
Glomerular Biomechanical Stress and Lipid Mediators during Cellular Changes Leading to Chronic Kidney Disease



Abstract
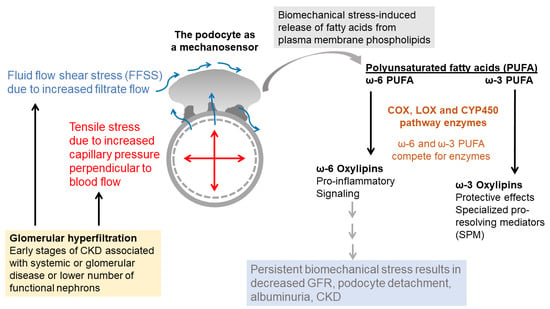
:
1. Introduction
1.1. Glomerular Hyperfiltration Is an Early Response That May Turn Maladaptive
1.2. Glomerular Hyperfiltration Is Associated with Several Pathophysiological Etiologies
1.3. Glomerular Hyperfiltration Is a Potential Predictor of CKD and Cardiovascular Disease
1.4. Glomerular Hyperfiltration Precedes Tissue Fibrosis and Organ Failure
1.5. Outline of the Article
2. Hyperfiltration and Biomechanical Forces
2.1. Fluid Flow Shear Stress (FFSS)
2.1.1. The Glomerular Filtration Barrier Function
2.1.2. Unilateral Nephrectomy in Rodent Models of Hyperfiltration Increases Single-Nephron Glomerular Filtration Rate (SNGFR)
2.1.3. FFSS Mediates the Early Effects of Hyperfiltration
2.2. Tensile Stress
Tensile Stress Alters the Actin Cytoskeleton Organization, Cell Adhesion, and Gene Expression of Podocytes
3. Tubulocentric and Podocentric Effects of Hyperfiltration
3.1. Tubular Function and Glomerular Hyperfiltration
3.2. Podocytes and Glomerular Hyperfiltration
Glomerular Hyperfiltration Results in Podocytes Loss
4. The Plasma Membrane as the Cellular Point of Contact with Biomechanical Forces
4.1. The Plasma Membrane Functions as a Sensor of Mechanical Stress
4.2. Membrane Lipid-Bound Fatty Acids Are Precursors of Signaling Mediators
4.3. Phospholipases Release Fatty Acids from Membrane Phospholipids
Mechanical Stress Activates Phospholipases
5. Arachidonic Acid (ω-6 PUFA) Generates Lipid Mediators through the Cyclooxygenase, Lipoxygenase and Cyto-Chrome P450 Pathways
5.1. Cyclooxygenases Catalyze the Conversion of Arachidonic Acid to Prostaglandins and Thromboxane
5.1.1. COX2 Expression Is Upregulated in Podocytes Exposed to FFSS
5.1.2. Arachidonic Acid Metabolite Prostaglandin E2 (PGE2) Is a Major Mediator of Biomechanical Stress
5.1.3. Arachidonic Acid Metabolite Prostacyclin (PGI2) and Biomechanical Stress
5.1.4. Thromboxane A2 (TXA2) Is Associated with Inflammation during Hyperfiltration
5.2. Lipoxygenases Catalyze the Conversion of Arachidonic Acid to Leukotrienes (LT) and Hydroxyeicosatetraenoic Acids (HETE)
5.2.1. Leukotrienes Mediate Glomerular Injury
5.2.2. Urinary Leukotriene Metabolites Indicate Tubular Injury
5.3. Cytochrome P450 Enzymes Catalyze the Conversion of Arachidonic Acid to Hydroxy- and Epoxy-Oxylipins
5.3.1. 20-Hydroxyeicosatetraenoic Acid (20-HETE)
5.3.2. Epoxyeicosatrienoic Acids (EETs)
6. EPA and DHA (ω-3 PUFA) Generate Lipid Mediators through the Cyclooxygenase, Lipoxygenase and Cytochrome P450 Pathways
6.1. Protective Effects of ω-3 PUFA against Glomerular and Kidney Injury
6.2. Protective Effects of ω-3 PUFA Metabolites
7. Both ω-6 and ω-3 PUFA Yield Endocannabinoids
8. Both ω-6 and ω-3 PUFA Generate Specialized Pro-Resolving Mediators (SPM)
9. Lipids and Fatty Acids as Biomarkers of Hyperfiltration-The Early Stage of Renal Dysfunction
10. Current and Evolving Treatments to Modulate Hyperfiltration
10.1. The Renin-Angiotensin-Aldosterone System (RAAS)
10.2. Sodium–Glucose Transport Protein 2 (SGLT2) Inhibitors
10.3. Agonists and Antagonists of Prostaglandin E2 Receptors EP2, EP4
10.4. Compounds to Target the Cytochrome P450 Pathway
10.5. Novel Agonists of Peroxisome Proliferator-Activated Receptors (PPARs)
10.6. Lifestyle and Dietary Changes, Low Protein Diets, Plant-Based Diets, ω-3 PUFA-Rich Diets
10.6.1. Plant-Based Low-Protein Diets
10.6.2. Dietary ω-3 PUFA
10.7. Flavonoids
10.8. Novel Compounds to Target the Endocannabinoid System (ECS)
10.9. Other Drugs and Novel Biologicals
11. Summary, Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Available online: https://www.kidney.org/sites/default/files/docs/11-10-1813_abe_patbro_gfr_b.pdf (accessed on 1 October 2021).
- Delanaye, P.; Schaeffner, E.; Ebert, N.; Cavalier, E.; Mariat, C.; Krzesinski, J.M.; Moranne, O. Normal reference values for glomerular filtration rate: What do we really know? Nephrol. Dial. Transplant. 2012, 27, 2664–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cachat, F.; Combescure, C.; Cauderay, M.; Girardin, E.; Chehade, H. A systematic review of glomerular hyperfiltration assessment and definition in the medical literature. Clin. J. Am. Soc. Nephrol. 2015, 6, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E.; Gao, X.; Bebu, I.; de Boer, I.H.; Lachin, J.; Paterson, A.; Perkins, B.; Saenger, A.K.; Steffes, M.; Zinman, B. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Research Group. Early Glomerular Hyperfiltration and Long-Term Kidney Outcomes in Type 1 Diabetes: The DCCT/EDIC Experience. Clin. J. Am. Soc. Nephrol. 2019, 14, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Porrini, E.L.; Gaspari, F.; Motterlini, N.; Cannata, A.; Carrara, F.; Cella, C.; Ferrari, S.; Stucchi, N.; Parvanova, A.; et al. Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care 2012, 35, 2061–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, K.P. Mechanisms of renal vasodilation and hyperfiltration during pregnancy. J. Soc. Gynecol. Investig. 2004, 11, 438–448. [Google Scholar] [CrossRef]
- Hussein, W.; Lafayette, R.A. Renal function in normal and disordered pregnancy. Curr. Opin. Nephrol. Hypertens. 2014, 23, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Frank, H.; Graf, J.; Amann-Gassner, U.; Bratke, R.; Daniel, H.; Heemann, U.; Hauner, H. Effect of short-term high-protein compared with normal-protein diets on renal hemodynamics and associated variables in healthy young men. Am. J. Clin. Nutr. 2009, 90, 1509–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwingshackl, L.; Hoffmann, G. Comparison of high vs. normal/low protein diets on renal function in subjects without chronic kidney disease: A systematic review and meta-analysis. PLoS ONE 2014, 9, e97656. [Google Scholar] [CrossRef] [Green Version]
- Bankir, L.; Roussel, R.; Bouby, N. Protein- and diabetes-induced glomerular hyperfiltration: Role of glucagon, vasopressin, and urea. Am. J. Physiol. Renal. Physiol. 2015, 309, F2–F23. [Google Scholar] [CrossRef] [Green Version]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Bosma, R.J.; Krikken, J.A.; Homan van der Heide, J.J.; de Jong, P.E.; Navis, G.J. Obesity and renal hemodynamics. Contrib. Nephrol. 2006, 151, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Delanaye, P.; Weekers, L.; Dubois, B.E.; Cavalier, E.; Detry, O.; Squifflet, J.P.; Krzesinski, J.M. Outcome of the living kidney donor. Nephrol. Dial. Transplant. 2012, 27, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blantz, R.C.; Steiner, R.W. Benign hyperfiltration after living kidney donation. J. Clin. Investig. 2015, 125, 972–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, T.; Thiagarajan, G.; Alon, U.S.; Sharma, R.; El-Meanawy, A.; McCarthy, E.T.; Savin, V.J.; Sharma, M. Role of biomechanical forces in hyperfiltration-mediated glomerular injury in congenital anomalies of the kidney and urinary tract. Nephrol. Dial. Transplant. 2017, 32, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, T.; Hariharan, S.; Alon, U.S.; McCarthy, E.T.; Sharma, R.; El-Meanawy, A.; Savin, V.J.; Sharma, M. Hyperfiltration-mediated Injury in the Remaining Kidney of a Transplant Donor. Transplantation 2018, 102, 1624–1635. [Google Scholar] [CrossRef]
- Bosch, J.P.; Saccaggi, A.; Lauer, A.; Ronco, C.; Belledonne, M.; Glabman, S. Renal functional reserve in humans. Effect of protein intake on glomerular filtration rate. Am. J. Med. 1983, 75, 943–950. [Google Scholar] [CrossRef]
- Lin, J.; Hu, F.B.; Curhan, G.C. Associations of diet with albuminuria and kidney function decline. Clin. J. Am. Soc. Nephrol. 2010, 5, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Kalantar-Zadeh, K.; Kramer, H.M.; Fouque, D. High-protein diet is bad for kidney health: Unleashing the taboo. Nephrol. Dial. Transplant. 2020, 35, 1–4. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/nchs/fastats/obesity-overweight.htm (accessed on 1 October 2021).
- Sasson, A.N.; Cherney, D.Z. Renal hyperfiltration related to diabetes mellitus and obesity in human disease. World J. Diabetes 2012, 3, 1–6. [Google Scholar] [CrossRef]
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular hyperfiltration: Definitions, mechanisms and clinical implications. Nat. Rev. Nephrol. 2012, 8, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Mascali, A.; Franzese, O.; Nisticò, S.; Campia, U.; Lauro, D.; Cardillo, C.; Di Daniele, N.; Tesauro, M. Obesity and kidney disease: Beyond the hyperfiltration. Int. J. Immunopathol. Pharmacol. 2016, 29, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingerman, B.; Herman-Edelstein, M.; Erman, A.; Bar Sheshet Itach, S.; Ori, Y.; Rozen-Zvi, B.; Gafter, U.; Chagnac, A. Effect of Acetazolamide on Obesity-Induced Glomerular Hyperfiltration: A Randomized Controlled Trial. PLoS ONE 2015, 10, e0137163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabayashi, Y.; Tsuboi, N.; Sasaki, T.; Haruhara, K.; Kanzaki, G.; Koike, K.; Miyazaki, Y.; Kawamura, T.; Ogura, M.; Yokoo, T. Glomerulopathy Associated with Moderate Obesity. Kidney Int. Rep. 2016, 1, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Chagnac, A.; Zingerman, B.; Rozen-Zvi, B.; Herman-Edelstein, M. Consequences of Glomerular Hyperfiltration: The Role of Physical Forces in the Pathogenesis of Chronic Kidney Disease in Diabetes and Obesity. Nephron 2019, 143, 38–42. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Edwards, A.; Christensen, E.I.; Unwin, R.J.; Norden, A.G.W. Obesity-Related Glomerulopathy: Hyperfiltration May Contribute to Early Proteinuria. Kidney Int. Rep. 2021, 6, 867. [Google Scholar] [CrossRef]
- Mogensen, C.E. Glomerular filtration rate and renal plasma flow in short-term and long-term juvenile diabetes mellitus. Scand. J. Clin. Lab. Investig. 1971, 28, 91–100. [Google Scholar] [CrossRef]
- Hostetter, T.H.; Troy, J.L.; Brenner, B.M. Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int. 1981, 19, 410–415. [Google Scholar] [CrossRef] [Green Version]
- Kreisberg, J.I.; Patel, P.Y. The effects of insulin, glucose and diabetes on prostaglandin production by rat kidney glomeruli and cultured glomerular mesangial cells. Prostaglandins Leukot. Med. 1983, 11, 431–442. [Google Scholar] [CrossRef]
- Christiansen, J.S.; Gammelgaard, J.; Frandsen, M.; Parving, H.H. Increased kidney size, glomerular filtration rate and renal plasma flow in short-term insulin-dependent diabetics. Diabetologia 1981, 20, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Quilley, J.; McGiff, J.C. Arachidonic acid metabolism and urinary excretion of prostaglandins and thromboxane in rats with experimental diabetes mellitus. J. Pharmacol. Exp. Ther. 1985, 234, 211–216. [Google Scholar] [PubMed]
- Nelson, R.G.; Bennett, P.H.; Beck, G.J.; Tan, M.; Knowler, W.C.; Mitch, W.E.; Hirschman, G.H.; Myers, B.D. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N. Engl. J. Med. 1996, 335, 1636–1642. [Google Scholar] [CrossRef]
- Komers, R.; Lindsley, J.N.; Oyama, T.T.; Schutzer, W.E.; Reed, J.F.; Mader, S.L.; Anderson, S. Immunohistochemical and functional correlations of renal cyclooxygenase-2 in experimental diabetes. J. Clin. Investig. 2001, 107, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Li, J.; Quilley, J. Deficient renal 20-HETE release in the diabetic rat is not the result of oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2305–H2312. [Google Scholar] [CrossRef] [Green Version]
- Magee, G.M.; Bilous, R.W.; Cardwell, C.R.; Hunter, S.J.; Kee, F.; Fogarty, D.G. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 2009, 52, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.K.; Maahs, D.M.; Perkins, B.A.; Cherney, D.Z. Renal hyperfiltration and systemic blood pressure in patients with uncomplicated type 1 diabetes mellitus. PLoS ONE 2013, 8, e68908. [Google Scholar] [CrossRef] [Green Version]
- Mora-Fernandez, C.; Domínguez-Pimentel, V.; de Fuentes, M.M.; Górriz, J.L.; Martínez-Castelao, A.; Navarro-González, J.F. Diabetic kidney disease: From physiology to therapeutics. J. Physiol. 2014, 592, 3997–4012. [Google Scholar] [CrossRef]
- Trevisan, R.; Dodesini, A.R. The Hyperfiltering Kidney in Diabetes. Nephron 2017, 136, 277–280. [Google Scholar] [CrossRef]
- Tuttle, K.R. Back to the Future: Glomerular Hyperfiltration and the Diabetic Kidney. Diabetes 2017, 66, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Tonneijck, L.; Muskiet, M.H.; Smits, M.M.; van Bommel, E.J.; Heerspink, H.J.; van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef] [Green Version]
- Penno, G.; Orsi, E.; Solini, A.; Bonora, E.; Fondelli, C.; Trevisan, R.; Vedovato, M.; Cavalot, F.; Gruden, G.; Laviola, L.; et al. Renal Insufficiency And Cardiovascular Events (RIACE) Study Group. Renal hyperfiltration is independently associated with increased all-cause mortality in individuals with type 2 diabetes: A prospective cohort study. BMJ Open Diabetes Res. Care 2020, 8, e001481. [Google Scholar] [CrossRef]
- Brenner, B.M.; Garcia, D.L.; Anderson, S. Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertens. 1988, 1, 335–347. [Google Scholar] [CrossRef]
- Kotchen, T.A.; Piering, A.W.; Cowley, A.W.; Grim, C.E.; Gaudet, D.; Hamet, P.; Kaldunski, M.L.; Kotchen, J.M.; Roman, R.J. Glomerular hyperfiltration in hypertensive African Americans. Hypertension 2000, 35, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Hoy, W.E.; Hughson, M.D.; Bertram, J.F.; Douglas-Denton, R.; Amann, K. Nephron number, hypertension, renal disease, and renal failure. J. Am. Soc. Nephrol. 2005, 16, 2557–2564. [Google Scholar] [CrossRef]
- Palatini, P.; Mormino, P.; Dorigatti, F.; Santonastaso, M.; Mos, L.; De Toni, R.; Winnicki, M.; Dal Follo, M.; Biasion, T.; Garavelli, G.; et al. Glomerular hyperfiltration predicts the development of microalbuminuria in stage 1 hypertension: The HARVEST. Kidney Int. 2006, 70, 578–584. [Google Scholar] [CrossRef]
- Kanzaki, G.; Tsuboi, N.; Haruhara, K.; Koike, K.; Ogura, M.; Shimizu, A.; Yokoo, T. Factors associated with a vicious cycle involving a low nephron number, hypertension and chronic kidney disease. Hypertens. Res. 2015, 38, 633–641. [Google Scholar] [CrossRef]
- Pugh, D.; Gallacher, P.J.; Dhaun, N. Management of Hypertension in Chronic Kidney Disease. Drugs 2019, 79, 365–379. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, M.E.; Nadeau-Fredette, A.C.; Madore, F.; Agharazii, M.; Goupil, R. Association of Glomerular Hyperfiltration and Cardiovascular Risk in Middle-Aged Healthy Individuals. JAMA Netw. Open 2020, 3, e202377. [Google Scholar] [CrossRef]
- Rossitto, G.; Maiolino, G.; Lerco, S.; Ceolotto, G.; Blackburn, G.; Mary, S.; Antonelli, G.; Berton, C.; Bisogni, V.; Cesari, M.; et al. High sodium intake, glomerular hyperfiltration, and protein catabolism in patients with essential hypertension. Cardiovasc. Res. 2021, 117, 1372–1381. [Google Scholar] [CrossRef]
- Rossi, G.P.; Sacchetto, A.; Visentin, P.; Canali, C.; Graniero, G.R.; Palatini, P.; Pessina, A.C. Changes in left ventricular anatomy and function in hypertension and primary aldosteronism. Hypertension 1996, 27, 1039–1045. [Google Scholar] [CrossRef]
- Ribstein, J.; Du Cailar, G.; Fesler, P.; Mimran, A. Relative glomerular hyperfiltration in primary aldosteronism. J. Am. Soc. Nephrol. 2005, 16, 1320–1325. [Google Scholar] [CrossRef]
- Sechi, L.A.; Novello, M.; Lapenna, R.; Baroselli, S.; Nadalini, E.; Colussi, G.L.; Catena, C. Long-term renal outcomes in patients with primary aldosteronism. JAMA 2006, 295, 2638–2645. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.C.; Wu, V.C.; Tsai, C.W.; Wu, K.D. Taiwan Primary Aldosteronism Investigation (TAIPAI) Study Group). Relative kidney hyperfiltration in primary aldosteronism: A meta-analysis. J. Renin Angiotensin Aldosterone Syst. 2011, 12, 113–122. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Cohney, S.; De Michieli, F.; Pinach, S.; Saba, F.; Gambino, R. Fatty Liver and Chronic Kidney Disease: Novel Mechanistic Insights and Therapeutic Opportunities. Diabetes Care 2016, 39, 1830–1845. [Google Scholar] [CrossRef] [Green Version]
- Kiapidou, S.; Liava, C.; Kalogirou, M.; Akriviadis, E.; Sinakos, E. Chronic kidney disease in patients with non-alcoholic fatty liver disease: What the Hepatologist should know? Ann. Hepatol. 2020, 19, 134–144. [Google Scholar] [CrossRef]
- Yodoshi, T.; Arce-Clachar, A.C.; Sun, Q.; Fei, L.; Bramlage, K.; Xanthakos, S.A.; Flores, F.; Mouzaki, M. Glomerular Hyperfiltration Is Associated with Liver Disease Severity in Children with Nonalcoholic Fatty Liver Disease. J. Pediatr. 2020, 222, 127–133. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD as a driver of chronic kidney disease. J. Hepatol. 2020, 72, 785–801. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Leon, E.; Cioffi, C.E.; Vos, M.B. Perspectives on youth-onset nonalcoholic fatty liver disease. Endocrinol. Diabetes Metab. 2020, 3, e00184. [Google Scholar] [CrossRef]
- Abbate, M.; Mascaró, C.M.; Montemayor, S.; Casares, M.; Gómez, C.; Ugarriza, L.; Tejada, S.; Abete, I.; Zulet, M.A.; Sureda, A.; et al. Non-Alcoholic Fatty Liver Disease Is Associated with Kidney Glomerular Hyperfiltration in Adults with Metabolic Syndrome. J. Clin. Med. 2021, 10, 1717. [Google Scholar] [CrossRef]
- Hakim, R.M.; Goldszer, R.C.; Brenner, B.M. Hypertension and proteinuria: Long-term sequelae of uninephrectomy in humans. Kidney Int. 1984, 25, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Kasiske, B.L.; Anderson-Haag, T.; Israni, A.K.; Kalil, R.S.; Kimmel, P.L.; Kraus, E.S.; Kumar, R.; Posselt, A.A.; Pesavento, T.E.; Rabb, H.; et al. A prospective controlled study of living kidney donors: Three-year follow-up. Am. J. Kidney Dis. 2015, 66, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, H.N.; Foley, R.N.; Reule, S.A.; Spong, R.; Kukla, A.; Issa, N.; Berglund, D.M.; Sieger, G.K.; Matas, A.J. Renal Function Profile in White Kidney Donors: The First 4 Decades. J. Am. Soc. Nephrol. 2016, 27, 2885–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chagnac, A.; Weinstein, T.; Korzets, A.; Ramadan, E.; Hirsch, J.; Gafter, U. Glomerular hemodynamics in severe obesity. Am. J. Physiol. Renal. Physiol. 2000, 278, F817–F822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenihan, C.R.; Busque, S.; Derby, G.; Blouch, K.; Myers, B.D.; Tan, J.C. Longitudinal study of living kidney donor glomerular dynamics after nephrectomy. J. Clin. Investig. 2015, 125, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Grams, M.E.; Sang, Y.; Levey, A.S.; Matsushita, K.; Ballew, S.; Chang, A.R.; Chow, E.K.; Kasiske, B.L.; Kovesdy, C.P.; Nadkarni, G.N.; et al. Chronic Kidney Disease Prognosis Consortium. Kidney-Failure Risk Projection for the Living Kidney-Donor Candidate. N. Engl. J. Med. 2016, 374, 411–421. [Google Scholar] [CrossRef]
- Schreuder, M.F.; Langemeijer, M.E.; Bökenkamp, A.; Delemarre-Van de Waal, H.A.; Van Wijk, J.A. Hypertension and microalbuminuria in children with congenital solitary kidneys. J. Paediatr. Child Health 2008, 44, 363–368. [Google Scholar] [CrossRef]
- Sanna-Cherchi, S.; Ravani, P.; Corbani, V.; Parodi, S.; Haupt, R.; Piaggio, G.; Innocenti, M.L.; Somenzi, D.; Trivelli, A.; Caridi, G.; et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009, 76, 528–533. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.J.; Moxey-Mims, M.; Jerry-Fluker, J.; Warady, B.A.; Furth, S.L. CKiD (CKD in children) prospective cohort study: A review of current findings. Am. J. Kidney Dis. 2012, 60, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westland, R.; Schreuder, M.F.; van Goudoever, J.B.; Sanna-Cherchi, S.; van Wijk, J.A. Clinical implications of the solitary functioning kidney. Clin. J. Am. Soc. Nephrol. 2014, 9, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, T.; Celsi, G.E.; Sharma, M.; Dai, H.; McCarthy, E.T.; Ruiz, M.; Cudmore, P.A.; Alon, U.S.; Sharma, R.; Savin, V.J. Fluid flow shear stress over podocytes is increased in the solitary kidney. Nephrol. Dial. Transplant. 2014, 29, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myrvang, H. Polycystic kidney disease: Glomerular hyperfiltration may be a risk factor for progression of ADPKD. Nat. Rev. Nephrol. 2011, 7, 608. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Han, B.G.; Choi, S.O.; Cha, S.K. Secondary Focal Segmental Glomerulosclerosis: From Podocyte Injury to Glomerulosclerosis. BioMed Res. Int. 2016, 2016, 1630365. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef] [Green Version]
- Shabaka, A.; Tato Ribera, A.; Fernández-Juárez, G. Focal Segmental Glomerulosclerosis: State-of-the-Art and Clinical Perspective. Nephron 2020, 144, 413–427. [Google Scholar] [CrossRef]
- KDIGO Clinical Practice Guideline for Glomerulonephriti. Available online: https://kdigo.org/wp-content/uploads/2017/02/KDIGO-2012-GN-Guideline-English.pdf (accessed on 10 October 2021).
- Bhathena, D.B.; Sondheimer, J.H. The glomerulopathy of homozygous sickle hemoglobin (SS) disease: Morphology and pathogenesis. J. Am. Soc. Nephrol. 1991, 1, 1241–1252. [Google Scholar] [CrossRef]
- Hirschberg, R. Glomerular hyperfiltration in sickle cell disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 748–749. [Google Scholar] [CrossRef] [Green Version]
- Aygun, B.; Mortier, N.A.; Smeltzer, M.P.; Hankins, J.S.; Ware, R.E. Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatr. Nephrol. 2011, 26, 1285–1290. [Google Scholar] [CrossRef] [Green Version]
- Olaniran, K.O.; Allegretti, A.S.; Zhao, S.H.; Achebe, M.M.; Eneanya, N.D.; Thadhani, R.I.; Nigwekar, S.U.; Kalim, S. Kidney Function Decline among Black Patients with Sickle Cell Trait and Sickle Cell Disease: An Observational Cohort Study. J. Am. Soc. Nephrol. 2020, 31, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Spear, G.S. The glomerulus in cyanotic congenital heart disease and primary pulmonary hypertension. a review. Nephron 1964, 1, 238–248. [Google Scholar] [CrossRef]
- Flanagan, M.F.; Hourihan, M.; Keane, J.F. Incidence of renal dysfunction in adults with cyanotic congenital heart disease. Am. J. Cardiol. 1991, 68, 403–406. [Google Scholar] [CrossRef]
- Hida, K.; Wada, J.; Yamasaki, H.; Nagake, Y.; Zhang, H.; Sugiyama, H.; Shikata, K.; Makino, H. Cyanotic congenital heart disease associated with glomerulomegaly and focal segmental glomerulosclerosis: Remission of nephrotic syndrome with angiotensin converting enzyme inhibitor. Nephrol. Dial. Transplant. 2002, 17, 144–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inatomi, J.; Matsuoka, K.; Fujimaru, R.; Nakagawa, A.; Iijima, K. Mechanisms of development and progression of cyanotic nephropathy. Pediatr. Nephrol. 2006, 21, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Ossa Galvis, M.M.; Bhakta, R.T.; Tarmahomed, A. Cyanotic Heart Disease; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK500001/ (accessed on 7 November 2021).
- Altay, S.; Onat, A.; Özpamuk-Karadeniz, F.; Karadeniz, Y.; Kemaloğlu-Öz, T.; Can, G. Renal “hyperfiltrators” are at elevated risk of death and chronic diseases. BMC Nephrol. 2014, 15, 160. [Google Scholar] [CrossRef] [Green Version]
- Onat, A.; Can, G. Enhanced proinflammatory state and autoimmune activation: A breakthrough to understanding chronic diseases. Curr. Pharm. Des. 2014, 20, 575–584. [Google Scholar] [CrossRef]
- Arestegui, A.H.; Fuquay, R.; Sirota, J.; Swenson, E.R.; Schoene, R.B.; Jefferson, J.A.; Chen, W.; Yu, X.Q.; Kelly, J.P.; Johnson, R.J.; et al. High altitude renal syndrome (HARS). J. Am. Soc. Nephrol. 2011, 22, 1963–1968. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.W.; Park, S.; Lee, S.; Lee, Y.; Cho, S.; Han, K.; Cho, H.; Kim, Y.; Kim, Y.C.; Han, S.S.; et al. Glomerular hyperfiltration is associated with dementia: A nationwide population-based study. PLoS ONE 2020, 15, e0228361. [Google Scholar] [CrossRef]
- Nistrup Holmegaard, S.; Christoffersen, H.; Haase, J. Albuminuria, intermittent hyperfiltration and salt wasting in patients with stroke: A pilot study. Scand. J. Clin. Lab. Investig. 2006, 66, 437–749. [Google Scholar] [CrossRef]
- Putaala, J.; Haapaniemi, E.; Gordin, D.; Liebkind, R.; Groop, P.H.; Kaste, M.; Tatlisumak, T. Factors associated with impaired kidney function and its impact on long-term outcome in young ischemic stroke. Stroke 2011, 42, 2459–2964. [Google Scholar] [CrossRef] [Green Version]
- Cherney, D.Z.; Scholey, J.W.; Miller, J.A. Insights into the regulation of renal hemodynamic function in diabetic mellitus. Curr. Diabetes Rev. 2008, 4, 280–290. [Google Scholar] [CrossRef]
- Park, M.; Yoon, E.; Lim, Y.H.; Kim, H.; Choi, J.; Yoon, H.J. Renal hyperfiltration as a novel marker of all-cause mortality. J. Am. Soc. Nephrol. 2015, 26, 1426–1433. [Google Scholar] [CrossRef]
- Kanbay, M.; Ertuglu, L.A.; Afsar, B.; Ozdogan, E.; Kucuksumer, Z.S.; Ortiz, A.; Covic, A.; Kuwabara, M.; Cherney, D.Z.I.; van Raalte, D.H.; et al. Renal hyperfiltration defined by high estimated glomerular filtration rate: A risk factor for cardiovascular disease and mortality. Diabetes Obes. Metab. 2019, 21, 2368–2383. [Google Scholar] [CrossRef] [PubMed]
- Ould Setti, M.; Kacimi, S.E.O.; Niskanen, L.; Tuomainen, T.P. Mortality-based definition of renal hyperfiltration in middle-aged men: A 35-year cohort from Finland. Int. Urol. Nephrol. 2021, 1–8. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Holt, S.G.; Smith, E.R. Progression of Tubulointerstitial Fibrosis and the Chronic Kidney Disease Phenotype-Role of Risk Factors and Epigenetics. Front. Pharmacol. 2017, 8, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, H.; Yanagita, M. Janus-Faced: Molecular Mechanisms and Versatile Nature of Renal Fibrosis. KIDNEY360 2020, 1, 697–704. [Google Scholar] [CrossRef]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef]
- Brosius, F.C., 3rd. New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev. Endocr. Metab. Disord. 2008, 9, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Valero, B.; Cachofeiro, V.; Martínez-Martínez, E. Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved. Cells 2021, 10, 1824. [Google Scholar] [CrossRef]
- Endlich, N.; Sunohara, M.; Nietfeld, W.; Wolski, E.W.; Schiwek, D.; Kränzlin, B.; Gretz, N.; Kriz, W.; Eickhoff, H.; Endlich, K. Analysis of differential gene expression in stretched podocytes: Osteopontin enhances adaptation of podocytes to mechanical stress. FASEB J. 2002, 16, 1850–1852. [Google Scholar] [CrossRef] [PubMed]
- Endlich, N.; Endlich, K. Stretch, tension and adhesion-adaptive mechanisms of the actin cytoskeleton in podocytes. Eur. J. Cell Biol. 2006, 85, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.; Endlich, N.; Kriz, W.; Endlich, K. Podocytes are sensitive to fluid shear stress in vitro. Am. J. Physiol. Renal. Physiol. 2006, 291, F856–F865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endlich, N.; Endlich, K. The challenge and response of podocytes to glomerular hypertension. Semin. Nephrol. 2012, 32, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Celsi, G.; Larsson, L.; Seri, I.; Savin, V.J.; Aperia, A. Glomerular adaptation in uninephrectomized young rats. Pediatr. Nephrol. 1989, 3, 280–285. [Google Scholar] [CrossRef]
- Celsi, G.; Savin, V.J.; Henter, J.I.; Sohtell, M. The contribution of ultrafiltration pressure for glomerular hyperfiltration in young nephrectomized rats. Acta Physiol. Scand. 1991, 141, 483–487. [Google Scholar] [CrossRef]
- Bank, N.; Alterman, L.; Aynedjian, H.S. Selective deep nephron hyperfiltration in uninephrectomized spontaneously hypertensive rats. Kidney Int. 1983, 24, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Löwen, J.; Federico, G.; van den Born, J.; Gröne, E.; Gröne, H.J. Accumulation of worn-out GBM material substantially contributes to mesangial matrix expansion in diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2017, 312, F1101–F1111. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, T.; McCarthy, E.T.; Sharma, R.; Cudmore, P.A.; Sharma, M.; Johnson, M.L.; Bonewald, L.F. Prostaglandin E(2) is crucial in the response of podocytes to fluid flow shear stress. J. Cell Commun. Signal. 2010, 4, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endlich, N.; Kress, K.R.; Reiser, J.; Uttenweiler, D.; Kriz, W.; Mundel, P.; Endlich, K. Podocytes respond to mechanical stress in vitro. J. Am. Soc. Nephrol. 2001, 12, 413–422. [Google Scholar] [CrossRef]
- Neal, C.R. Podocytes … What’s Under Yours? (Podocytes and Foot Processes and How They Change in Nephropathy). Front. Endocrinol. 2015, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Endlich, K.; Kliewe, F.; Endlich, N. Stressed podocytes-mechanical forces, sensors, signaling and response. Pflugers Arch. 2017, 469, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Durvasula, R.V.; Shankland, S.J. Mechanical strain increases SPARC levels in podocytes: Implications for glomerulosclerosis. Am. J. Physiol. Renal. Physiol. 2005, 289, F577–F584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schordan, S.; Schordan, E.; Endlich, K.; Endlich, N. AlphaV-integrins mediate the mechanoprotective action of osteopontin in podocytes. Am. J. Physiol. Renal. Physiol. 2011, 300, F119–F132. [Google Scholar] [CrossRef] [PubMed]
- Martineau, L.C.; McVeigh, L.I.; Jasmin, B.J.; Kennedy, C.R. p38 MAP kinase mediates mechanically induced COX-2 and PG EP4 receptor expression in podocytes: Implications for the actin cytoskeleton. Am. J. Physiol. Renal. Physiol. 2004, 286, F693–F701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, A.T.; Pippin, J.; Durvasula, R.; Pichler, R.; Hiromura, K.; Monkawa, T.; Couser, W.G.; Shankland, S.J. Mechanical stretch induces podocyte hypertrophy in vitro. Kidney Int. 2005, 67, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fields, T.A.; Pazmino, K.; Dai, Q.; Burchette, J.L.; Howell, D.N.; Coffman, T.M.; Spurney, R.F. Activation of Galpha q-coupled signaling pathways in glomerular podocytes promotes renal injury. J. Am. Soc. Nephrol. 2005, 16, 3611–3622. [Google Scholar] [CrossRef] [Green Version]
- Faour, W.H.; Thibodeau, J.F.; Kennedy, C.R. Mechanical stretch and prostaglandin E2 modulate critical signaling pathways in mouse podocytes. Cell Signal. 2010, 22, 1222–1230. [Google Scholar] [CrossRef]
- Eekhoff, A.; Bonakdar, N.; Alonso, J.L.; Hoffmann, B.; Goldmann, W.H. Glomerular podocytes: A study of mechanical properties and mechano-chemical signaling. Biochem. Biophys. Res. Commun. 2011, 406, 229–233. [Google Scholar] [CrossRef]
- Janmey, P.A.; Miller, R.T. Mechanisms of mechanical signaling in development and disease. J. Cell Sci. 2011, 124, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Holstein-Rathlou, N.H. Dynamic aspects of the tubuloglomerular feedback mechanism. Dan. Med. Bull. 1992, 39, 134–154. [Google Scholar]
- Panchapakesan, U.; Pollock, C. The primary cilia in diabetic kidney disease: A tubulocentric view? Int. J. Biochem. Cell Biol. 2020, 122, 105718. [Google Scholar] [CrossRef]
- Repetti, R.; Majumder, N.; De Oliveira, K.C.; Meth, J.; Yangchen, T.; Sharma, M.; Srivastava, T.; Rohatgi, R. Unilateral nephrectomy stimulates erk and is associated with enhanced Na transport. Front. Physiol. 2021, 12, 583453. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Blantz, R.C.; Thomson, S. Glomerular hyperfiltration and the salt paradox in early [corrected] type 1 diabetes mellitus: A tubulo-centric view. J. Am. Soc. Nephrol. 2003, 14, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [CrossRef]
- Fiseha, T.; Tamir, Z. Urinary markers of tubular injury in early diabetic nephropathy. Int. J. Nephrol. 2016, 2016, 4647685. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves-Dias, C.; Morello, J.; Correia, M.J.; Coelho, N.R.; Antunes, A.M.M.; Macedo, M.P.; Monteiro, E.C.; Soto, K.; Pereira, S.A. Mercapturate Pathway in the Tubulocentric Perspective of Diabetic Kidney Disease. Nephron 2019, 143, 17–23. [Google Scholar] [CrossRef]
- Marshall, C.B. Rethinking glomerular basement membrane thickening in diabetic nephropathy: Adaptive or pathogenic? Am. J. Physiol. Renal. Physiol. 2016, 311, F831–F843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler Sekulic, S.; Sekulic, M. Rheological influence upon the glomerular podocyte and resultant mechanotransduction. Kidney Blood Press. Res. 2015, 40, 176–187. [Google Scholar] [CrossRef]
- Srivastava, T.; McCarthy, E.T.; Sharma, R.; Kats, A.; Carlton, C.G.; Alon, U.S.; Cudmore, P.A.; El-Meanawy, A.; Sharma, M. Fluid flow shear stress upregulates prostanoid receptor EP2 but not EP4 in murine podocytes. Prostaglandins Other Lipid Mediat. 2013, 104–105, 49–57. [Google Scholar] [CrossRef]
- Trimarchi, H. Podocyturia: What is in a name? J. Transl. Int. Med. 2015, 3, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Yanagihara, T.; Kihara, I. Urinary podocytes in primary focal segmental glomerulosclerosis. Nephron 2001, 89, 342–347. [Google Scholar] [CrossRef]
- Nakamura, T.; Ushiyama, C.; Suzuki, S.; Hara, M.; Shimada, N.; Sekizuka, K.; Ebihara, I.; Koide, H. Effects of angiotensin-converting enzyme inhibitor, angiotensin II receptor antagonist and calcium antagonist on urinary podocytes in patients with IgA nephropathy. Am. J. Nephrol. 2000, 20, 373–379. [Google Scholar] [CrossRef]
- Nakamura, T.; Ushiyama, C.; Osada, S.; Hara, M.; Shimada, N.; Koide, H. Pioglitazone reduces urinary podocyte excretion in type 2 diabetes patients with microalbuminuria. Metabolism 2001, 50, 1193–1196. [Google Scholar] [CrossRef]
- Wight, T.N.; Kinsella, M.G.; Qwarnström, E.E. The role of proteoglycans in cell adhesion, migration and proliferation. Curr. Opin. Cell Biol. 1992, 4, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liang, Y.; Wang, D.; Xu, D.; Joshi, T. A dynamic programing approach to integrate gene expression data and network information for pathway model generation. Bioinformatics 2020, 36, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.; Joshi, T.; Jiang, Y.; Heruth, D.P.; Rezaiekhaligh, M.H.; Novak, J.; Staggs, V.S.; Alon, U.S.; Garola, R.E.; El-Meanawy, A.; et al. Upregulated proteoglycan-related signaling pathways in fluid flow shear stress-treated podocytes. Am. J. Physiol. Renal. Physiol. 2020, 319, F312–F322. [Google Scholar] [CrossRef]
- Stamenović, D.; Wang, N. Stress transmission within the cell. Compr. Physiol. 2011, 1, 499–524. [Google Scholar] [CrossRef] [Green Version]
- Lamparter, L.; Galic, M. Cellular Membranes, a Versatile Adaptive Composite Material. Front. Cell Dev. Biol. 2020, 8, 684. [Google Scholar] [CrossRef]
- Bhatia, T.; Cornelius, F.; Ipsen, J.H. Exploring the raft-hypothesis by probing planar bilayer patches of free-standing giant vesicles at nanoscale resolution, with and without Na, K-ATPase. Biochim. Biophys. Acta 2016, 1858, 3041–3049. [Google Scholar] [CrossRef] [Green Version]
- Sturzenegger, F.; Robinson, T.; Hess, D.; Dittrich, P.S. Membranes under shear stress: Visualization of non-equilibrium domain patterns and domain fusion in a microfluidic device. Soft Matter 2016, 12, 5072–5076. [Google Scholar] [CrossRef]
- Yamamoto, K.; Nogimori, Y.; Imamura, H.; Ando, J. Shear stress activates mitochondrial oxidative phosphorylation by reducing plasma membrane cholesterol in vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2020, 117, 33660–33667. [Google Scholar] [CrossRef]
- Kang, J.X. Balance of omega-6/omega-3 essential fatty acids is important for health. The evidence from gene transfer studies. World Rev. Nutr. Diet. 2005, 95, 93–102. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef]
- Murakami, M. Novel functions of phospholipase A2s: Overview. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 763–765. [Google Scholar] [CrossRef]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating Phospholipase A2 Biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef]
- Balboa, M.A.; Balsinde, J. Phospholipases: From Structure to Biological Function. Biomolecules 2021, 11, 428. [Google Scholar] [CrossRef]
- Christie, W.W.; Harwood, J.L. Oxidation of polyunsaturated fatty acids to produce lipid mediators. Essays Biochem. 2020, 64, 401–421. [Google Scholar] [CrossRef]
- Huang, C.; Bruggeman, L.A.; Hydo, L.M.; Miller, R.T. Shear stress induces cell apoptosis via a c-Src-phospholipase D-mTOR signaling pathway in cultured podocytes. Exp. Cell Res. 2012, 318, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Ziembicki, J.; Tandon, R.; Schelling, J.R.; Sedor, J.R.; Miller, R.T.; Huang, C. Mechanical force-activated phospholipase D is mediated by Galpha12/13-Rho and calmodulin-dependent kinase in renal epithelial cells. Am. J. Physiol. Renal. Physiol. 2005, 289, F826–F834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, S.M.; Liddle, R.A. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J. Biol. Chem. 2021, 296, 100171. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Shi, H.; Caligiuri, S.P.; Wu, Y.; Declercq, V.; Taylor, C.G.; Zahradka, P.; Ogborn, M.R.; Aukema, H.M. Trans-10,cis-12-conjugated linoleic acid worsens renal pathology and alters cyclooxygenase derived oxylipins in obesity-associated nephropathy. J. Nutr. Biochem. 2015, 26, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Hassid, A.; Konieczkowski, M.; Dunn, M.J. Prostaglandin synthesis in isolated rat kidney glomeruli. Proc. Natl. Acad. Sci. USA 1979, 76, 1155–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebel, M.; Grose, J.H. Abnormal renal prostaglandin production during the evolution of chronic nephropathy. Am. J. Nephrol. 1986, 6, 96–100. [Google Scholar] [CrossRef]
- Zook, T.E.; Strandhoy, J.W. Mechanisms of the natriuretic and diuretic effects of prostaglandin F2 alpha. J. Pharmacol. Exp. Ther. 1981, 217, 674–680. [Google Scholar]
- Uehara, Y.; Hirawa, N.; Kawabata, Y.; Akie, Y.; Ichikawa, A.; Funahashi, N.; Goto, A.; Omata, M. Lipid metabolism and renal protection by chronic cicletanine treatment in Dahl salt-sensitive rats with salt-induced hypertension. Blood Press. 1997, 6, 180–187. [Google Scholar] [CrossRef]
- Zhang, A.; Dong, Z.; Yang, T. Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am. J. Physiol. Renal. Physiol. 2006, 291, F1332–F1342. [Google Scholar] [CrossRef] [Green Version]
- Reilly, C.M.; Oates, J.C.; Cook, J.A.; Morrow, J.D.; Halushka, P.V.; Gilkeson, G.S. Inhibition of mesangial cell nitric oxide in MRL/lpr mice by prostaglandin J2 and proliferator activation receptor-gamma agonists. J. Immunol. 2000, 164, 1498–1504. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, P.K.; Patel, N.S.; Cuzzocrea, S.; Brown, P.A.; Stewart, K.N.; Mota-Filipe, H.; Britti, D.; Eberhardt, W.; Pfeilschifter, J.; Thiemermann, C. The cyclopentenone prostaglandin 15-deoxy-Delta(12,14)-prostaglandin J2 ameliorates ischemic acute renal failure. Cardiovasc. Res. 2004, 61, 630–643. [Google Scholar] [CrossRef] [Green Version]
- Kömhoff, M.; Grone, H.J.; Klein, T.; Seyberth, H.W.; Nüsing, R.M. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: Implication for renal function. Am. J. Physiol. 1997, 272, F460–F468. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, S.; Jo, Y.I.; Hao, C.M.; Zhang, M.; Fan, X.; Kennedy, C.; Breyer, M.D.; Moeckel, G.W.; Harris, R.C. Overexpression of cyclooxygenase-2 predisposes to podocyte injury. J. Am. Soc. Nephrol. 2007, 18, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FitzGerald, G.A. The choreography of cyclooxygenases in the kidney. J. Clin. Investig. 2002, 110, 33–34. [Google Scholar] [CrossRef]
- Cheng, H.; Fan, X.; Guan, Y.; Moeckel, G.W.; Zent, R.; Harris, R.C. Distinct roles for basal and induced COX-2 in podocyte injury. J. Am. Soc. Nephrol. 2009, 20, 1953–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Breyer, M.D.; Davis, L.; Jacobson, H.R.; Breyer, R.M. Differential localization of prostaglandin E receptor subtypes in human kidney. Am. J. Physiol. 1996, 270, F912–F918. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid receptors: Structures, properties, and functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar] [CrossRef]
- Frölich, S.; Olliges, A.; Kern, N.; Schreiber, Y.; Narumiya, S.; Nüsing, R.M. Temporal expression of the PGE2 synthetic system in the kidney is associated with the time frame of renal developmental vulnerability to cyclooxygenase-2 inhibition. Am. J. Physiol. Renal. Physiol. 2012, 303, F209–F219. [Google Scholar] [CrossRef] [Green Version]
- Breyer, M.D.; Jacobson, H.R.; Breyer, R.M. Functional and molecular aspects of renal prostaglandin receptors. J. Am. Soc. Nephrol. 1996, 7, 8–17. [Google Scholar] [CrossRef]
- Mangelsen, E.; Rothe, M.; Schulz, A.; Kourpa, A.; Panáková, D.; Kreutz, R.; Bolbrinker, J. Concerted EP2 and EP4 Receptor Signaling Stimulates Autocrine Prostaglandin E2 Activation in Human Podocytes. Cells 2020, 9, 1256. [Google Scholar] [CrossRef]
- Srivastava, T.; Heruth, D.P.; Duncan, R.S.; Rezaiekhaligh, M.H.; Garola, R.E.; Priya, L.; Zhou, J.; Boinpelly, V.C.; Novak, J.; Ali, M.F.; et al. Transcription actor β-Catenin plays a key role in fluid flow shear stress-mediated glomerular injury in solitary kidney. Cells 2021, 10, 1253. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Namba, T.; Shigemoto, R.; Negishi, M.; Ichikawa, A.; Narumiya, S. Distinct cellular localization of mRNAs for three subtypes of prostaglandin E receptor in kidney. Am. J. Physiol. 1994, 266, F823–F828. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinbaum, S.; Duan, Y.; Satlin, L.M.; Wang, T.; Weinstein, A.M. Mechanotransduction in the renal tubule. Am. J. Physiol. Renal. Physiol. 2010, 299, F1220–F1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Flores, D.; Carrisoza-Gaytán, R.; Rohatgi, R. Cholesterol affects flow-stimulated cyclooxygenase-2 expression and prostanoid secretion in the cortical collecting duct. Am. J. Physiol. Renal. Physiol. 2015, 308, F1229–F1237. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, R.; Battini, L.; Kim, P.; Israeli, S.; Wilson, P.D.; Gusella, G.L.; Satlin, L.M. Mechanoregulation of intracellular Ca2+ in human autosomal recessive polycystic kidney disease cyst-lining renal epithelial cells. Am. J. Physiol. Renal. Physiol. 2008, 294, F890–F899. [Google Scholar] [CrossRef]
- Morimoto, T.; Liu, W.; Woda, C.; Carattino, M.D.; Wei, Y.; Hughey, R.P.; Apodaca, G.; Satlin, L.M.; Kleyman, T.R. Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. Am. J. Physiol. Renal. Physiol. 2006, 291, F663–F669. [Google Scholar] [CrossRef]
- Carrisoza-Gaytan, R.; Liu, Y.; Flores, D.; Else, C.; Lee, H.G.; Rhodes, G.; Sandoval, R.M.; Kleyman, T.R.; Lee, F.Y.; Molitoris, B.; et al. Effects of biomechanical forces on signaling in the cortical collecting duct (CCD). Am. J. Physiol. Renal. Physiol. 2014, 307, F195–F204. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Flores, D.; Carrisoza-Gaytán, R.; Rohatgi, R. Biomechanical regulation of cyclooxygenase-2 in the renal collecting duct. Am. J. Physiol. Renal. Physiol. 2014, 306, F214–F223. [Google Scholar] [CrossRef] [Green Version]
- Flores, D.; Battini, L.; Gusella, G.L.; Rohatgi, R. Fluid shear stress induces renal epithelial gene expression through polycystin-2-dependent trafficking of extracellular regulated kinase. Nephron Physiol. 2011, 117, 27–36. [Google Scholar] [CrossRef]
- Alexander, L.D.; Alagarsamy, S.; Douglas, J.G. Cyclic stretch-induced cPLA2 mediates ERK 1/2 signaling in rabbit proximal tubule cells. Kidney Int. 2004, 65, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Gotoh, N.; Yan, Q.; Du, Z.; Weinstein, A.M.; Wang, T.; Weinbaum, S. Shear-induced reorganization of renal proximal tubule cell actin cytoskeleton and apical junctional complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 11418–11423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenghat, F.J.; Nauli, S.M.; Kolb, R.; Zhou, J.; Ingber, D.E. Global cytoskeletal control of mechanotransduction in kidney epithelial cells. Exp. Cell Res. 2004, 301, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Midgett, C.; Stitham, J.; Martin, K.; Hwa, J. Prostacyclin receptor regulation--from transcription to trafficking. Curr. Mol. Med. 2011, 11, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, R.; Hébert, R.L. Prostacyclin signaling in the kidney: Implications for health and disease. Am. J. Physiol. Renal. Physiol. 2005, 289, F235–F246. [Google Scholar] [CrossRef] [Green Version]
- Wise, H.; Jones, R.L. Focus on prostacyclin and its novel mimetics. Trends Pharmacol. Sci. 1996, 17, 17–21. [Google Scholar] [CrossRef]
- Lipsky, P.E.; Brooks, P.; Crofford, L.J.; DuBois, R.; Graham, D.; Simon, L.S.; van de Putte, L.B.; Abramson, S.B. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch. Intern. Med. 2000, 160, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Zambraski, E.J. The effects of nonsteroidal anti-inflammatory drugs on renal function: Experimental studies in animals. Semin. Nephrol. 1995, 15, 205–213. [Google Scholar]
- Friedman, A.N. High-protein diets: Potential effects on the kidney in renal health and disease. Am. J. Kidney Dis. 2004, 44, 950–962. [Google Scholar] [CrossRef]
- Cupisti, A.; Giannese, D.; Moriconi, D.; D’Alessandro, C.; Torreggiani, M.; Piccoli, G.B. Nephroprotection by SGLT2i in CKD Patients: May It Be Modulated by Low-Protein Plant-Based Diets? Front. Med. 2020, 7, 622593. [Google Scholar] [CrossRef]
- Wang, L.; Chang, J.H.; Paik, S.Y.; Tang, Y.; Eisner, W.; Spurney, R.F. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol. Endocrinol. 2011, 25, 1376–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnahan, B.A.; Le Breton, G.C.; Lianos, E.A. Localization of authentic thromboxane A2/prostaglandin H2 receptor in the rat kidney. Kidney Int. 1996, 49, 1207–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahata, N. Thromboxane A2: Physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol. Ther. 2008, 118, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Craven, P.A.; Caines, M.A.; DeRubertis, F.R. Sequential alterations in glomerular prostaglandin and thromboxane synthesis in diabetic rats: Relationship to the hyperfiltration of early diabetes. Metabolism 1987, 36, 95–103. [Google Scholar] [CrossRef]
- Purkerson, M.L.; Joist, J.H.; Yates, J.; Valdes, A.; Morrison, A.; Klahr, S. Inhibition of thromboxane synthesis ameliorates the progressive kidney disease of rats with subtotal renal ablation. Proc. Natl. Acad. Sci. USA 1985, 82, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Saleem, M.A.; Nam, J.A.; Ha, T.S.; Shin, J.I. Effects of interleukin-13 and montelukast on the expression of zonula occludens-1 in human podocytes. Yonsei Med. J. 2015, 56, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, Y.; Tanji, M.; Takano, K.; Fukuda, Y.; Isome, M.; Nozawa, R.; Suzuki, H.; Hosoya, M. The leukotriene B4 receptor antagonist ONO-4057 inhibits mesangioproliferative changes in anti-Thy-1 nephritis. Nephrol. Dial. Transplant. 2005, 20, 2697–2703. [Google Scholar] [CrossRef] [Green Version]
- Doi, K.; Hamasaki, Y.; Noiri, E.; Nosaka, K.; Suzuki, T.; Toda, A.; Shimizu, T.; Fujita, T.; Nakao, A. Role of leukotriene B4 in accelerated hyperlipidaemic renal injury. Nephrology 2011, 16, 304–309. [Google Scholar] [CrossRef]
- Gambaro, G.; Perazella, M.A. Adverse renal effects of anti-inflammatory agents: Evaluation of selective and nonselective cyclooxygenase inhibitors. J. Intern. Med. 2003, 253, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Badr, K.F.; Lakkis, F.G. Lipoxygenase products in normal and diseased glomeruli. Ann. N. Y. Acad. Sci. 1994, 744, 216–228. [Google Scholar] [CrossRef]
- Badr, K.F.; Baylis, C.; Pfeffer, J.M.; Pfeffer, M.A.; Soberman, R.J.; Lewis, R.A.; Austen, K.F.; Corey, E.J.; Brenner, B.M. Renal and systemic hemodynamic responses to intravenous infusion of leukotriene C4 in the rat. Circ. Res. 1984, 54, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Spurney, R.F.; Ibrahim, S.; Butterly, D.; Klotman, P.E.; Sanfilippo, F.; Coffman, T.M. Leukotrienes in renal transplant rejection in rats. Distinct roles for leukotriene B4 and peptidoleukotrienes in the pathogenesis of allograft injury. J. Immunol. 1994, 152, 867–876. [Google Scholar] [PubMed]
- Petric, R.; Ford-Hutchinson, A.W. Elevated cysteinyl leukotriene excretion in experimental glomerulonephritis. Kidney Int. 1994, 46, 1322–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.; Dvash, E. Leukotrienes and kidney diseases. Curr. Opin. Nephrol. Hypertens. 2018, 27, 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, V.; Gilani, A.; Shkolnik, B.; Pandey, V.; Zhang, F.F.; Dakarapu, R.; Gandham, S.K.; Reddy, N.R.; Graves, J.P.; Gruzdev, A.; et al. 20-HETE Signals Through G-Protein-Coupled Receptor GPR75 (Gq) to Affect Vascular Function and Trigger Hypertension. Circ. Res. 2017, 120, 1776–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.K.; McGiff, J.C.; Carroll, M.A. Renal arterial 20-hydroxyeicosatetraenoic acid levels: Regulation by cyclooxygenase. Am. J. Physiol. Renal. Physiol. 2003, 284, F474–F479. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Puri, N.; Sodhi, K.; Falck, J.R.; Abraham, N.G.; Shapiro, J.; Schwartzman, M.L. Cyclooxygenase-2 dependent metabolism of 20-HETE increases adiposity and adipocyte enlargement in mesenchymal stem cell-derived adipocytes. J. Lipid Res. 2013, 54, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Fan, F.; Muroya, Y.; Roman, R.J. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 37–46. [Google Scholar] [CrossRef]
- Zhang, C.; Booz, G.W.; Yu, Q.; He, X.; Wang, S.; Fan, F. Conflicting roles of 20-HETE in hypertension and renal end organ damage. Eur. J. Pharmacol. 2018, 833, 190–200. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.T.; Sharma, R.; Sharma, M. Protective effect of 20-hydroxyeicosatetraenoic acid (20-HETE) on glomerular protein permeability barrier. Kidney Int. 2005, 67, 152–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, E.T.; Zhou, J.; Eckert, R.; Genochio, D.; Sharma, R.; Oni, O.; De, A.; Srivastava, T.; Sharma, R.; Savin, V.J.; et al. Ethanol at low concentrations protects glomerular podocytes through alcohol dehydrogenase and 20-HETE. Prostaglandins Other Lipid Mediat. 2015, 116–117, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rand, A.A.; Barnych, B.; Morisseau, C.; Cajka, T.; Lee, K.S.S.; Panigrahy, D.; Hammock, B.D. Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc. Natl. Acad. Sci. USA 2017, 114, 4370–4375. [Google Scholar] [CrossRef] [Green Version]
- Rand, A.A.; Rajamani, A.; Kodani, S.D.; Harris, T.R.; Schlatt, L.; Barnych, B.; Passerini, A.G.; Hammock, B.D. Epoxyeicosatrienoic acid (EET)-stimulated angiogenesis is mediated by epoxy hydroxyeicosatrienoic acids (EHETs) formed from COX-2. J. Lipid Res. 2019, 60, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Herrnreiter, A.; Pfister, S.L.; Gauthier, K.M.; Falck, B.A.; Falck, J.R.; Campbell, W.B. GPR40 is a low-affinity epoxyeicosatrienoic acid receptor in vascular cells. J. Biol. Chem. 2018, 293, 10675–10691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, A.A.; Kim, H.Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Biophys. Acta 2015, 1851, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imig, J.D. Epoxyeicosanoids in hypertension. Physiol. Res. 2019, 68, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Zhou, Y.; Chang, H.H.; Zhang, J.; Seki, T.; Wang, C.Y.; Inscho, E.W.; Wang, M.H. Glomerular 20-HETE, EETs, and TGF-beta1 in diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2009, 296, F556–F563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.; Chen, C. The Role of Epoxyeicosatrienoic Acids in Cardiac Remodeling. Front. Physiol. 2021, 12, 642470. [Google Scholar] [CrossRef]
- Sharma, M.; McCarthy, E.T.; Reddy, D.S.; Patel, P.K.; Savin, V.J.; Medhora, M.; Falck, J.R. 8,9-Epoxyeicosatrienoic acid protects the glomerular filtration barrier. Prostaglandins Other Lipid Mediat. 2009, 89, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Imig, J.D. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol. Rev. 2012, 92, 101–130. [Google Scholar] [CrossRef] [Green Version]
- Imig, J.D. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol. Ther. 2018, 192, 1–19. [Google Scholar] [CrossRef]
- Spector, A.A.; Norris, A.W. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 2007, 292, C996–C1012. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. 2009, 50, S52–S56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wei, Y.; Falck, J.R.; Atcha, K.R.; Wang, W.H. Arachidonic acid inhibits basolateral K channels in the cortical collecting duct via cytochrome P-450 epoxygenase-dependent metabolic pathways. Am. J. Physiol. Renal. Physiol. 2008, 294, F1441–F1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Zarate, R.; El Jaber-Vazdekis, N.; Tejera, N.; Pérez, J.A.; Rodríguez, C. Significance of long chain polyunsaturated fatty acids in human health. Clin. Transl. Med. 2017, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Knapp, H.R. Hypotensive effects of omega 3 fatty acids: Mechanistic aspects. World Rev. Nutr. Diet. 1991, 66, 313–328. [Google Scholar]
- Cicero, A.F.; Ertek, S.; Borghi, C. Omega-3 polyunsaturated fatty acids: Their potential role in blood pressure prevention and management. Curr. Vasc. Pharmacol. 2009, 7, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Ulu, A.; Harris, T.R.; Morisseau, C.; Miyabe, C.; Inoue, H.; Schuster, G.; Dong, H.; Iosif, A.M.; Liu, J.Y.; Weiss, R.H.; et al. Anti-inflammatory effects of ω-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J. Cardiovasc. Pharmacol. 2013, 62, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Katsiki, N.; Mikhailidis, D.P.; Banach, M. Lipid-lowering agents for concurrent cardiovascular and chronic kidney disease. Expert Opin. Pharmacother. 2019, 20, 2007–2017. [Google Scholar] [CrossRef]
- Okada, T.; Morino, K.; Nakagawa, F.; Tawa, M.; Kondo, K.; Sekine, O.; Imamura, T.; Okamura, T.; Ugi, S.; Maegawa, H. N-3 Polyunsaturated Fatty Acids Decrease the Protein Expression of Soluble Epoxide Hydrolase via Oxidative Stress-Induced P38 Kinase in Rat Endothelial Cells. Nutrients 2017, 9, 654. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, H.; Theilla, M.; Attal-Singer, J.; Singer, P. Effects of polyunsaturated fatty acid consumption in diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Adler, A.I. Recent findings on the effects of marine-derived n-3 polyunsaturated fatty acids on urinary albumin excretion and renal function. Curr. Atheroscler. Rep. 2012, 14, 535–541. [Google Scholar] [CrossRef]
- Donadio, J.V., Jr. Omega-3 polyunsaturated fatty acids: A potential new treatment of immune renal disease. Mayo Clin. Proc. 1991, 66, 1018–1028. [Google Scholar] [CrossRef]
- Zivkovic, A.M.; Yang, J.; Georgi, K.; Hegedus, C.; Nording, M.L.; O’Sullivan, A.; German, J.B.; Hogg, R.J.; Weiss, R.H.; Bay, C.; et al. Serum oxylipin profiles in IgA nephropathy patients reflect kidney functional alterations. Metabolomics 2012, 8, 1102–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmocker, C.; Zhang, I.W.; Kiesler, S.; Kassner, U.; Ostermann, A.I.; Steinhagen-Thiessen, E.; Schebb, N.H.; Weylandt, K.H. Effect of Omega-3 Fatty Acid Supplementation on Oxylipins in a Routine Clinical Setting. Int. J. Mol. Sci. 2018, 19, 180. [Google Scholar] [CrossRef] [Green Version]
- Konkel, A.; Schunck, W.H. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim. Biophys. Acta 2011, 1814, 210–222. [Google Scholar] [CrossRef]
- Arnold, C.; Markovic, M.; Blossey, K.; Wallukat, G.; Fischer, R.; Dechend, R.; Konkel, A.; von Schacky, C.; Luft, F.C.; Muller, D.N.; et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}-3 fatty acids. J. Biol. Chem. 2010, 285, 32720–32733. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.; Konkel, A.; Fischer, R.; Schunck, W.H. Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids. Pharmacol. Rep. 2010, 62, 536–547. [Google Scholar] [CrossRef]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Song, Y.; Zhang, X.; Wang, C. Effect of ω-3 Polyunsaturated Fatty Acids-Derived Bioactive Lipids on Metabolic Disorders. Front. Physiol. 2021, 12, 646491. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: Cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 2011, 111, 5899–5921. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going? Neuropsychopharmacology 2018, 43, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.E.; Kim, J.S.; Das, A. Emerging class of omega-3 fatty acid endocannabinoids & their derivatives. Prostaglandins Other Lipid Mediat. 2019, 143, 106337. [Google Scholar] [CrossRef]
- de Bus, I.; Witkamp, R.; Zuilhof, H.; Albada, B.; Balvers, M. The role of n-3 PUFA-derived fatty acid derivatives and their oxygenated metabolites in the modulation of inflammation. Prostaglandins Other Lipid Mediat. 2019, 144, 106351. [Google Scholar] [CrossRef]
- Koura, Y.; Ichihara, A.; Tada, Y.; Kaneshiro, Y.; Okada, H.; Temm, C.J.; Hayashi, M.; Saruta, T. Anandamide decreases glomerular filtration rate through predominant vasodilation of efferent arterioles in rat kidneys. J. Am. Soc. Nephrol. 2004, 15, 1488–1494. [Google Scholar] [CrossRef] [Green Version]
- Jenkin, K.A.; McAinch, A.J.; Zhang, Y.; Kelly, D.J.; Hryciw, D.H. Elevated cannabinoid receptor 1 and G protein-coupled receptor 55 expression in proximal tubule cells and whole kidney exposed to diabetic conditions. Clin. Exp. Pharmacol. Physiol. 2015, 42, 256–262. [Google Scholar] [CrossRef]
- Hinden, L.; Tam, J. Do Endocannabinoids Regulate Glucose Reabsorption in the Kidney? Nephron 2019, 143, 24–27. [Google Scholar] [CrossRef]
- Nam, D.H.; Lee, M.H.; Kim, J.E.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Kim, S.H.; et al. Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology 2012, 153, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, T.; Szanda, G.; Rosenberg, A.Z.; Tam, J.; Earley, B.J.; Godlewski, G.; Cinar, R.; Liu, Z.; Liu, J.; Ju, C.; et al. Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2014, 111, E5420–E5428. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, T.; Park, J.K.; Varga, Z.V.; Pálóczi, J.; Coffey, N.J.; Rosenberg, A.Z.; Godlewski, G.; Cinar, R.; Mackie, K.; Pacher, P.; et al. Cannabinoid-1 receptor deletion in podocytes mitigates both glomerular and tubular dysfunction in a mouse model of diabetic nephropathy. Diabetes Obes. Metab. 2018, 20, 698–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barutta, F.; Bellini, S.; Mastrocola, R.; Gambino, R.; Piscitelli, F.; di Marzo, V.; Corbetta, B.; Vemuri, V.K.; Makriyannis, A.; Annaratone, L.; et al. Reversal of albuminuria by combined AM6545 and perindopril therapy in experimental diabetic nephropathy. Br. J. Pharmacol. 2018, 175, 4371–4385. [Google Scholar] [CrossRef] [Green Version]
- Barutta, F.; Piscitelli, F.; Pinach, S.; Bruno, G.; Gambino, R.; Rastaldi, M.P.; Salvidio, G.; Di Marzo, V.; Cavallo Perin, P.; Gruden, G. Protective role of cannabinoid receptor type 2 in a mouse model of diabetic nephropathy. Diabetes 2011, 60, 2386–2396. [Google Scholar] [CrossRef] [Green Version]
- Barutta, F.; Grimaldi, S.; Franco, I.; Bellini, S.; Gambino, R.; Pinach, S.; Corbelli, A.; Bruno, G.; Rastaldi, M.P.; Aveta, T.; et al. Deficiency of cannabinoid receptor of type 2 worsens renal functional and structural abnormalities in streptozotocin-induced diabetic mice. Kidney Int. 2014, 86, 979–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Locatelli, M.; Corna, D.; Villa, S.; Rottoli, D.; Nava, V.; Verde, R.; Piscitelli, F.; Di Marzo, V.; Fingerle, J.; et al. Therapy with a Selective Cannabinoid Receptor Type 2 Agonist Limits Albuminuria and Renal Injury in Mice with Type 2 Diabetic Nephropathy. Nephron 2016, 132, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Vartak, T.; Godson, C.; Brennan, E. Therapeutic potential of pro-resolving mediators in diabetic kidney disease. Adv. Drug Deliv. Rev. 2021, 178, 113965. [Google Scholar] [CrossRef]
- Brennan, E.; Kantharidis, P.; Cooper, M.E.; Godson, C. Pro-resolving lipid mediators: Regulators of inflammation, metabolism and kidney function. Nat. Rev. Nephrol. 2021, 17, 725–739. [Google Scholar] [CrossRef]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: An update and role in anti-inflammation and pro-resolution. Prostaglandins Other Lipid Mediat. 2002, 68, 433–455. [Google Scholar] [CrossRef]
- Brennan, E.P.; Mohan, M.; McClelland, A.; Tikellis, C.; Ziemann, M.; Kaspi, A.; Gray, S.P.; Pickering, R.; Tan, S.M.; Ali-Shah, S.T.; et al. Lipoxins Regulate the Early Growth Response-1 Network and Reverse Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1437–1448. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvall, M.G.; Levy, B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2016, 785, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qu, X.; Sun, Y.B.; Caruana, G.; Bertram, J.F.; Nikolic-Paterson, D.J.; Li, J. Resolvin D1 protects podocytes in adriamycin-induced nephropathy through modulation of 14-3-3β acetylation. PLoS ONE 2013, 8, e67471. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Lu, Y. Omega-3 fatty acid-derived resolvins and protectins in inflammation resolution and leukocyte functions: Targeting novel lipid mediator pathways in mitigation of acute kidney injury. Front. Immunol. 2013, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.V.; Vik, A.; Serhan, C.N. The Protectin Family of Specialized Pro-resolving Mediators: Potent Immunoresolvents Enabling Innovative Approaches to Target Obesity and Diabetes. Front. Pharmacol. 2019, 9, 1582. [Google Scholar] [CrossRef]
- Maciejewska-Markiewicz, D.; Stachowska, E.; Hawryłkowicz, V.; Stachowska, L.; Prowans, P. The Role of Resolvins, Protectins and Marensins in Non-Alcoholic Fatty Liver Disease (NAFLD). Biomolecules 2021, 11, 937. [Google Scholar] [CrossRef]
- Tang, S.; Gao, C.; Long, Y.; Huang, W.; Chen, J.; Fan, F.; Jiang, C.; Xu, Y. Maresin 1 Mitigates High Glucose-Induced Mouse Glomerular Mesangial Cell Injury by Inhibiting Inflammation and Fibrosis. Mediat. Inflamm. 2017, 2017, 2438247. [Google Scholar] [CrossRef]
- Tang, S.; Wan, M.; Huang, W.; Stanton, R.C.; Xu, Y. Maresins: Specialized Proresolving Lipid Mediators and Their Potential Role in Inflammatory-Related Diseases. Mediat. Inflamm. 2018, 2018, 2380319. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Covic, M.; Huth, C.; Rommel, M.; Adam, J.; Zukunft, S.; Prehn, C.; Wang, L.; Nano, J.; Scheerer, M.F.; et al. Validation of Candidate Phospholipid Biomarkers of Chronic Kidney Disease in Hyperglycemic Individuals and Their Organ-Specific Exploration in Leptin Receptor-Deficient db/db Mouse. Metabolites 2021, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, S.W.; Scherl, T.; Stölcker, B.; Bergler, T.; Hoffmann, U.; Weingart, C.; Banas, M.C.; Kollins, D.; Kammerl, M.C.; Krüger, B.; et al. Lipoxygenase products in the urine correlate with renal function and body temperature but not with acute transplant rejection. Lipids 2013, 48, 167–175. [Google Scholar] [CrossRef]
- Dvash, E.; Har-Tal, M.; Barak, S.; Meir, O.; Rubinstein, M. Leukotriene C4 is the major trigger of stress-induced oxidative DNA damage. Nat. Commun. 2015, 6, 10112. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Sun, S.; Huo, Y.; Yun, L.; Huang, S.; Li, G.; Yan, S. Effects of ACEIs Versus ARBs on Proteinuria or Albuminuria in Primary Hypertension: A Meta-Analysis of Randomized Trials. Medicine 2015, 94, e1560. [Google Scholar] [CrossRef]
- Yanai, K.; Ishibashi, K.; Morishita, Y. Systematic Review and Meta-Analysis of Renin-Angiotensin-Aldosterone System Blocker Effects on the Development of Cardiovascular Disease in Patients With Chronic Kidney Disease. Front. Pharmacol. 2021, 12, 662544. [Google Scholar] [CrossRef] [PubMed]
- Škrtić, M.; Cherney, D.Z. Sodium-glucose cotransporter-2 inhibition and the potential for renal protection in diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2015, 24, 96–103. [Google Scholar] [CrossRef] [PubMed]
- van den Belt, S.M.; Heerspink, H.J.L.; Kirchner, M.; Gracchi, V.; Thurn-Valsassina, D.; Bayazit, A.K.; Niemirska, A.; Canpolat, N.; Kaplan Bulut, I.; Azukaitis, K.; et al. Discontinuation of RAAS Inhibition in Children with Advanced CKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Groen In ‘t Woud, S.; Westland, R.; Feitz, W.F.J.; Roeleveld, N.; van Wijk, J.A.E.; van der Zanden, L.F.M.; Schreuder, M.F. Clinical Management of Children with a Congenital Solitary Functioning Kidney: Overview and Recommendations. Eur. Urol. Open Sci. 2021, 25, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Jandeleit-Dahm, K.; Bonnet, F. Beyond Glycosuria: Exploring the intrarenal effects of SGLT−2 inhibition in diabetes. Diabetes Metab. 2014, 40, S17–S22. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef]
- Zeni, L.; Norden, A.G.W.; Cancarini, G.; Unwin, R.J. A more tubulocentric view of diabetic kidney disease. J. Nephrol. 2017, 30, 701–717. [Google Scholar] [CrossRef]
- Fattah, H.; Vallon, V. The Potential Role of SGLT2 Inhibitors in the Treatment of Type 1 Diabetes Mellitus. Drugs 2018, 78, 717–726. [Google Scholar] [CrossRef]
- Michael, A.; Andreucci, M.; Arturi, F. Sodium-Glucose Co-transporter-2 Inhibitors and Nephroprotection in Diabetic Patients: More Than a Challenge. Front. Med. 2021, 8, 654557. [Google Scholar] [CrossRef]
- Herrington, W.G.; Preiss, D.; Haynes, R.; von Eynatten, M.; Staplin, N.; Hauske, S.J.; George, J.T.; Green, J.B.; Landray, M.J.; Baigent, C.; et al. The potential for improving cardio-renal outcomes by sodium-glucose co-transporter-2 inhibition in people with chronic kidney disease: A rationale for the EMPA-KIDNEY study. Clin. Kidney J. 2018, 11, 749–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, C.C.J.; Gansevoort, R.T.; Heerspink, H.J.L. New Diabetes Therapies and Diabetic Kidney Disease Progression: The Role of SGLT-2 Inhibitors. Curr. Diab. Rep. 2018, 18, 27. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; van Raalte, D.H. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: A promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef]
- Piperidou, A.; Loutradis, C.; Sarafidis, P. SGLT-2 inhibitors and nephroprotection: Current evidence and future perspectives. J. Hum. Hypertens. 2021, 35, 12–25. [Google Scholar] [CrossRef]
- De Nicola, L.; Gabbai, F.B.; Garofalo, C.; Conte, G.; Minutolo, R. Nephroprotection by SGLT2 Inhibition: Back to the Future? J. Clin. Med. 2020, 9, 2243. [Google Scholar] [CrossRef] [PubMed]
- af Forselles, K.J.; Root, J.; Clarke, T.; Davey, D.; Aughton, K.; Dack, K.; Pullen, N. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2; receptor antagonist. Br. J. Pharmacol. 2011, 164, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Aoki, T.; Tsuruyama, T.; Narumiya, S. Definition of Prostaglandin E2-EP2 Signals in the Colon Tumor Microenvironment That Amplify Inflammation and Tumor Growth. Cancer Res. 2015, 75, 2822–2832. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, K.; Saji, T.; Murata, T.; Nagamachi, M.; Matsuoka, T.; Segi, E.; Tsuboi, K.; Sugimoto, Y.; Kobayashi, T.; Miyachi, Y.; et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J. Clin. Investig. 2002, 109, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Thieme, K.; Majumder, S.; Brijmohan, A.S.; Batchu, S.N.; Bowskill, B.B.; Alghamdi, T.A.; Advani, S.L.; Kabir, M.G.; Liu, Y.; Advani, A. EP4 inhibition attenuates the development of diabetic and non-diabetic experimental kidney disease. Sci. Rep. 2017, 7, 3442. [Google Scholar] [CrossRef] [PubMed]
- Edson, K.Z.; Rettie, A.E. CYP4 enzymes as potential drug targets: Focus on enzyme multiplicity, inducers and inhibitors, and therapeutic modulation of 20-hydroxyeicosatetraenoic acid (20-HETE) synthase and fatty acid ω-hydroxylase activities. Curr. Top. Med. Chem. 2013, 13, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Y. Inhibition of Soluble Epoxide Hydrolase for Renal Health. Front. Pharmacol. 2019, 9, 1551. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.P.; Zhang, X.Y.; Morisseau, C.; Hwang, S.H.; Zhang, Z.J.; Hammock, B.D.; Ma, X.C. Discovery of Soluble Epoxide Hydrolase Inhibitors from Chemical Synthesis and Natural Products. J. Med. Chem. 2021, 64, 184–215. [Google Scholar] [CrossRef]
- Luther, J.M.; Brown, N.J. Epoxyeicosatrienoic acids and glucose homeostasis in mice and men. Prostaglandins Other Lipid Mediat. 2016, 125, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Bobiński, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.L.; Lin, J.J.; Goldfine, I.D. Novel approach to treat insulin resistance, type 2 diabetes, and the metabolic syndrome: Simultaneous activation of PPARalpha, PPARgamma, and PPARdelta. Curr. Diabetes Rev. 2005, 1, 299–307. [Google Scholar] [CrossRef]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef]
- Apostoli, A.J.; Nicol, C.J.B. Pharmacological and Toxicological Advances in PPAR-Related Medicines. PPAR Res. 2012, 2012, 940964. [Google Scholar] [CrossRef] [Green Version]
- Kiss-Tóth, E.; Roszer, T. PPARgamma in Kidney Physiology and Pathophysiology. PPAR Res. 2008, 2008, 183108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrales, P.; Izquierdo-Lahuerta, A.; Medina-Gómez, G. Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma. Int. J. Mol. Sci. 2018, 19, 2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.C.; Deleuze, S.; Zuo, Y.; Potthoff, S.A.; Ma, L.J.; Fogo, A.B. The PPARgamma agonist pioglitazone ameliorates aging-related progressive renal injury. J. Am. Soc. Nephrol. 2009, 20, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Shi, M.; Wang, Y.; Liu, J. PPARγ and Its Agonists in Chronic Kidney Disease. Int. J. Nephrol. 2020, 2020, 2917474. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, A.; Satoh, M.; Tomita, N.; Sasaki, T.; Kashihara, N. Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia 2010, 53, 2056–2065. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Marco, R.; Codoñer-Franch, P.; Pons Morales, S.; Del Castillo Villaescusa, C.; Boix García, L.; Valls Bellés, V. Oxidant/antioxidant status and hyperfiltration in young patients with type 1 diabetes mellitus. Pediatr. Nephrol. 2009, 24, 121–127. [Google Scholar] [CrossRef]
- Lew, Q.J.; Jafar, T.H.; Koh, H.W.; Jin, A.; Chow, K.Y.; Yuan, J.M.; Koh, W.P. Red Meat Intake and Risk of ESRD. J. Am. Soc. Nephrol. 2017, 28, 304–312. [Google Scholar] [CrossRef]
- Haring, B.; Selvin, E.; Liang, M.; Coresh, J.; Grams, M.E.; Petruski-Ivleva, N.; Steffen, L.M.; Rebholz, C.M. Dietary Protein Sources and Risk for Incident Chronic Kidney Disease: Results From the Atherosclerosis Risk in Communities (ARIC) Study. J. Ren. Nutr. 2017, 27, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Tantisattamo, E.; Dafoe, D.C.; Reddy, U.G.; Ichii, H.; Rhee, C.M.; Streja, E.; Landman, J.; Kalantar-Zadeh, K. Current Management of Patients With Acquired Solitary Kidney. Kidney Int. Rep. 2019, 4, 1205–1218. [Google Scholar] [CrossRef] [Green Version]
- Attini, R.; Leone, F.; Montersino, B.; Fassio, F.; Minelli, F.; Colla, L.; Rossetti, M.; Rollino, C.; Alemanno, M.G.; Barreca, A.; et al. Pregnancy, Proteinuria, Plant-Based Supplemented Diets and Focal Segmental Glomerulosclerosis: A Report on Three Cases and Critical Appraisal of the Literature. Nutrients 2017, 9, 770. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, D.J.; Kendall, C.W.; Marchie, A.; Jenkins, A.L.; Augustin, L.S.; Ludwig, D.S.; Barnard, N.D.; Anderson, J.W. Type 2 diabetes and the vegetarian diet. Am. J. Clin. Nutr. 2003, 78, 610S–616S. [Google Scholar] [CrossRef] [Green Version]
- Adair, K.E.; Bowden, R.G. Ameliorating Chronic Kidney Disease Using a Whole Food Plant-Based Diet. Nutrients 2020, 12, 1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Hashmi, S.; Shah, S.; Kalantar-Zadeh, K. Plant-based diets for prevention and management of chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Koppe, L.; Fouque, D. The Role for Protein Restriction in Addition to Renin-Angiotensin-Aldosterone System Inhibitors in the Management of CKD. Am. J. Kidney Dis. 2019, 73, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M. Omega-3 fatty acid biochemistry: Perspectives from human nutrition. Mil. Med. 2014, 179, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, Z.; Zhang, H. Omega-3 fatty acid supplementation as an adjunctive therapy in the treatment of chronic kidney disease: A meta-analysis. Clinics 2017, 72, 58–64. [Google Scholar] [CrossRef]
- Fazelian, S.; Moradi, F.; Agah, S.; Hoseini, A.; Heydari, H.; Morvaridzadeh, M.; Omidi, A.; Pizarro, A.B.; Ghafouri, A.; Heshmati, J. Effect of omega-3 fatty acids supplementation on cardio-metabolic and oxidative stress parameters in patients with chronic kidney disease: A systematic review and meta-analysis. BMC Nephrol. 2021, 22, 160. [Google Scholar] [CrossRef]
- Pazderka, C.W.; Oliver, B.; Murray, M.; Rawling, T. Omega-3 Polyunsaturated Fatty Acid Derived Lipid Mediators and their Application in Drug Discovery. Curr. Med. Chem. 2020, 27, 1670–1689. [Google Scholar] [CrossRef]
- Vargas, F.; Romecín, P.; García-Guillén, A.I.; Wangesteen, R.; Vargas-Tendero, P.; Paredes, M.D.; Atucha, N.M.; García-Estañ, J. Flavonoids in Kidney Health and Disease. Front. Physiol. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- Tam, J. The emerging role of the endocannabinoid system in the pathogenesis and treatment of kidney diseases. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Cuffe, J.S.M.; Hutchinson, D.S.; Headrick, J.P.; Perkins, A.V.; McAinch, A.J.; Hryciw, D.H. Peripheral modulation of the endocannabinoid system in metabolic disease. Drug Discov. Today 2018, 23, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Barutta, F.; Grimaldi, S.; Gambino, R.; Vemuri, K.; Makriyannis, A.; Annaratone, L.; di Marzo, V.; Bruno, G.; Gruden, G. Dual therapy targeting the endocannabinoid system prevents experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2017, 32, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Jenkin, K.A.; O’Keefe, L.; Simcocks, A.C.; Briffa, J.F.; Mathai, M.L.; McAinch, A.J.; Hryciw, D.H. Renal effects of chronic pharmacological manipulation of CB2 receptors in rats with diet-induced obesity. Br. J. Pharmacol. 2016, 173, 1128–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jin, D.; Kang, X.; Zhou, R.; Sun, Y.; Lian, F.; Tong, X. Signaling Pathways Involved in Diabetic Renal Fibrosis. Front. Cell Dev. Biol. 2021, 9, 696542. [Google Scholar] [CrossRef]
- Samuel, C.S.; Hewitson, T.D. Relaxin in cardiovascular and renal disease. Kidney Int. 2006, 69, 1498–1502. [Google Scholar] [CrossRef] [Green Version]
- Németh, Á.; Mózes, M.M.; Calvier, L.; Hansmann, G.; Kökény, G. The PPARγ agonist pioglitazone prevents TGF-β induced renal fibrosis by repressing EGR-1 and STAT3. BMC Nephrol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Zhang, M.J.; Spite, M. Resolvins: Anti-inflammatory and proresolving mediators derived from omega-3 polyunsaturated fatty acids. Annu. Rev. Nutr. 2012, 32, 203–227. [Google Scholar] [CrossRef]
- Mas, E.; Barden, A.; Burke, V.; Beilin, L.J.; Watts, G.F.; Huang, R.C.; Puddey, I.B.; Irish, A.B.; Mori, T.A. A randomized controlled trial of the effects of n-3 fatty acids on resolvins in chronic kidney disease. Clin. Nutr. 2016, 35, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]





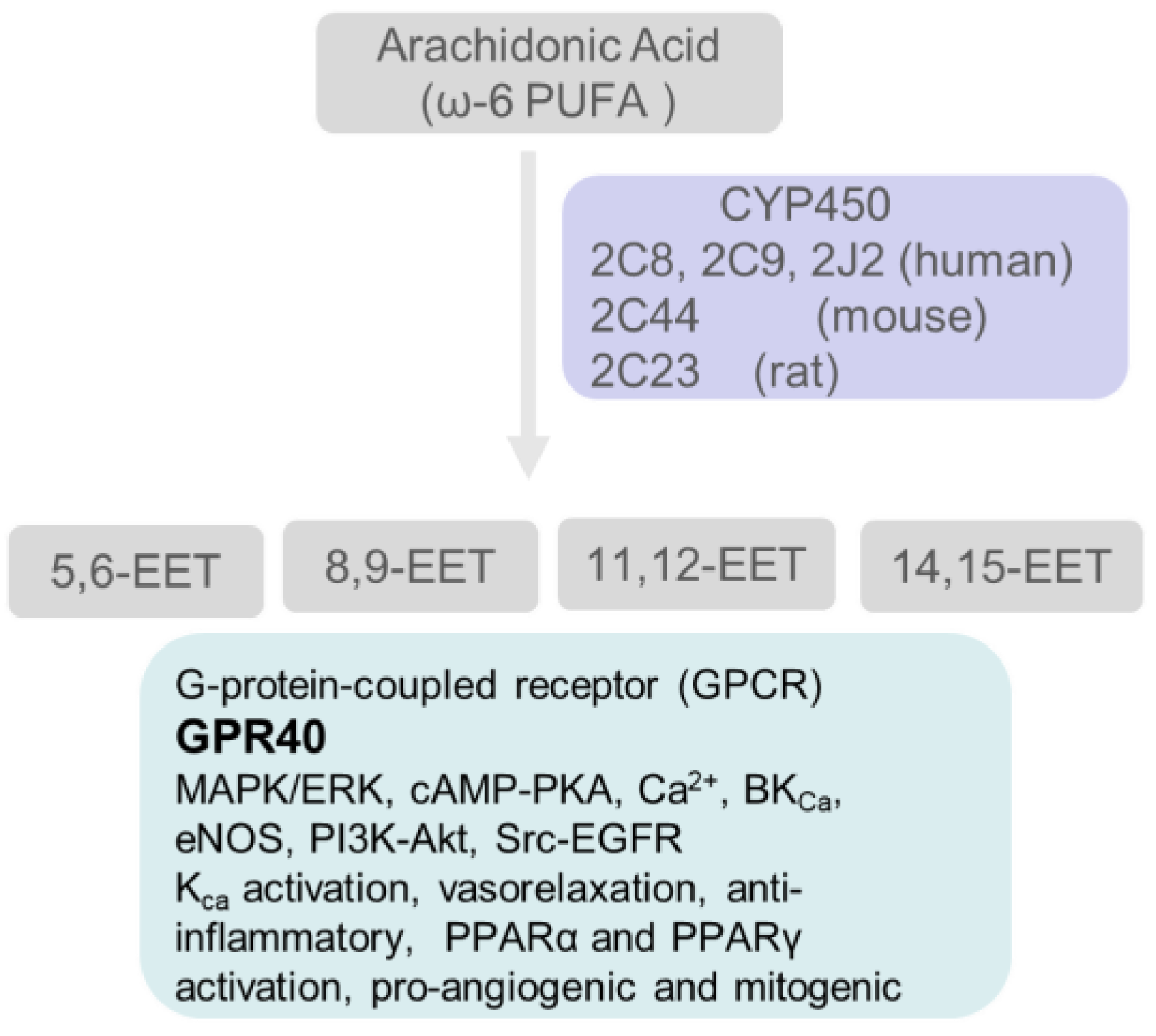

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathophysiology Associated with Hyperfiltration/Kidney Disease | References |
|---|---|
| High dietary protein consumption by vulnerable groups | [18,19,20] |
| Obesity | [21,22,23,24,25,26,27,28,29] |
| Diabetes | [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44] |
| Hypertension | [45,46,47,48,49,50,51,52] |
| Primary hyperaldosteronism | [53,54,55,56] |
| Non-alcoholic fatty liver disease (NAFLD) | [57,58,59,60,61,62] |
| CKD in Kidney donors | [14,17,63,64,65,66,67,68] |
| CKD in Children born with single functioning kidney or low number of functional nephrons due to other Congenital Anomalies of the Kidney and Urinary Tract (CAKUT) | [16,69,70,71,72,73] |
| Autosomal Dominant Polycystic Kidney Disease (ADPKD) | [74] |
| Secondary focal segmental glomerulosclerosis (FSGS) | [75,76,77,78] |
| Sickle cell Disease (SCD) and glomerular sclerosis | [79,80,81,82] |
| Cyanotic congenital heart disease/critical congenital heart disease (CCHD) | [83,84,85,86,87] |
| ‘Autoimmune activation’ and inflammation | [88,89] |
| High altitude renal syndrome | [90] |
| Dementia | [91] |
| Stroke | [92,93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; Singh, V.; Sharma, R.; Koul, A.; McCarthy, E.T.; Savin, V.J.; Joshi, T.; Srivastava, T. Glomerular Biomechanical Stress and Lipid Mediators during Cellular Changes Leading to Chronic Kidney Disease. Biomedicines 2022, 10, 407. https://doi.org/10.3390/biomedicines10020407
Sharma M, Singh V, Sharma R, Koul A, McCarthy ET, Savin VJ, Joshi T, Srivastava T. Glomerular Biomechanical Stress and Lipid Mediators during Cellular Changes Leading to Chronic Kidney Disease. Biomedicines. 2022; 10(2):407. https://doi.org/10.3390/biomedicines10020407
Chicago/Turabian StyleSharma, Mukut, Vikas Singh, Ram Sharma, Arnav Koul, Ellen T. McCarthy, Virginia J. Savin, Trupti Joshi, and Tarak Srivastava. 2022. "Glomerular Biomechanical Stress and Lipid Mediators during Cellular Changes Leading to Chronic Kidney Disease" Biomedicines 10, no. 2: 407. https://doi.org/10.3390/biomedicines10020407
APA StyleSharma, M., Singh, V., Sharma, R., Koul, A., McCarthy, E. T., Savin, V. J., Joshi, T., & Srivastava, T. (2022). Glomerular Biomechanical Stress and Lipid Mediators during Cellular Changes Leading to Chronic Kidney Disease. Biomedicines, 10(2), 407. https://doi.org/10.3390/biomedicines10020407