri, C. Apoptosis in human aortic endothelial cells induced by hyperglycemic condition involves mitochondrial depolarization and is prevented by N-acetyl-L-cysteine. Metabolism 2002, 51, 1384–1388. [Google Scholar] [CrossRef]
- Sheu, M.L.; Ho, F.M.; Yang, R.S.; Chao, K.F.; Lin, W.W.; Lin-Shiau, S.Y.; Liu, S.H. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Sheikh-Ali, M.; Sultan, S.; Alamir, A.R.; Haas, M.J.; Mooradian, A.D. Hyperglycemia-induced endoplasmic reticulum stress in endothelial cells. Nutrition 2010, 26, 1146–1150. [Google Scholar] [CrossRef]
- Sun, J.; Pu, Y.; Wang, P.; Chen, S.; Zhao, Y.; Liu, C.; Shang, Q.; Zhu, Z.; Liu, D. TRPV1-mediated UCP2 upregulation ameliorates hyperglycemia-induced endothelial dysfunction. Cardiovasc. Diabetol. 2013, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- De Nigris, V.; Pujadas, G.; La Sala, L.; Testa, R.; Genovese, S.; Ceriello, A. Short-term high glucose exposure impairs insulin signaling in endothelial cells. Cardiovasc. Diabetol. 2015, 14, 114. [Google Scholar] [CrossRef] [Green Version]
- Risso, A.; Mercuri, F.; Quagliaro, L.; Damante, G.; Ceriello, A. Intermittent high glucose enhances apoptosis in human umbilical vein endothelial cells in culture. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E924–E930. [Google Scholar] [CrossRef]
- Wu, N.; Shen, H.; Liu, H.; Wang, Y.; Bai, Y.; Han, P. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Dobler, D.; Ahmed, N.; Song, L.; Eboigbodin, K.E.; Thornalley, P.J. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes 2006, 55, 1961–1969. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Liu, F.Q.; Wang, J.; Wang, X.P.; Hou, X.G.; Sun, Y.; Qin, W.D.; Wei, S.J.; Zhang, Y.; Chen, L.; et al. Hyperglycemia induces apoptosis of pancreatic islet endothelial cells via reactive nitrogen species-mediated Jun N-terminal kinase activation. Biochim. Biophys. Acta 2011, 1813, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, F.; Hishikawa, K.; Katusic, Z.S.; Luscher, T.F. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 1997, 96, 25–28. [Google Scholar] [CrossRef]
- Hansen, C.; Olsen, K.; Pilegaard, H.; Bangsbo, J.; Gliemann, L.; Hellsten, Y. High metabolic substrate load induces mitochondrial dysfunction in rat skeletal muscle microvascular endothelial cells. Physiol. Rep. 2021, 9, e14855. [Google Scholar] [CrossRef]
- Yamagishi, S.; Okamoto, T.; Amano, S.; Inagaki, Y.; Koga, K.; Koga, M.; Choei, H.; Sasaki, N.; Kikuchi, S.; Takeuchi, M.; et al. Palmitate-induced apoptosis of microvascular endothelial cells and pericytes. Mol. Med. 2002, 8, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Artwohl, M.; Roden, M.; Waldhausl, W.; Freudenthaler, A.; Baumgartner-Parzer, S.M. Free fatty acids trigger apoptosis and inhibit cell cycle progression in human vascular endothelial cells. FASEB J. 2004, 18, 146–148. [Google Scholar] [CrossRef]
- Staiger, K.; Staiger, H.; Weigert, C.; Haas, C.; Haring, H.U.; Kellerer, M. Saturated, but not unsaturated, fatty acids induce apoptosis of human coronary artery endothelial cells via nuclear factor-kappaB activation. Diabetes 2006, 55, 3121–3126. [Google Scholar] [CrossRef]
- Artwohl, M.; Lindenmair, A.; Sexl, V.; Maier, C.; Rainer, G.; Freudenthaler, A.; Huttary, N.; Wolzt, M.; Nowotny, P.; Luger, A.; et al. Different mechanisms of saturated versus polyunsaturated FFA-induced apoptosis in human endothelial cells. J. Lipid Res. 2008, 49, 2627–2640. [Google Scholar] [CrossRef] [Green Version]
- Esenabhalu, V.E.; Schaeffer, G.; Graier, W.F. Free fatty acid overload attenuates Ca2+ signaling and NO production in endothelial cells. Antioxid. Redox Signal. 2003, 5, 147–153. [Google Scholar] [CrossRef]
- Kim, F.; Tysseling, K.A.; Rice, J.; Pham, M.; Haji, L.; Gallis, B.M.; Baas, A.S.; Paramsothy, P.; Giachelli, C.M.; Corson, M.A.; et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.L.; Zhang, L.; Youker, K.; Zhang, M.X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Shen, Y.H. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 2006, 55, 2301–2310. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhao, W.; Gao, X.; Huang, F.; Kou, J.; Liu, B. Diosgenin ameliorates palmitate-induced endothelial dysfunction and insulin resistance via blocking IKKbeta and IRS-1 pathways. Atherosclerosis 2012, 223, 350–358. [Google Scholar] [CrossRef]
- Ye, M.; Qiu, H.; Cao, Y.; Zhang, M.; Mi, Y.; Yu, J.; Wang, C. Curcumin Improves Palmitate-Induced Insulin Resistance in Human Umbilical Vein Endothelial Cells by Maintaining Proteostasis in Endoplasmic Reticulum. Front. Pharmacol. 2017, 8, 148. [Google Scholar] [CrossRef] [Green Version]
- Piro, S.; Spampinato, D.; Spadaro, L.; Oliveri, C.E.; Purrello, F.; Rabuazzo, A.M. Direct apoptotic effects of free fatty acids on human endothelial cells. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 96–104. [Google Scholar] [CrossRef]
- Zhu, P.; Chen, G.; You, T.; Yao, J.; Jiang, Q.; Lin, X.; Shen, X.; Qiao, Y.; Lin, L. High FFA-induced proliferation and apoptosis in human umbilical vein endothelial cell partly through Wnt/beta-catenin signal pathway. Mol. Cell. Biochem. 2010, 338, 123–131. [Google Scholar] [CrossRef]
| Cells | Culture Conditions | FFA Duration and Concentrations, Other Essentials | Assessed Parameters | Major Findings/Mechanism | Reference |
|---|---|---|---|---|---|
| HUAEC (arterial) | M199, 10% FCS, 10% HS, ECGS, passage 2–4 | 0.5 mM oleate, 50 μM carnitine | FAO, ATP generation | Carnitine stimulates FAO and ATP production by ~2.5-fold | [37] |
| HUVEC | EBM-2 (Clonetics), 50 μM carnitine (in metabolic studies), passage 3–5 | 0.11 mM palmitate in metabolic studies; 0.2–2 mM AICAR, 0.5–2 h | FAO vs. Glucose metabolism | In the presence of carnitine, AMPK activation by AICAR reverts energy production from glycolysis to FAO | [39] |
| HUVEC | EBM-2 (Clonetics), 50 μM carnitine, passage 3–5 | 0, 5, or 30 mM glucose, 2 h; 0.1 mM palmitate, 24 h in metabolic studies | FAO vs. Glucose metabolism | AICAR reduces malonyl-CoA and glycolysis, but increases FAO ~3-fold only in the presence of carnitine | [40] |
| HUVEC | EGM-2 (Clonetics) (for cell growth); M199, 10% FBS (for assays); 50 μM carnitine (in metabolic studies), passage 5–6 | 5 vs. 30 mM glucose, 24–72 h, 1 mM AICAR, 24 h; 0.1 mM palmitate, 24 h in metabolic studies | Apoptosis (TUNEL), caspase-3, P-Akt (S473, WB), FAO vs. glucose oxidation, ATP, DAG, ceramide, malonyl-CoA | AICAR or AMPK forced expression inhibits apoptosis, decreases malonyl-CoA and DAG, increases FAO and P-Akt, reduces lactate, pyruvate, and glucose oxidation | [23] |
| HUVEC | EBM-2 (Cambrex), M199, 5% FBS, 50 μM carnitine with FFA, passage 4–6 | 5 vs. 25 mM glucose, 24 h, 0.4 mM palmitate, 16–24 h | NF-kB, VCAM-1 expression | Palmitate (but not HG) increases, but AICAR decreases both NF-kB activation and VCAM-1 expression | [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorotnikov, A.V.; Khapchaev, A.Y.; Nickashin, A.V.; Shirinsky, V.P. In Vitro Modeling of Diabetes Impact on Vascular Endothelium: Are Essentials Engaged to Tune Metabolism? Biomedicines 2022, 10, 3181. https://doi.org/10.3390/biomedicines10123181
Vorotnikov AV, Khapchaev AY, Nickashin AV, Shirinsky VP. In Vitro Modeling of Diabetes Impact on Vascular Endothelium: Are Essentials Engaged to Tune Metabolism? Biomedicines. 2022; 10(12):3181. https://doi.org/10.3390/biomedicines10123181
Chicago/Turabian StyleVorotnikov, Alexander V., Asker Y. Khapchaev, Alexey V. Nickashin, and Vladimir P. Shirinsky. 2022. "In Vitro Modeling of Diabetes Impact on Vascular Endothelium: Are Essentials Engaged to Tune Metabolism?" Biomedicines 10, no. 12: 3181. https://doi.org/10.3390/biomedicines10123181