
Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease


Abstract
1. Introduction
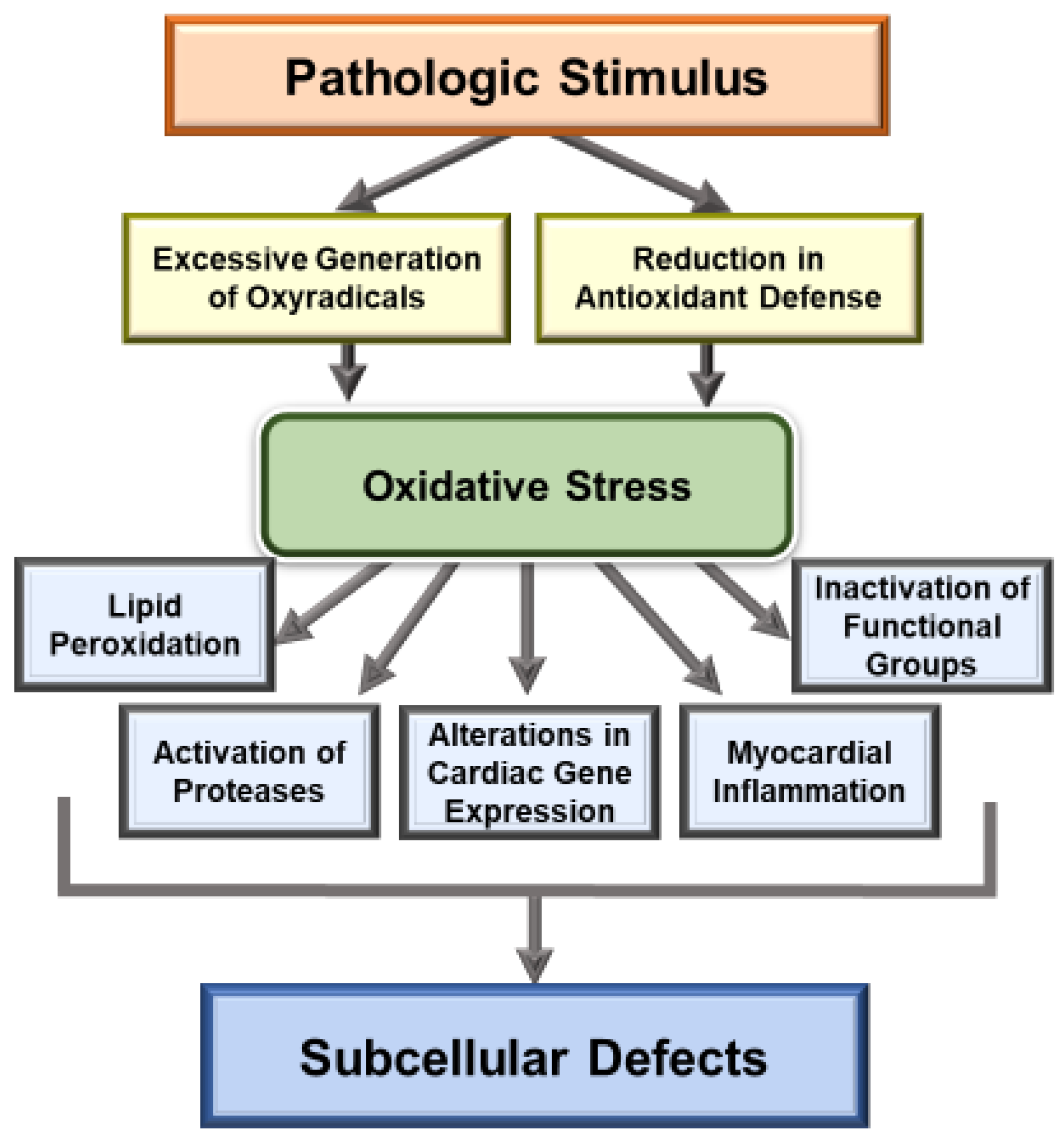
2. Generation of Oxidative Stress in Heart Disease
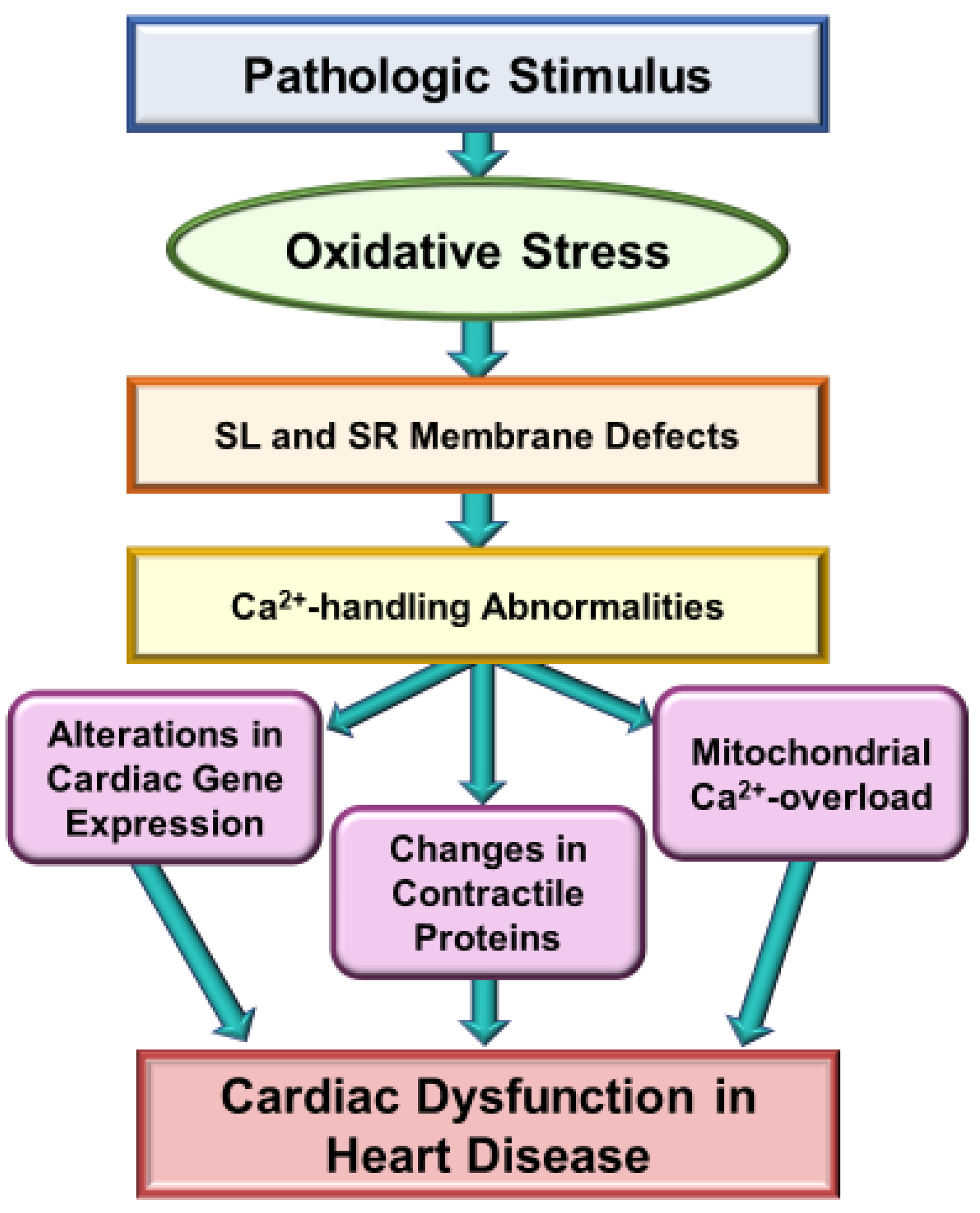
3. Implications of Oxidative Stress in Heart Disease
4. Evidence for the Direct Action of Oxidative Stress on Subcellular Organelles
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Zou, Y.; Hasegawa, H.; Akazawa, H.; Nagai, T.; Komuro, I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: Involvement of ROS in heart diseases. Antioxid. Redox. Signal 2003, 5, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Molavi, B.; Mehta, J.L. Oxidative stress in cardiovascular disease: Molecular basis of its deleterious effects, its detection, and therapeutic considerations. Curr. Opin. Cardiol. 2004, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kusano, K.F.; Matsubara, H.; Nakamura, Y.; Miura, A.; Nishii, N.; Banba, K.; Nagase, S.; Miyaji, K.; Morita, H.; et al. Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J. Card. Fail. 2005, 11, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Vijaya Lakshmi, S.V.; Padmaja, G.; Kuppusamy, P.; Kutala, V.K. Oxidative stress in cardiovascular disease. Indian J. Biochem. Biophys. 2009, 46, 421–440. [Google Scholar]
- Hecker, P.A.; Galvao, T.F.; O’Shea, K.M.; Brown, B.H.; Henderson Jr, R.; Riggle, H.; Gupte, S.A.; Stanley, W. High-sugar intake does not exacerbate metabolic abnormalities or cardiac dysfunction in genetic cardiomyopathy. Nutrition 2012, 28, 520–526. [Google Scholar] [CrossRef][Green Version]
- Munzel, T.; Gori, T.; Keaney Jr, J.F.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef]
- Munzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of oxidative stress on the heart and vasculature: Part 2 of a 3-part series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Steinhorn, B.; Sorrentino, A.; Badole, S.; Bogdanova, Y.; Belousov, V.; Michel, T. Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nat. Commun. 2018, 9, 4044. [Google Scholar] [CrossRef]
- Romuk, E.; Wojciechowska, C.; Jachec, W.; Nowak, J.; Niedziela, J.; Malinowska-Borowska, J.; Glogowska-Gruszka, A.; Birkner, W.; Rozentryt, P. Comparison of oxidative stress parameters in heart failure patients depending on ischaemic or nonischaemic aetiology. Oxid. Med. Cell. Longev. 2019, 2019, 7156038. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, I.; Aimo, A.; Grigoratos, C.; Castiglione, V.; Gentile, F.; Saccaro, L.F.; Arzilli, C.; Cardinale, D.; Passino, C.; Emdin, M. Oxidative stress and inflammation: Determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail. Rev. 2021, 26, 881–890. [Google Scholar] [CrossRef]
- Du, Y.; Demillard, L.J.; Ren, J. Catecholamine-induced cardiotoxicity: A critical element in the pathophysiology of stroke-induced heart injury. Life Sci. 2021, 287, 120106. [Google Scholar] [CrossRef]
- Hafstad, A.D.; Nabeebaccus, A.A.; Shah, A.M. Novel aspects of ROS signaling in heart failure. Basic Res. Cardiol. 2013, 108, 359. [Google Scholar] [CrossRef] [PubMed]
- Dekleva, M.; Celic, V.; Pencic, B.; Ivanovic, A.M.; Caparevic, Z. Left ventricular diastolic dysfunction is related to oxidative stress and exercise capacity in hypertensive patients with preserved systolic function. Cardiology 2007, 108, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Wold, L.E.; Ceylan-Isik, A.F.; Ren, J. Oxidative stress and stress signaling: Menace of diabetic cardiomyopathy. Acta Pharmacol. Sin. 2005, 26, 908–917. [Google Scholar] [CrossRef]
- Kayama, Y.; Raaz, U.; Jagger, A.; Matti, A.; Schellinger, I.N.; Sakamoto, M.; Suzuki, H.; Toyama, K.; Spin, J.M.; Tsao, P.S. Diabetic cardiovascular disease induced by oxidative stress. Int. J. Mol. Sci. 2015, 16, 25234–25263. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Dong, S.; Zhang, A.; Lu, Y.; Li, Y.; Lv, S.; Zhang, J. Role of oxidative stress in cardiotoxicity of antineoplastic drugs. Life Sci. 2019, 232, 116526. [Google Scholar] [CrossRef]
- Sterba, M.; Popelova, O.; Vavrova, A.; Jirkovsky, E.; Kovarikova, P.; Gersl, V.; Simunek, T. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox. Signal 2013, 18, 899–929. [Google Scholar] [CrossRef]
- Kumar, S.; Enjamoori, R.; Jaiswal, A.; Ray, R.; Seth, S.; Maulik, S.K. Catecholamine-induced myocardial fibrosis and oxidative stress is attenuated by Terminalia arjuna (roxb.). J. Pharm. Pharmacol. 2009, 61, 1529–1536. [Google Scholar] [CrossRef]
- Costa, V.M.; Carvalho, F.; Bastos, M.L.; Carvalho, R.A.; Remiao, F. Contribution of catecholamine reactive intermediates and oxidative stress to the pathologic features of heart diseases. Curr. Med. Chem. 2011, 18, 2272–2314. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Iwase, M.; Kanazawa, H.; Ichihara, S.; Ichihara, G.; Nagata, K.; Obata, K.; Kitaichi, K.; Yokoi, T.; Watanabe, M.; et al. Serial alterations of beta-adrenergic signaling in dilated cardiomyopathic hamsters: Possible role of myocardial oxidative stress. Circ. J. 2004, 68, 1051–1060. [Google Scholar] [CrossRef][Green Version]
- Escobales, N.; Crespo, M.J. Angiotensin II-dependent vascular alterations in young cardiomyopathic hamsters: Role for oxidative stress. Vascul. Pharmacol. 2006, 44, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Bruckdorfer, K.R. Antioxidants and CVD. Proc. Nutr. Soc. 2008, 67, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Mehra, N.K.; Swarnakar, N.K. Role of antioxidant for the treatment of cardiovascular diseases: Challenges and opportunities. Curr. Pharm. Des. 2015, 21, 4441–4455. [Google Scholar] [CrossRef]
- Mattera, R.; Benvenuto, M.; Giganti, M.G.; Tresoldi, I.; Pluchinotta, F.R.; Bergante, S.; Tettamanti, G.; Masueilli, L.; Manzari, V.; Modesti, A.; et al. Effects of polyphenols on oxidative stress-mediated injury in cardiomyocytes. Nutrients 2017, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Riba, A.; Deres, L.; Sumegi, B.; Toth, K.; Szabados, E.; Halmosi, R. Cardioprotective effect of resveratrol in a postinfarction heart failure model. Oxid. Med. Cell. Longev. 2017, 2017, 6819281. [Google Scholar] [CrossRef]
- Reyes, D.R.A.; Gomes, M.J.; Rosa, C.M.; Pagan, L.U.; Damatto, F.C.; Damatto, R.L.; Depra, I.; Campos, D.H.; Fernandez, A.A.H.; Martinez, P.F.; et al. N-acetylcysteine influence on oxidative stress and cardiac remodeling in rats during transition from compensated left ventricular hypertrophy to heart failure. Cell Physiol. Biochem. 2017, 44, 2310–2321. [Google Scholar] [CrossRef]
- Bunsawat, K.; Ratchford, S.M.; Alpenglow, J.K.; Park, S.H.; Jarett, C.L.; Stehlik, J.; Drakos, S.G.; Richardson, R.S.; Wray, D.W. Chronic antioxidant administrations restores macrovascular function in patients with heart failure with reduced ejection fraction. Exp. Physiol. 2020, 105, 1384–1395. [Google Scholar] [CrossRef]
- Bartekova, M.; Adameova, A.; Gorbe, A.; Ferenczyova, K.; Pechanova, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef] [PubMed]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schluter, K.D. Specific mechanisms underlying right heart failure: The missing upregulation of superoxide dismutase-2 and its decisive role in antioxidant defense. Antioxid. Redox. Signal 2015, 23, 1220–1232. [Google Scholar] [CrossRef]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Khaper, N.; Kaur, K.; Li, T.; Farahmand, F.; Singal, P.K. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol. Cell. Biochem. 2003, 251, 9–15. [Google Scholar] [CrossRef]
- Yan, Q.; He, B.; Hao, G.; Liu, Z.; Tang, J.; Fu, Q.; Jiang, C.X. KLF9 aggravates ischemic injury in cardiomyocytes through augmenting oxidative stress. Life Sci. 2019, 233, 116641. [Google Scholar] [CrossRef]
- Wang, W.; Li, S.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Horvath, G.; Wang, Q.; Yamamoto, M.; Janicki, J.S.; et al. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J. Mol. Cell. Cardiol. 2014, 72, 305–315. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H928–H939. [Google Scholar] [CrossRef]
- Ungvari, Z.; Gupte, S.A.; Recchia, F.A.; Batkai, S.; Pacher, P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr. Vasc. Pharmacol. 2005, 3, 221–229. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Pacher, P.; Schulz, R.; Liaudet, L.; Szabo, C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol. Sci. 2005, 26, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, M.; Merli, E.; Malagutti, P.; Soukhomovskaia, O.; Cicchitelli, G.; Antelli, A.; Canistro, D.; Francolini, G.; Macri, G.; Mastrorilli, F.; et al. Hydroxyl radical generation, levels of tumor necrosis factor-alpha, and progression to heart failure after acute myocardial infarction. J. Am. Coll. Cardiol. 2004, 43, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Guardigli, G.; Mele, D.; Percoco, G.F.; Ceconi, C.; Curello, S. Oxidative stress during myocardial ischemia and heart failure. Curr. Pharm. Des. 2004, 10, 1699–1711. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, F.; Zhou, H.; Hu, Y.; Guo, D.; Fang, X.; Chen, Y. Interplay of TNF-α, soluble TNF receptors and oxidative stress in coronary chronic total occlusion of the oldest patients with coronary heart disease. Cytokine 2020, 125, 154836. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, E.; Magno, F.; Gnemmi, I.; Carbone, M.; Colombo, M.; La Rocca, G.; Anzalone, R.; Genta, F.T.; Zummo, G.; Di Stefano, A.; et al. Role of oxidative stress and nitrosative stress biomarkers in chronic heart failure. Front. Biosci. 2009, 14, 2230–2237. [Google Scholar] [CrossRef]
- Huet, O.; Dupic, L.; Harrois, A.; Duranteau, J. Oxidative stress and endothelial dysfunction during sepsis. Front. Biosci. 2011, 16, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Davidoff, M.N. Oxidative stress and endothelial dysfunction in heart failure. Congest. Heart Fail. 2002, 8, 165–172. [Google Scholar] [CrossRef]
- Indik, J.H.; Goldman, S.; Gaballa, M.A. Oxidative stress contributes to vascular endothelial dysfunction in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1767–H1770. [Google Scholar] [CrossRef]
- Warnholtz, A.; Wendt, M.; August, M.; Munzel, T. Clinical aspects of reactive oxygen and nitrogen. Biochem. Soc. Symp. 2004, 71, 121–133. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Okonko, D.O.; Shah, A.M. Mitochondrial dysfunction and oxidative stress in CHF. Nat. Rev. Cardiol. 2015, 12, 6–8. [Google Scholar] [CrossRef]
- Anatoliotakis, N.; Deftereos, S.; Bouras, G.; Giannopoulos, G.; Tsounis, D.; Angelidis, C.; Koukis, A.; Stefanadis, C. Myeloperoxidase: Expressing inflammation and oxidative stress in cardiovascular disease. Curr. Top. Med. Chem. 2013, 13, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Jane-Lise, S.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003, 41, 2164–2171. [Google Scholar] [CrossRef]
- Lauderic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef]
- Sawyer, D.B.; Colucci, W.S. Mitochondrial oxidative stress in heart failure. Circ. Res. 2000, 86, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Bayeva, M.; Ardehali, H. Mitochondrial dysfunction and oxidative damage to sarcomeric proteins. Curr. Hypertens. Rep. 2010, 12, 426–432. [Google Scholar] [CrossRef]
- Akhmendov, A.T.; Rybin, V.; Marin-Garcia, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail. Rev. 2015, 20, 227–249. [Google Scholar] [CrossRef]
- Nickel, A.; Kohlhass, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef]
- Tsuhima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational medicine modifications of AKAP121, DRP1, and OAP1 that promote mitochondrial fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintron, M.; Chen, T.; Marcinek, D.J.; Dorn 2nd, G.W.; Kang, Y.J.; Parolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Kang, D.; Hamasaki, N. Mitochondrial oxidative stress and mitochondrial DNA. Clin. Chem. Lab. Med. 2003, 41, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ. J. 2008, 72 (Suppl. A), A31–A37. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H. Mitochondrial oxidative stress and heart failure. Intern. Med. 2006, 45, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2009, 81, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef]
- Harrison, D.G.; Cai, H.; Landmesser, U.; Griendling, K.K. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 2003, 4, 51–61. [Google Scholar] [CrossRef]
- Cave, A.; Grieve, D.; Johar, S.; Zhang, M.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2327–2334. [Google Scholar] [CrossRef]
- Sorescu, D.; Griendling, K.K. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest. Heart Fail. 2002, 8, 132–140. [Google Scholar] [CrossRef]
- Nediani, C.; Borchi, E.; Giordano, C.; Baruzzo, S.; Pnziani, V.; Sebastiani, M.; Nassi, P.; Mugelli, A.; d’Amati, G.; Cerbai, E. NADPH oxidase-dependent redox signalling in human heart failure: Relationship between the left and right ventricle. J. Moll. Cell. Cardiol. 2007, 42, 826–834. [Google Scholar] [CrossRef]
- Nabeebaccus, A.; Zhang, M.; Shah, A.M. NADPH oxidase and cardiac remodeling. Heart Fail. Rev. 2011, 16, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kuroda, J.; Matsushima, S.; Ago, T.; Sadoshima, J. Regulation of myocardial growth and death by NADPH oxidase. J. Moll. Cell. Cardiol. 2011, 50, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Sadoshima, J. NADPH oxidase and cardiac failure. J. Cardiovasc. Transl. Res. 2010, 3, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Barca, E.; Subramnyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NADPH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS ONE 2016, 11, e145750. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.D.; Canugovi, C.; Vendrov, A.E.; Hayami, T.; Bowles, D.E.; Krause, K.H.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates inflammation in ischemic heart failure: Role of soluble epoxide hydrolase. Antioxid. Redox. Signal 2019, 31, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matshushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (NOX4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef]
- Qin, F. Inhibition of NADPH oxidase reduce myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free Radic. Biol. Med. 2007, 43, 271–281. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, H.; Xia, W.; Tang, Y.; Li, H.; Huang, C. NADPH oxidase inhibition ameliorates cardiac dysfunction in rabbits with heart failure. Mol. Cell. Biochem. 2010, 343, 143–153. [Google Scholar] [CrossRef]
- Kaludercic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef]
- Bianchi, P.; Pimentel, D.R.; Murphy, M.P.; Colucci, W.S.; Parini, A. A new hypertrophic mechanism of serotonin in cardiac myocytes: Receptor-independent ROS generation. FASEB J. 2005, 19, 641–643. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Nueral. Transm. 2018, 125, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutuar, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’h, F.; et al. Oxidative stress by monoamine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid. Redox. Signal 2016, 25, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Manni, E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: An intriguing therapeutic target. Oxid. Med. Cell. Longev. 2016, 2016, 4375418. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Laim, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine oxidase A–mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Duicu, O.M.; Lighezan, R.; Sturza, A.; Ceausu, R.A.; Borza, C.; Vaduva, A.; Noveanu, L.; Gaspar, M.; Ionac, A.; Feier, H.; et al. Monoamine oxidase as potential contributors to oxidative stress in diabetes: Time for study in patients undergoing heart surgery. Biomed. Res. Int. 2015, 2015, 515437. [Google Scholar] [CrossRef] [PubMed]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tian, J.; Sun, Y.; Xu, T.R.; Chi, R.F.; Zhang, X.L.; Zhang, Y.A.; Qin, F.Z.; Zhang, W.F. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochim. Biophys. Acta 2015, 1852, 805–815. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Adameova, A.; Shah, A.K.; Dhalla, N.S. Role of oxidative stress in the genesis of ventricular arrhythmias. Int. J. Mol. Sci. 2020, 21, 4200. [Google Scholar] [CrossRef]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Moll. Cell. Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Houser, S.R. Abnormal Ca2+ cycling in failing ventricular myocytes: Role of NOS1-mediated nitroso-redox balance. Antioxid. Redox. Signal. 2014, 21, 2044–2059. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Dudley Jr, S.C. Heart failure, oxidative stress, and ion channel modulation. Congest. Heart Fail. 2002, 8, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.; Wen, Y.; Hegemann, N.; Primessnig, U.; Parwani, A.; Boldt, L.H.; Pieske, B.M.; Heinzel, F.R.; Hohendanner, F. Oxidative stress and inflammatory modulation of Ca2+ handling in metabolic HFpEF-related left atrial cardiomyopathy. Antioxidants 2020, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Yan, C.; Patel, R.; Liu, W.; Dong, E. Vitamins C and E attenuates apoptosis, β-adrenergic receptor desensitization, and sarcoplasmic reticular Ca2+ ATPase downregulation after myocardial infarction. Free Radic. Biol. Med. 2006, 40, 1827–1842. [Google Scholar] [CrossRef]
- Liu, W.; Chen, P.; Deng, J.; Lv, J.; Liu, J. Resveratrol and polydatin as modulators of Ca2+ mobilization in the cardiovascular system. Ann. New York Acad. Sci. 2017, 1403, 82–91. [Google Scholar] [CrossRef]
- Lacerda, D.; Turck, P.; Campos-Carraro, C.; Hickmann, A.; Ortiz, V.; Bianchi, S.; Bello-Klein, A.; de Castro, A.L.; Bassani, V.L.; da Rosa Araujo, A.S. Pterostilbene improves cardiac function in a rat model of right heart failure through modulation of calcium handling proteins and oxidative stress. Appl. Physiol. Nutr. Metab. 2020, 45, 987–995. [Google Scholar] [CrossRef]
- Wu, T.; Yao, H.; Zhang, B.; Zhou, S.; Hou, P.; Chen, K. κ Opioid receptor agonist inhibits myocardial injury in heart failure rats through activating Nrf2/HO-1 pathway and regulating Ca2+-SERCA2a. Oxid. Med. Cell. Longev. 2021, 2021, 7328437. [Google Scholar] [CrossRef]
- Luo, J.; Xuan, Y.T.; Gu, Y.; Prabhu, S.D. Prolonged oxidative stress inverts the cardiac force-frequency relation: Role of altered calcium handling and myofilament calcium responsiveness. J. Mol. Cell. Cardiol. 2006, 40, 64–75. [Google Scholar] [CrossRef]
- Ahmed, M.; Gladden, J.D.; Litovsky, S.H.; Llyod, S.G.; Gupta, H.; Inusah, S.; Denny, T., Jr.; Powell, P.; McGiffin, D.C.; Dell’ltalia, L.J. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction >60%. J. Am. Coll. Cardiol. 2010, 55, 671–679. [Google Scholar] [CrossRef]
- Canton, M.; Menazza, S.; Sheeran, F.L.; de Laureto, P.P.; Di Lisa, F.; Pepe, S. Oxidation of myofibrillar proteins in human heart. J. Am. Coll. Cardiol. 2011, 57, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.G.; Ravi, R.; Stull, L.B.; Murphy, A.M. Chronic xanthine oxidase inhibition prevents myofibrillar protein oxidation and preserves cardiac function in a transgenic mouse model of cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1512–H1518. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Ren, B.; Elimban, V.; Tappia, P.S.; Takeda, N.; Dhalla, N.S. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger in heart failure by blockade of renin-angiotensin system. Am. J. Heart Physiol. Circ. Physiol. 2005, 288, H2637–H2646. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L. Inhibiting Na+-K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS ONE 2014, 9, e93928. [Google Scholar] [CrossRef]
- Kaneko, M.; Panagia, V.; Paolillo, G.; Majumder, S.; Ou, C.; Dhalla, N.S. Inhibiton of cardiac phosphatidylethanolamine N-methylation by oxygen free radicals. Biochim. Biophys. Acta 1990, 1021, 33–38. [Google Scholar] [CrossRef]
- Tappia, P.S.; Dent, M.R.; Dhalla, N.S. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic. Biol. Med. 2006, 41, 349–361. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Maack, C. Interplay of defective excitation-contraction coupling, energy starvation, and oxidative stress in heart failure. Trends. Cardiovasc. Med. 2011, 21, 69–73. [Google Scholar] [CrossRef]
- Nojiri, H.; Shimizu, T.; Funakoshi, M.; Yamaguchi, O.; Zhou, H.; Kawakami, S.; Ohta, Y.; Sami, M.; Ishikawa, H.; Kurosawa, H.; et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J. Biol. Chem. 2006, 281, 33789–33801. [Google Scholar] [CrossRef]
- Shaheen, M.; Cheema, Y.; Shahbaz, A.U.; Bhattacharya, S.K.; Weber, K.T. Intracellular calcium overloading and oxidative stress in cardiomyocyte necrosis via a mitochondriocentric signal-transducer-effector pathway. Exp. Clin. Cardiol. 2011, 16, 109–115. [Google Scholar]
- Dietl, A.; Maack, C. Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef]
- Cortassa, S.; Juhaszova, M.; Aon, M.A.; Zorov, D.B.; Sollott, S.J. Mitochondrial Ca2+, redox environment and ROS emission in heart failure: Two sides of the same coin? J. Mol. Cell. Cardiol. 2021, 151, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Pavez-Giani, M.G.; Sanchez-Aguilera, P.I.; Bomer, n.; Miyamota, S.; Booij, H.G.; Giraldo, P.; Oberdorf-Maass, S.U.; Nijholt, K.T.; Yurista, S.R.; Milting, H.; et al. ATPase inhibitory factor-1 disrputs mitochondrial Ca2+ handling and promotes pathological cardiac hypertrophy through CaMKIIδ. Int. J. Mol. Sci. 2021, 22, 4427. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Nickel, A.; Kohlhaas, M.; Hohl, M.; Sequeira, V.; Brune, C.; Schwemmlein, J.; Abeber, M.; Schuh, K.; Kutschka, I.; et al. Loss of mitochondrial Ca2+ uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in Barth syndrome cardiomyopathy. Circulation 2021, 144, 1694–1713. [Google Scholar] [CrossRef] [PubMed]
- Donoso, P.; Sanchez, G.; Bull, R.; Hidalgo, C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front. Biosci. 2011, 16, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Tokuhisa, T.; Yano, M.; Obayashi, M.; Noma, T.; Mochizuki, M.; Oda, T.; Okuda, S.; Doi, M.; Liu, J.; Ikeda, Y.; et al. AT1 receptor antagonist restores cardiac ryanodine receptor function, rendering isoproterenol-induced failing heart less susceptible to Ca2+-leak induced by oxidative stress. Circ. J. 2006, 70, 777–786. [Google Scholar] [CrossRef]
- Terentyev, D.; Gyorke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; de Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef]
- Yano, M.; Okuda, S.; Oda, T.; Tokuhisa, T.; Tateishi, H.; Mochizuki, M.; Noma, T.; Doi, M.; Kobayashi, S.; Yamamoto, T.; et al. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation 2005, 112, 3633–3643. [Google Scholar] [CrossRef]
- Koitabashi, N.; Arai, M.; Tomaru, K.; Takizawa, T.; Watanabe, A.; Niwano, K.; Yokoyama, T.; Wuytack, F.; Periasamy, M.; Nagai, R.; et al. Carvedilol effectively blocks oxidative stress-mediated downregulation of sarcoplasmic reticulum Ca2+-ATPase 2 gene transcription through modification of SP1 binding. Biochem. Biophys. Res. Commun. 2005, 328, 116–124. [Google Scholar] [CrossRef]
- Kaneko, M.; Elimban, V.; Dhalla, N.S. Mechanism for depression of heart sarcolemmal Ca2+ pump by oxygen free radicals. Am. J. Physiol. 1989, 257, H804–H811. [Google Scholar] [CrossRef]
- Kaneko, M.; Beamish, R.E.; Dhalla, N.S. Depression of heart sarcolemmal Ca2+-pump activity by oxygen free radicals. Am. J. Physiol. 1989, 256, H368–H374. [Google Scholar] [CrossRef]
- Kaneko, M.; Lee, S.L.; Wolf, C.M.; Dhalla, N.S. Reduction of calcium channel antagonist binding sites by oxygen free radicals in rat heart. J. Mol. Cell. Cardiol. 1989, 21, 935–943. [Google Scholar] [CrossRef]
- Kaneko, M.; Singal, P.K.; Dhalla, N.S. Alterations in heart sarcolemmal Ca2+ ATPase and Ca2+-binding activities due to oxygen free radicals. Basic Res. Cardiol. 1990, 85, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Relationship between mechanical dysfunction and depression of sarcolemmal Ca2+-pump activity in hearts perfused with oxygen free radicals. Mol. Cell. Biochem. 1996, 160–161, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Mochizuki, S.; Saini, H.K.; Elimban, V.; Dhalla, N.S. Modification of alterations in cardiac function and sarcoplasmic reticulum by vanadate in ischemic-reperfused rat hearts. J. Appl. Physiol. 2005, 99, 999–1005. [Google Scholar] [CrossRef]
- Suzuki, S.; Kaneko, M.; Chapman, D.C.; Dhalla, N.S. Alterations in cardiac contractile proteins due to oxygen free radicals. Biochim. Biophys. Acta 1991, 1074, 95–100. [Google Scholar] [CrossRef]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondria function in ischemic-reperfused hearts. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef]
- Kato, K.; Shao, Q.; Elimban, V. Mechanism of depression in cardiac sarcolemmal Na+-K+ ATPase by hypochlorous acid. Am. J. Physiol. 1998, 275, C826–C831. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parameters | Control | X + XO | 0.5 mM H2O2 | 0.1 mM H2O2 + 0.05 mM Fe2+ |
|---|---|---|---|---|
| Na+-K+ ATPase (µmol Pi/mg/h) | 14.27 ± 1.07 | 6.81 ± 1.03 * | 8.64 ± 1.04 * | 6.98 ± 0.13 * |
| Na+-Ca2+ exchange (nmol/mg/2 s) | 4.59 ± 0.08 | 1.97 ± 0.13 * | 2.97 ± 0.11 * | 3.02 ± 0.14 * |
| MDA content (nmol/mg protein) | 61.67 ± 3.67 | 81.85 ± 2.54 * | 78.77 ± 3.23 * | 88.26 ± 3.07 * |
| SH-group content (nmol/mg protein) | 72.61 ± 2.37 | 37.59 ± 4.41 * | 42.36 ± 2.75 * | 40.86 ± 3.54 * |
| Parameters | ATP-Dependent Ca2+ Accumulation (nmol Ca2+/mg/5 min) | Mg2+ ATPase (µmol/mg/h) | Ca2+-Stimulated ATPase (µmol/mg/h) |
|---|---|---|---|
| Control | 27.0 ± 1.7 | 195 ± 3 | 13.6 ± 0.7 |
| X + XO treated | 9.5 ± 0.8 * | 176 ± 2 * | 2.6 ± 0.5 * |
| X + XO + 80 µg/mL SOD | 21.8 ± 0.8 † | 192 ± 2 † | 10.1 ± 0.4 † |
| 0.5 mM H2O2 treated | 4.7 ± 1.3 * | 165 ± 6 * | 2.9 ± 0.4 * |
| 0.5 mM H2O2 + 10 µg/mL catalase | 20.6 ± 1.1 † | 190 ± 5 † | 8.4 ± 0.3 † |
| 0.1 mM H2O2 + 0.2 mM Fe2+ treated | 6.7 ± 0.5 * | 169 ± 4 * | 4.0 ± 0.3 * |
| 0.1 mM H2O2 + 0.2 mM Fe2+ + 20 mM mannitol | 17.2 ± 0.8 † | 184 ± 2 † | 8.0 ± 0.5 † |
| Parameters | Control | X+XO | 1 mM H2O2 | 0.1 mM H2O2 + 0.2 mM Fe2+ |
|---|---|---|---|---|
| A. Ca2+-channel binding | ||||
| Kd (nM) | 0.231 ± 0.011 | 0.252 ± 0.011 | 0.254 ± 0.018 | 0.267 ± 0.017 |
| Bmax (fmol/mg) | 199 ± 12 | 139 ± 7.0 * | 142 ± 8.0 * | 157 ± 9.0 * |
| B. ATP-binding | ||||
| Low affinity (1.25 mM Ca2+) | 97.8 ± 4.3 | 147.2 ± 6.1 * | 141.3 ± 5.4 * | 41.4 ± 4.9 * |
| High affinity (50 µM Ca2+) | 7.95 ± 0.32 | 12.08 ± 0.68 * | 13.92 ± 0.66 * | 4.08 ± 0.24 * |
| C. Ca2+-ecto ATPase | ||||
| (µmol Pi/mg/h) | 44.3 ± 1.1 | 57.7 ± 1.4 * | 57.0 ± 1.2 * | 31. 4 ± 1.3 * |
| Parameters | Control | X+XO | 1 mM H2O2 |
|---|---|---|---|
| A. Sarcoplasmic reticulum: | |||
| Ca2+-release (nmol Ca2+/mg/15 s) | 8.5 ± 1.4 | 4.2 ± 0.8 * | 3.9 ± 0.7 * |
| Ca2+-uptake (nmol Ca2+/mg/min) | 29.6 ± 2.4 | 15.9 ± 1.6 * | 12.7 ± 1.5 * |
| Ca2+-pump ATPase (µmol Pi/mg/h) | 14.7 ± 1.3 | 6.4 ± 0.8 * | 5.7 ± 0.9 * |
| B. Myofibrils: | |||
| Mg2+-ATPase (µmol/mg/h) | 2.53 ± 0.13 | 4.97 ± 0.16 * | 5.46 ± 0.18 * |
| Ca2+-stimulated ATPase (µmol/mg/h) | 10.29 ± 0.17 | 6.48 ± 0.18 * | 5.92 ± 0.38 * |
| Sulfhydryl group content (nmol/mg protein) | 67.0 ± 1.3 | 54.2 ± 1.6 * | 47.6 ± 2.06 * |
| C. Mitochondria | |||
| State 3 respiration (O/mg/min) | 293 ± 7.0 | 138 ± 7.0 * | 106 ± 4.0 * |
| RCI (State 3 to state 4 ratio) | 5.36 ± 0.13 | 2.66 ± 0.23 * | 1.89 ± 0.07 * |
| ADP to O ratio (nmol ADP/ng atom O) | 3.00 ± 0.15 | 2.55 ± 0.07 * | 2.37 ± 0.03 * |
| Parameters | Control | 0.1 mM HOCl | HOCl Plus 10 mM L-methionine |
|---|---|---|---|
| A. Sarcolemma: | |||
| Na+-K+ ATPase (µmol Pi/mg/h) | 18.86 ± 2.03 | 2.16 ± 1.05 * | 13.31 ± 2.44 † |
| MDA content (nmol/mg protein) | 51.64 ± 3.97 | 67.33 ± 3.97 * | 48.2 ± 3.59 † |
| Sulfhydryl group content (nmol/mg protein) | 64.84 ± 6.36 | 28.67 ± 4.40 * | 55.86 ± 5.72 † |
| B. Myofibrill ATPase (µmol/mg/h) | |||
| Mg2+ ATPase | 2.80 ± 0.12 | 9.51 ± 0.16 * | 3.79 ± 0.16 † |
| Ca2+-stimulated ATPase | 10.96 ± 0.15 | 5.73 ± 0.31 * | 11.64 ± 0.12 † |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines 2022, 10, 393. https://doi.org/10.3390/biomedicines10020393
Dhalla NS, Elimban V, Bartekova M, Adameova A. Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines. 2022; 10(2):393. https://doi.org/10.3390/biomedicines10020393
Chicago/Turabian StyleDhalla, Naranjan S., Vijayan Elimban, Monika Bartekova, and Adriana Adameova. 2022. "Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease" Biomedicines 10, no. 2: 393. https://doi.org/10.3390/biomedicines10020393
APA StyleDhalla, N. S., Elimban, V., Bartekova, M., & Adameova, A. (2022). Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines, 10(2), 393. https://doi.org/10.3390/biomedicines10020393