
Inflammation and Rho-Associated Protein Kinase-Induced Brain Changes in Vascular Dementia

 ,
,  , , , and
, , , and
Abstract
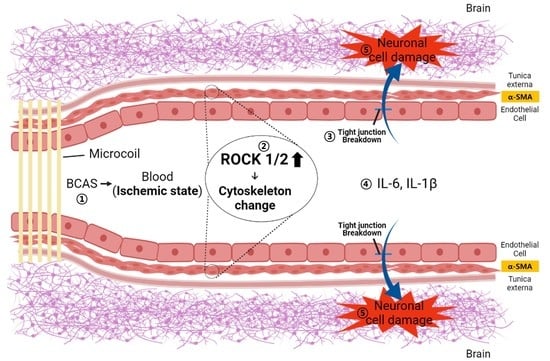
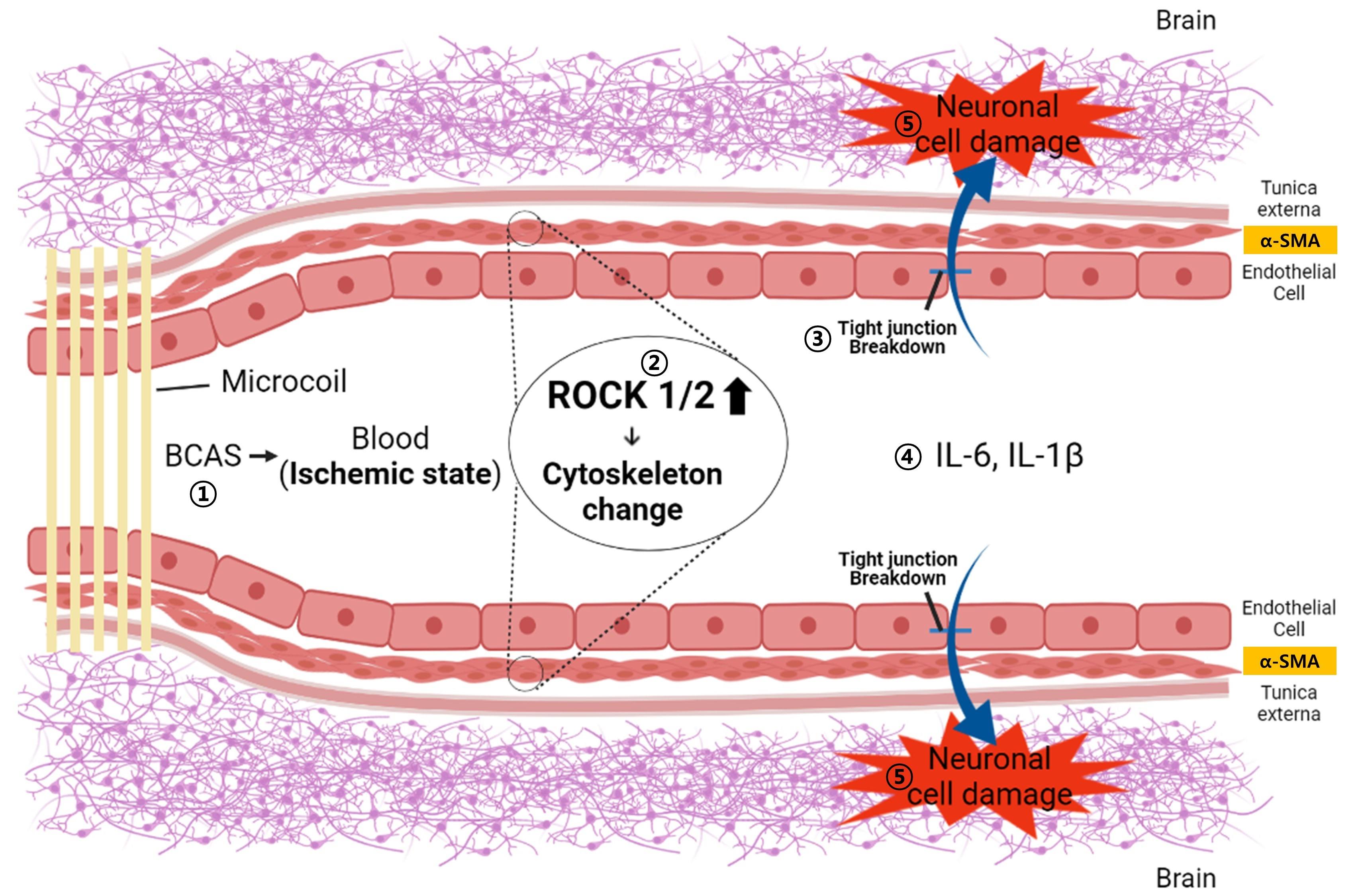
:
1. Introduction
2. Materials and Methods
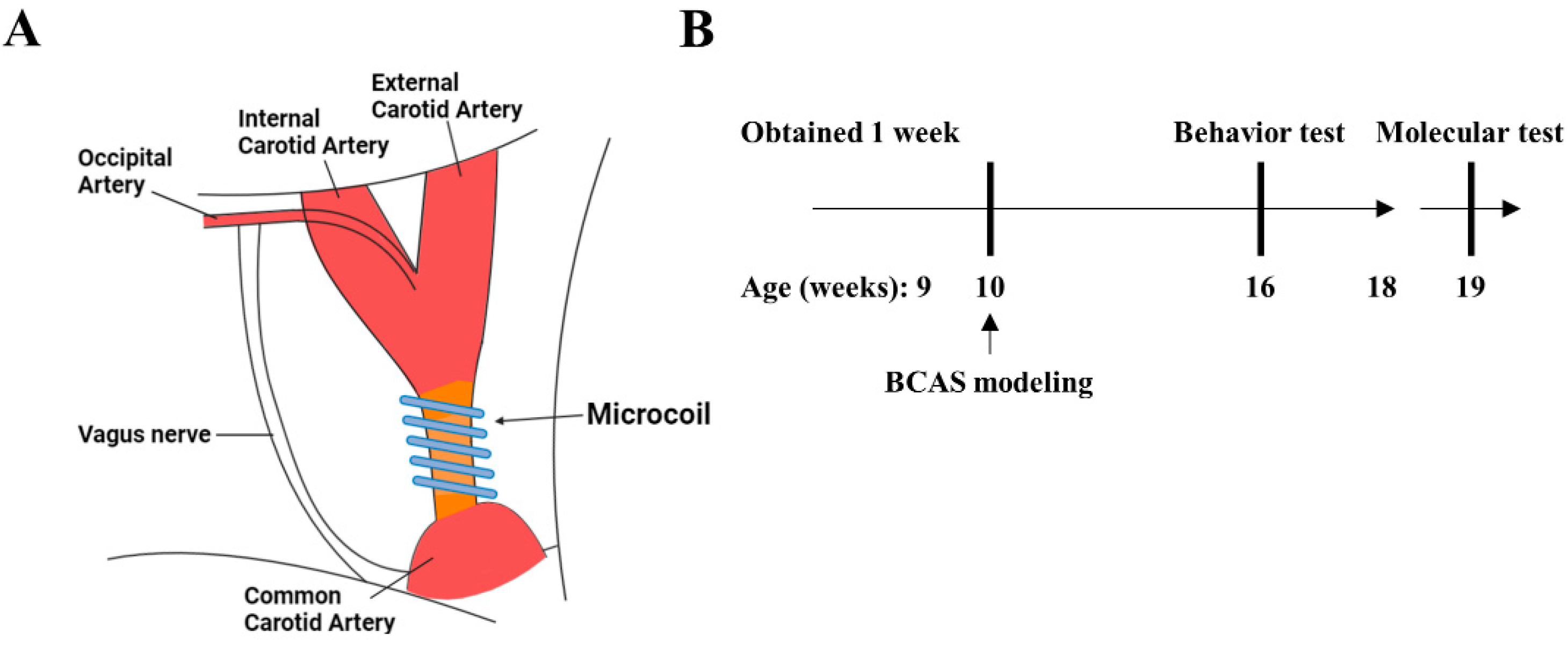
2.1. Animals
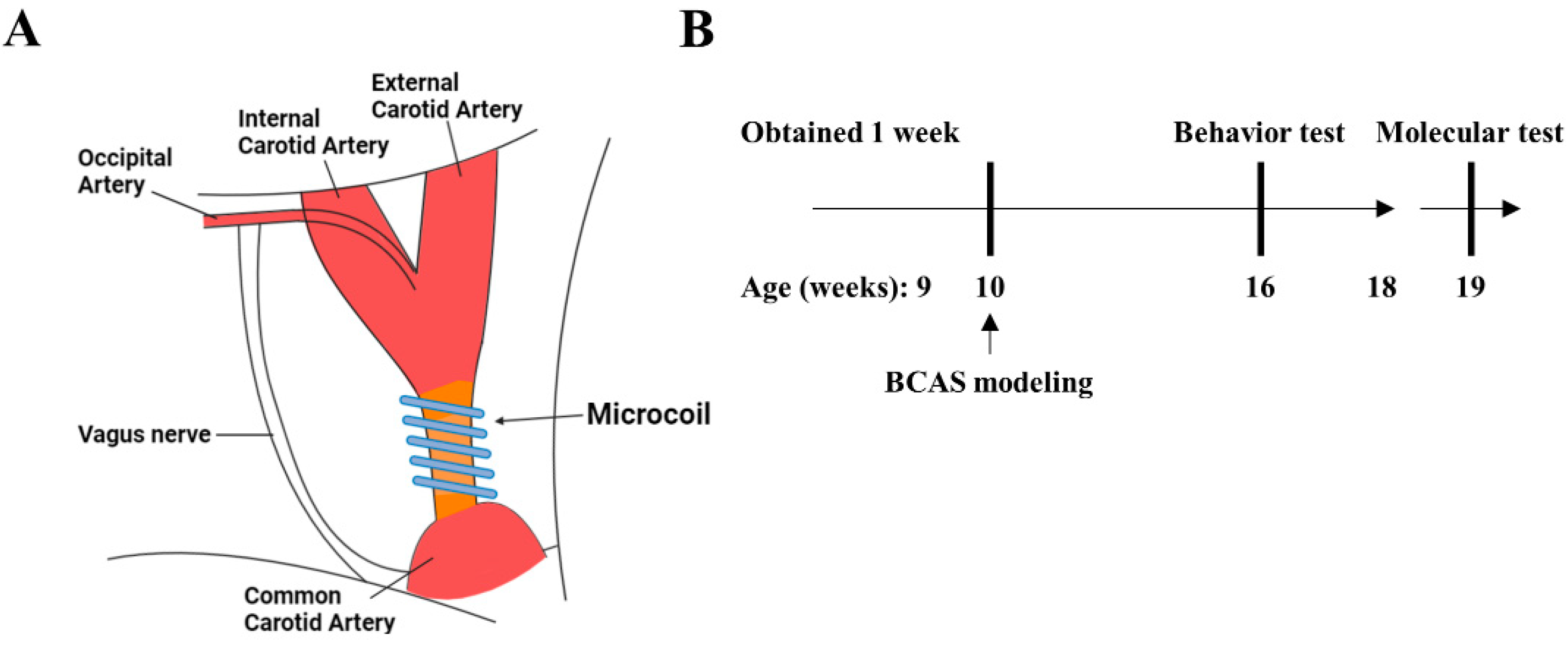
2.2. Experimental Design
2.3. Behavior Tests
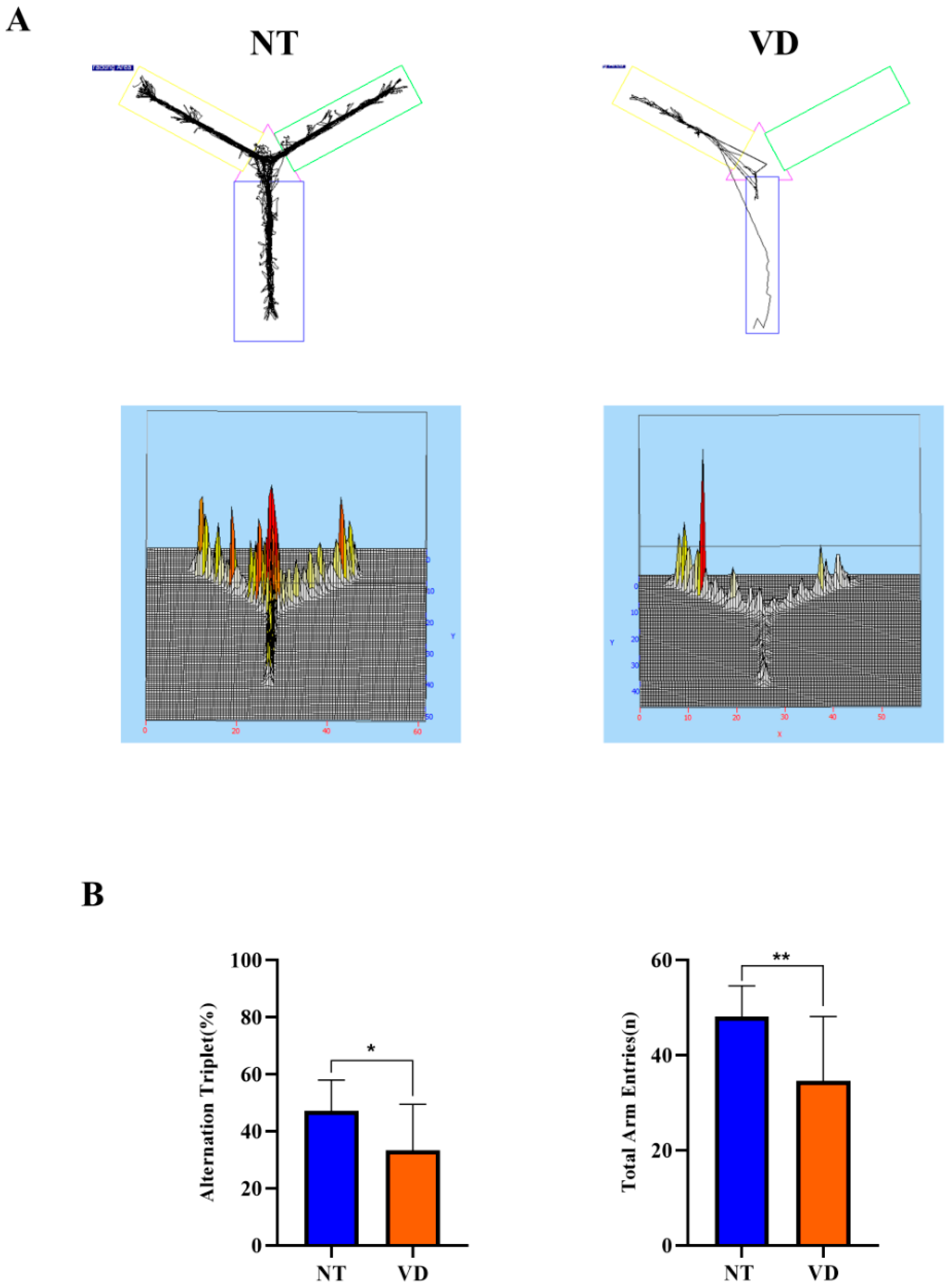
2.3.1. Y-Maze Test
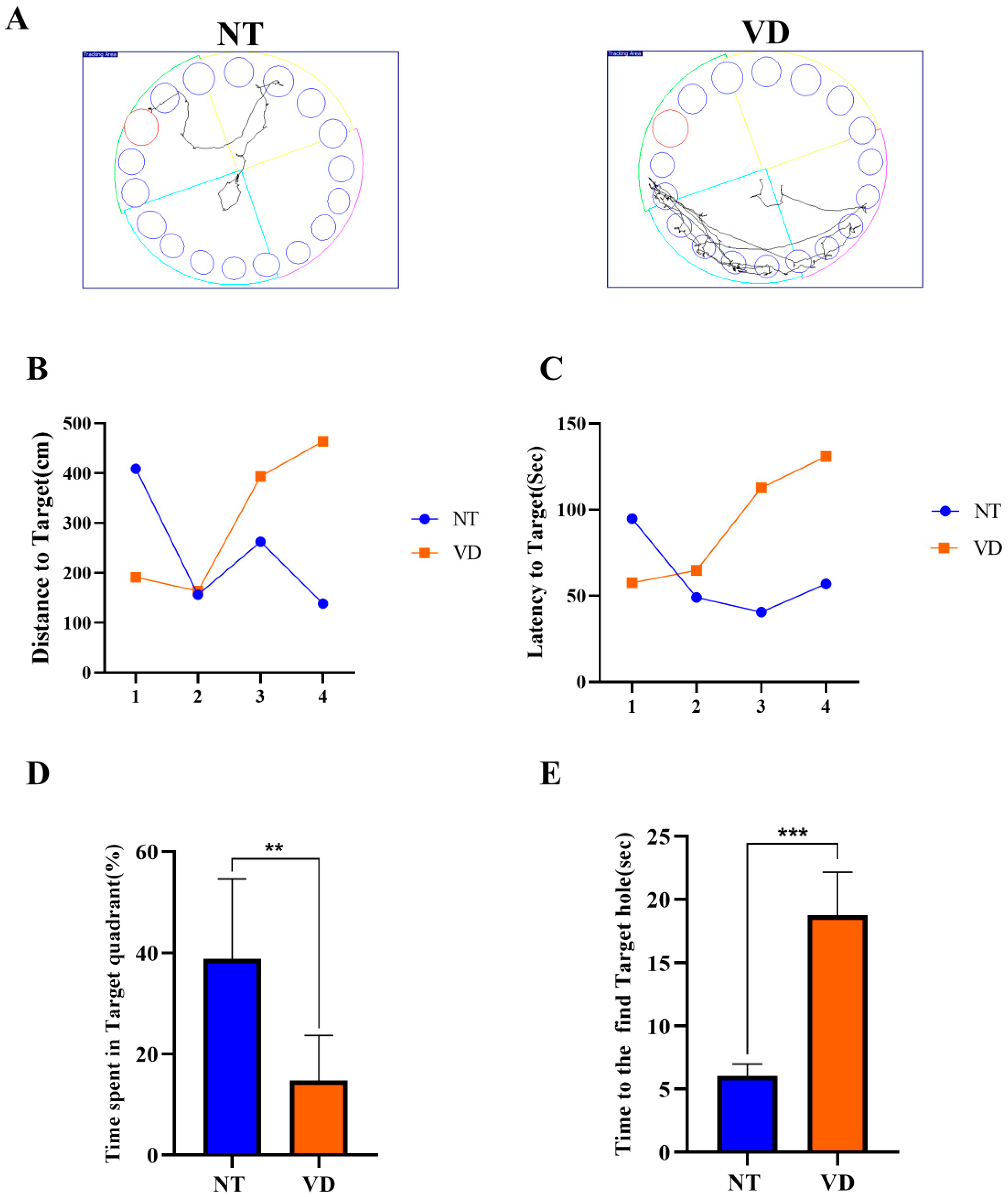
2.3.2. Barnes-Maze Test
2.3.3. Passive Avoidance Test
2.3.4. Open Field Test
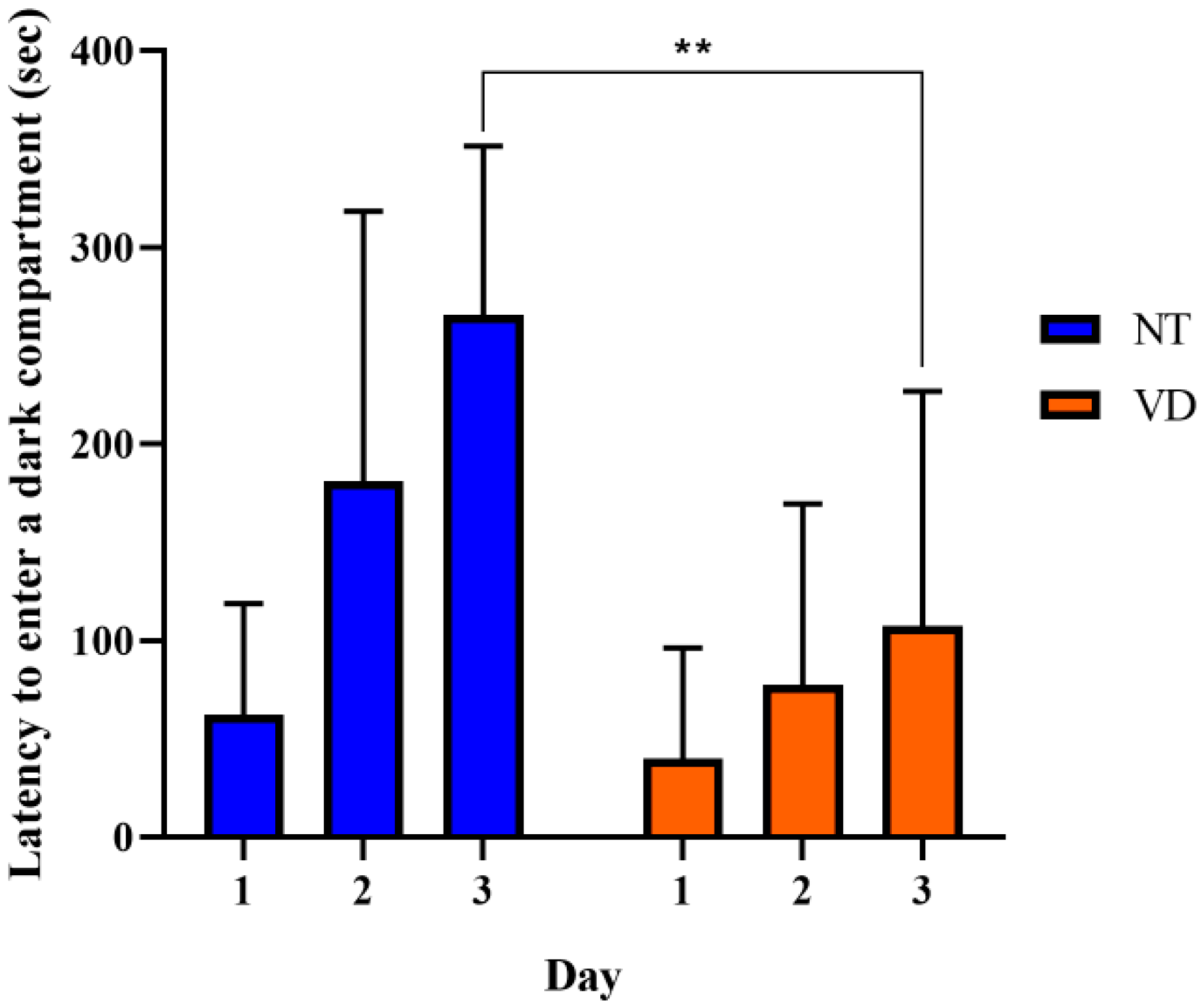
2.3.5. Light and Dark Test
2.4. Reverse-Transcription Quantitative Real-Time PCR
2.5. Immunohistochemical Staining
2.6. Luxol Fast Blue Staining
2.7. Statistical Analysis
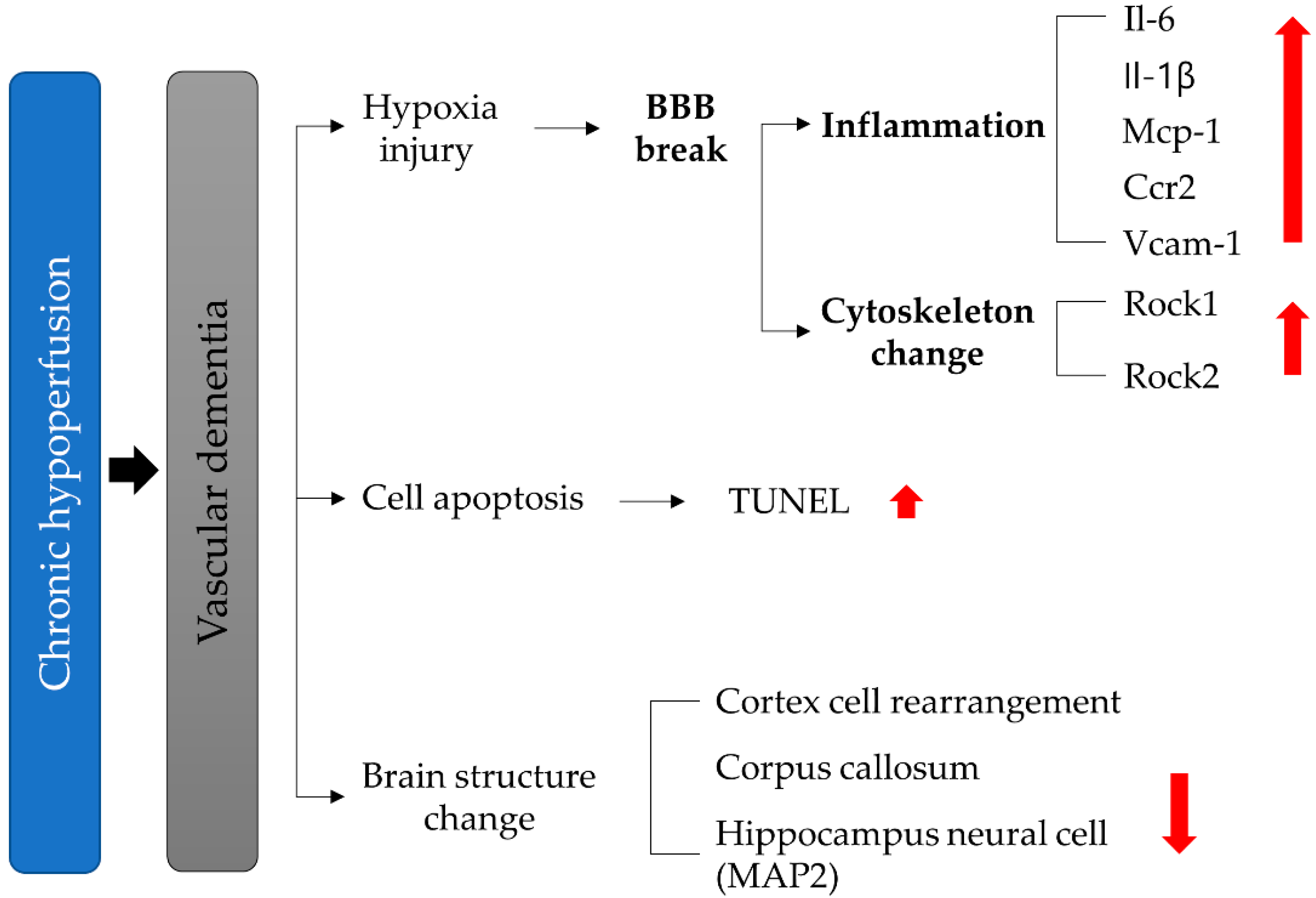
3. Results
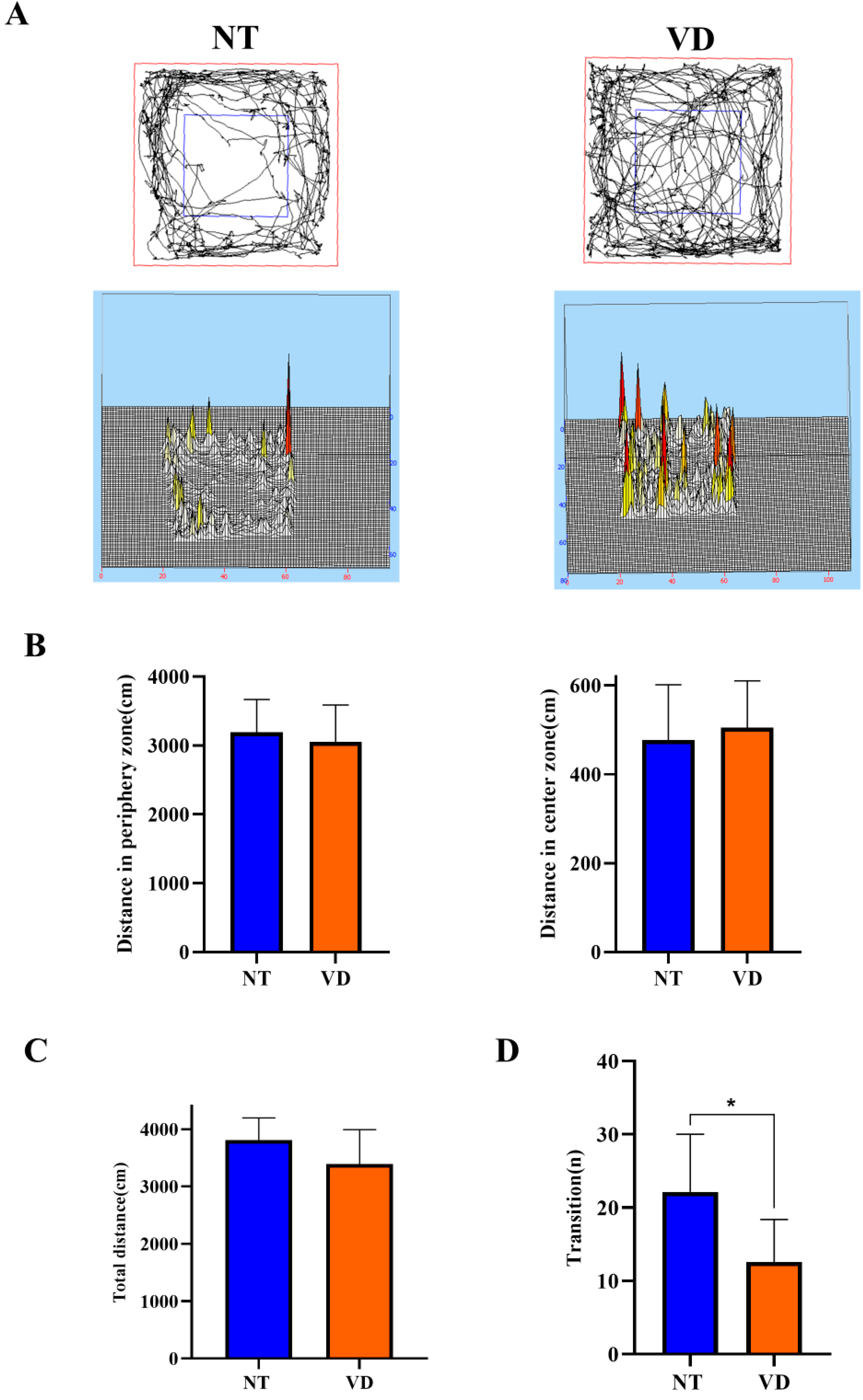
3.1. Behavior Testing Results
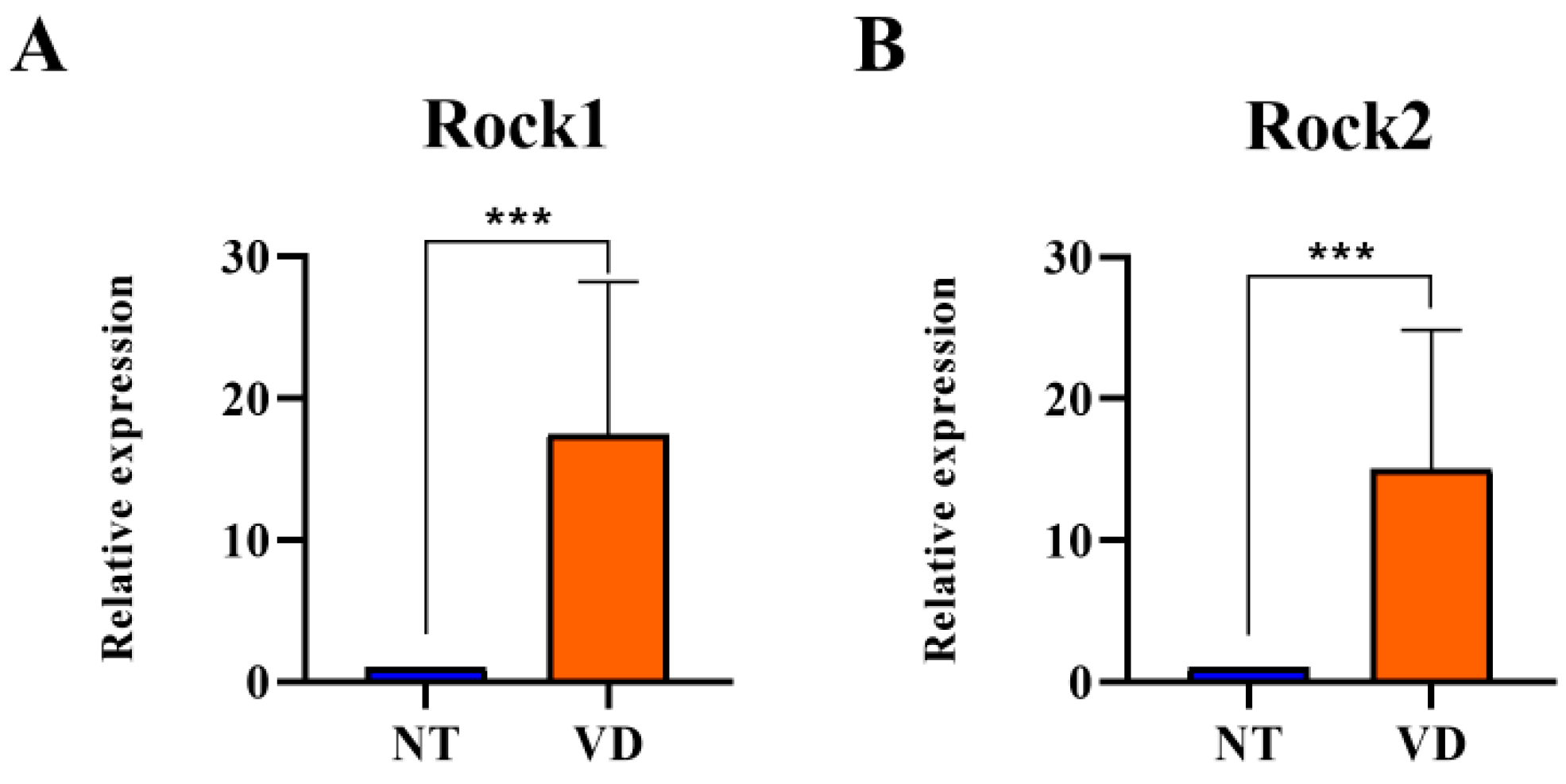
3.2. Increases in ROCK Expression in the Brains of VD Model Mice
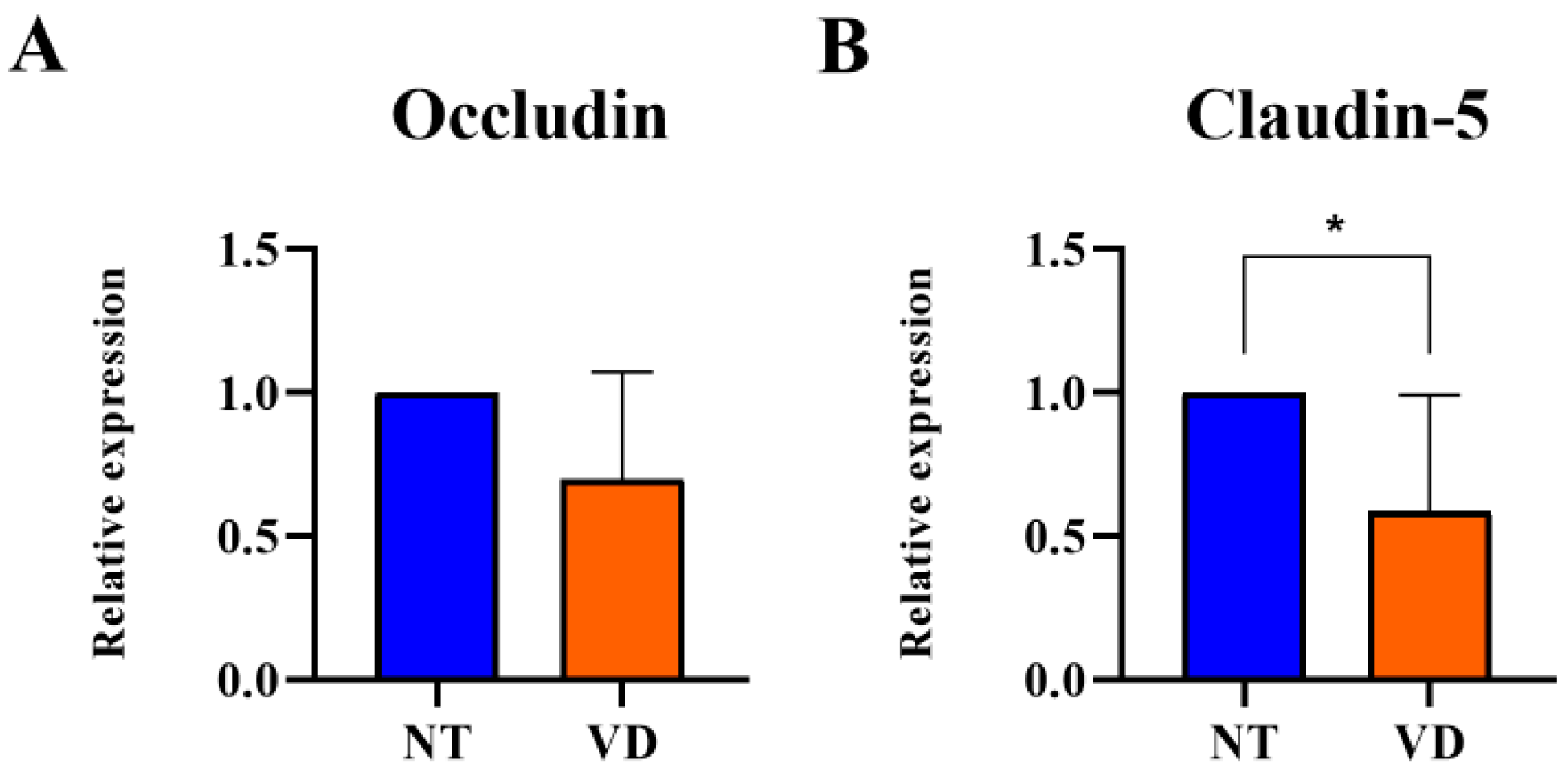
3.3. Reduction in TJ Protein Expression in the Brain of VD Model Mice
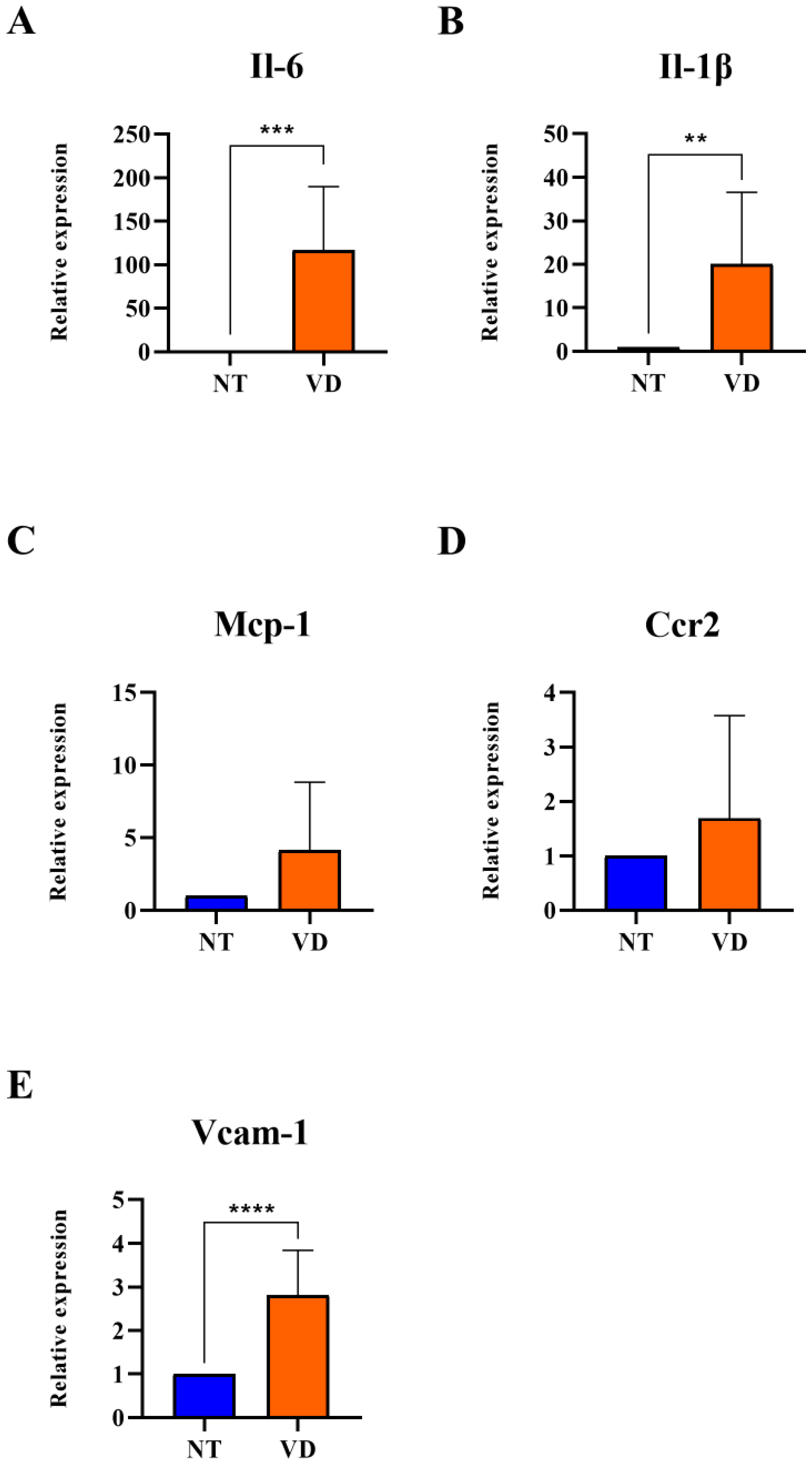
3.4. Increased Expression of Adhesion Molecules and Pro-Inflammatory Cytokines in VD Mice
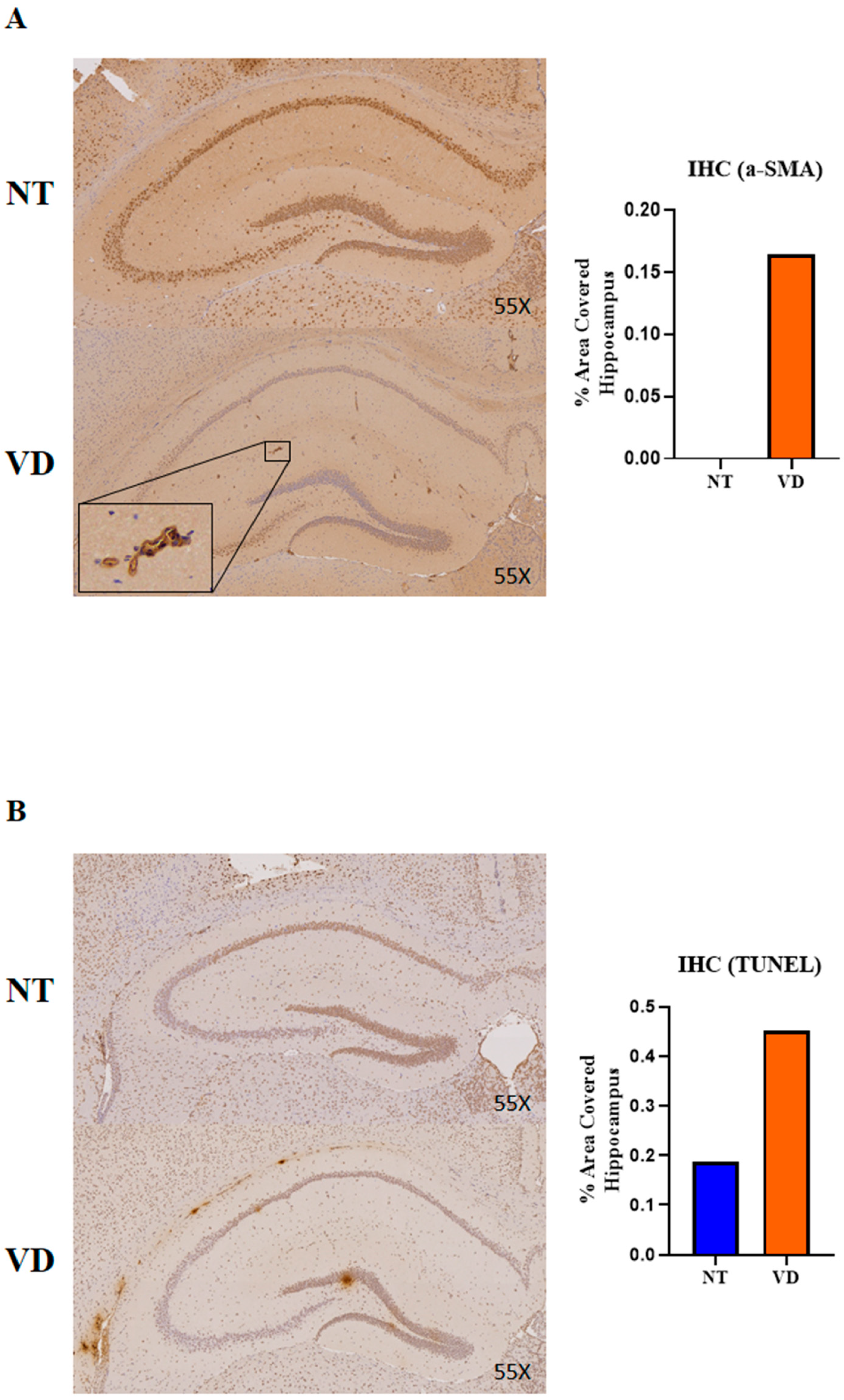
3.5. Vasoconstriction and Apoptosis in the VD Group
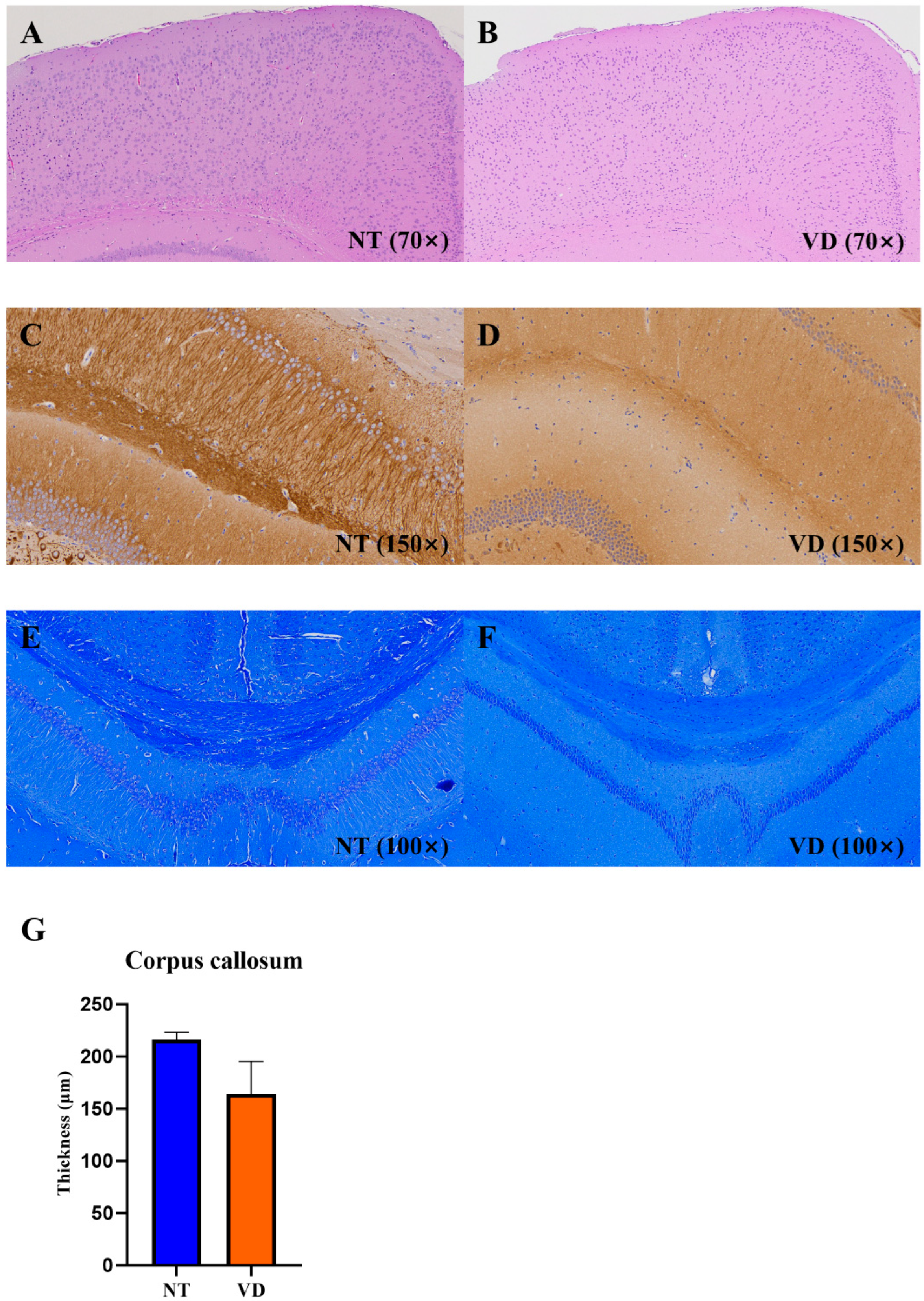
3.6. Changes in Brain Structure in the VD Group
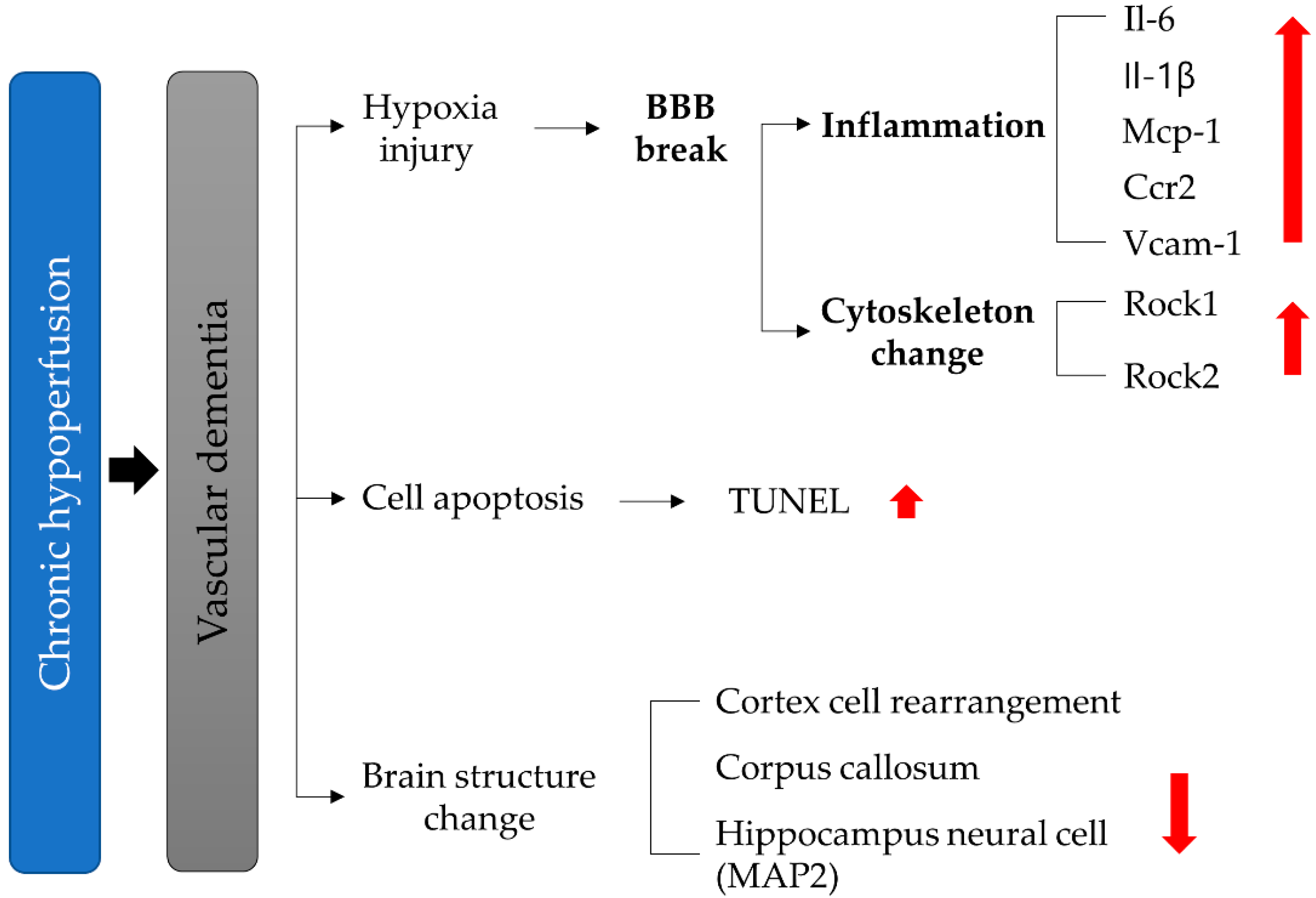
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- White, H.; Boden-Albala, B.; Wang, C.; Elkind, M.S.; Rundek, T.; Wright, C.B.; Sacco, R.L. Ischemic stroke subtype incidence among whites, blacks, and Hispanics: The Northern Manhattan Study. Circulation 2005, 111, 1327–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.A.; Dundas, R.; Howard, R.S.; Rudd, A.G.; Wolfe, C.D.A. Ethnic differences in incidence of stroke: Prospective study with stroke register. BMJ 1999, 318, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.H.; Bae, H.J.; Kwon, S.U.; Kang, D.W.; Hong, K.S.; Lee, Y.S.; Rha, J.H.; Koo, J.S.; Kim, J.S.; Kim, J.H.; et al. Analysis of 10,811 cases with acute ischemic stroke from Korean Stroke Registry: Hospital-based multicenter prospective registration study. J. Korean Neurol. Assoc. 2006, 24, 535–543. [Google Scholar]
- Wolters, F.J.; Ikram, M.A. Epidemiology of Vascular Dementia. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1542–1549. [Google Scholar] [CrossRef]
- Smith, E.E. Clinical presentations and epidemiology of vascular dementia. Clin. Sci. 2017, 131, 1059–1068. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Ballard, C. Overlap between pathology of Alzheimer disease and vascular dementia. In Alzheimer Disease and Associated Disorders; Lippincott Williams & Wilkins, Inc.: Philadelphia, PA, USA, 1999. [Google Scholar]
- Du, S.Q.; Wang, X.R.; Xiao, L.Y.; Tu, J.F.; Zhu, W.; He, T.; Liu, C.Z. Molecular Mechanisms of Vascular Dementia: What Can Be Learned from Animal Models of Chronic Cerebral Hypoperfusion? Mol. Neurobiol. 2017, 54, 3670–3682. [Google Scholar] [CrossRef]
- Bink, D.I.; Ritz, K.; Aronica, E.; Van Der Weerd, L.; Daemen, M.J. Mouse models to study the effect of cardiovascular risk factors on brain structure and cognition. J. Cereb. Blood Flow Metab. 2013, 33, 1666–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wu, B.; Nie, K.; Jia, Y.; Yu, J. Effects of acupuncture on declined cerebral blood flow, impaired mitochondrial respiratory function and oxidative stress in multi-infarct dementia rats. Neurochem. Int. 2014, 65, 23–29. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox. Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Bang, J.H.; Lee, J.; Han, J.S.; Kang, H.W.; Jeon, W.K. Fructus mume Ethanol Extract Prevents Inflammation and Normalizes the Septohippocampal Cholinergic System in a Rat Model of Chronic Cerebral Hypoperfusion. J. Med. Food 2016, 19, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Toldi, J.; Vecsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Lara, E.; Aguirre, S.; Clotet, N.; Sawkulycz, X.; Bartra, C.; Almenara-Fuentes, L.; Sunol, C.; Corpas, R.; Olah, P.; Tripon, F.; et al. Antibody Protection against Long-Term Memory Loss Induced by Monomeric C-Reactive Protein in a Mouse Model of Dementia. Biomedicines 2021, 9, 828. [Google Scholar] [CrossRef]
- Benkhalifa, M.; Ferreira, Y.J.; Chahine, H.; Louanjli, N.; Miron, P.; Merviel, P.; Copin, H. Mitochondria: Participation to infertility as source of energy and cause of senescence. Int. J. Biochem. Cell Biol. 2014, 55, 60–64. [Google Scholar] [CrossRef]
- Brunetti, D.; Catania, A.; Viscomi, C.; Deleidi, M.; Bindoff, L.A.; Ghezzi, D.; Zeviani, M. Role of PITRM1 in Mitochondrial Dysfunction and Neurodegeneration. Biomedicines 2021, 9, 833. [Google Scholar] [CrossRef]
- Jiwa, N.S.; Garrard, P.; Hainsworth, A.H. Experimental models of vascular dementia and vascular cognitive impairment: A systematic review. J. Neurochem. 2010, 115, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G.; Bari, F. Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 2007, 54, 162–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, M.; Tomimoto, H. Lessons from a mouse model characterizing features of vascular cognitive impairment with white matter changes. J. Aging Res. 2011, 2011, 978761. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.Z.; Zou, J.J.; Lei, P. Rodent Models of Vascular Cognitive Impairment. J. Mol. Neurosci. 2021, 71, 1–12. [Google Scholar] [CrossRef]
- Shibata, M.; Ohtani, R.; Ihara, M.; Tomimoto, H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 2004, 35, 2598–2603. [Google Scholar] [CrossRef] [Green Version]
- Gudapati, K.; Singh, A.; Clarkson-Townsend, D.; Feola, A.J.; Allen, R.S. Behavioral assessment of visual function via optomotor response and cognitive function via Y-maze in diabetic rats. JoVE J. Vis. Exp. 2020, 164, e61806. [Google Scholar] [CrossRef]
- Kang, S.; Ha, S.; Park, H.; Nam, E.; Suh, W.H.; Suh, Y.H.; Chang, K.A. Effects of a dehydroevodiamine-derivative on synaptic destabilization and memory impairment in the 5xFAD, Alzheimer’s disease mouse model. Front. Behav. Neurosci. 2018, 12, 273. [Google Scholar] [CrossRef]
- Doze, V.A.; Papay, R.S.; Goldenstein, B.L.; Gupta, M.K.; Collette, K.M.; Nelson, B.W.; Lyons, M.J.; Davis, B.A.; Luger, E.J.; Wood, S.G.; et al. Long-term α1A-adrenergic receptor stimulation improves synaptic plasticity, cognitive function, mood, and longevity. Mol. Pharmacol. 2011, 80, 747–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Yang, L.; Tucker, D.; Dong, Y.A.N.; Zhu, L.; Duan, R.U.I.; Liu, T.C.; Zhang, Q. Beneficial effects of exercise pretreatment in a sporadic Alzheimer’s rat model. Med. Sci. Sports Exerc. 2018, 50, 945. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.; Fang, J.; Xu, L.; Zhou, W.; Kang, D.; Xiong, W.; Jia, H.; Liu, A.L.; Du, G.H. DL0410 Ameliorates Memory and Cognitive Impairments Induced by Scopolamine via Increasing Cholinergic Neurotransmission in Mice. Molecules 2017, 22, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibenhener, M.L.; Wooten, M.C. Use of the open field maze to measure locomotor and anxiety-like behavior in mice. JoVE J. Vis. Exp. 2015, 96, e52434. [Google Scholar] [CrossRef] [Green Version]
- de Lombares, C.; Heude, E.; Alfama, G.; Fontaine, A.; Hassouna, R.; Vernochet, C.; de Chaumont, F.; Olivo-Marin, C.; Ey, E.; Parnaudeau, S.; et al. Dlx5 and Dlx6 expression in GABAergic neurons controls behavior, metabolism, healthy aging and lifespan. Aging 2019, 11, 6638–6656. [Google Scholar] [CrossRef]
- Shimada, T.; Matsumoto, K.; Osanai, M.; Matsuda, H.; Terasawa, K.; Watanabe, H. The modified light/dark transition test in mice: Evaluation of classic and putative anxiolytic and anxiogenic drugs. Gen. Pharmacol. 1995, 26, 205–210. [Google Scholar] [CrossRef]
- Kulesskaya, N.; Voikar, V. Assessment of mouse anxiety-like behavior in the light-dark box and open-field arena: Role of equipment and procedure. Physiol. Behav. 2014, 133, 30–38. [Google Scholar] [CrossRef]
- Supanc, V.; Biloglav, Z.; Kes, V.B.; Demarin, V. Role of cell adhesion molecules in acute ischemic stroke. Ann. Saudi Med. 2011, 31, 365–370. [Google Scholar] [CrossRef]
- Eidson, L.N.; Gao, Q.; Qu, H.; Kikuchi, D.S.; Campos, A.C.P.; Faidley, E.A.; Sun, Y.Y.; Kuan, C.Y.; Pagano, R.L.; Lassègue, B. Poldip2 controls leukocyte infiltration into the ischemic brain by regulating focal adhesion kinase-mediated VCAM-1 induction. Sci. Rep. 2021, 11, 5533. [Google Scholar] [CrossRef]
- Fang, W.; Sha, L.; Kodithuwakku, N.D.; Wei, J.; Zhang, R.; Han, D.; Mao, L.; Li, Y. Attenuated blood-brain barrier dysfunction by XQ-1H following ischemic stroke in hyperlipidemic rats. Mol. Neurobiol. 2015, 52, 162–175. [Google Scholar] [CrossRef]
- Hosoki, S.; Tanaka, T.; Ihara, M. Diagnostic and prognostic blood biomarkers in vascular dementia: From the viewpoint of ischemic stroke. Neurochem. Int. 2021, 146, 105015. [Google Scholar] [CrossRef]
- Schüz, A.; Preiβl, H. Basic connectivity of the cerebral cortex and some considerations on the corpus callosum. Neurosci. Biobehav. Rev. 1996, 20, 567–570. [Google Scholar] [CrossRef]
- Westerhausen, R.; Luders, E.; Specht, K.; Ofte, S.H.; Toga, A.W.; Thompson, P.M.; Helland, T.; Hugdahl, K. Structural and functional reorganization of the corpus callosum between the age of 6 and 8 years. Cereb. Cortex 2011, 21, 1012–1017. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Sullivan, N.; Esiri, M.M. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke 2001, 32, 1162–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.R.; Kim, D.H.; Back, D.B.; Kang, C.H.; Moon, W.J.; Han, J.S.; Choi, D.H.; Kwon, K.J.; Shin, C.Y.; Kim, B.R.; et al. Characterization of White Matter Injury in a Rat Model of Chronic Cerebral Hypoperfusion. Stroke 2016, 47, 542–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hase, Y.; Craggs, L.; Hase, M.; Stevenson, W.; Slade, J.; Lopez, D.; Mehta, R.; Chen, A.; Liang, D.; Oakley, A.; et al. Effects of environmental enrichment on white matter glial responses in a mouse model of chronic cerebral hypoperfusion. J. Neuroinflammation 2017, 14, 81. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.H.; Si, W.; Yu, Q.; Wang, Y.; Wu, Y. Therapeutic effects of combination environmental enrichment with necrostatin-1 on cognition following vascular cognitive impairment in mice. Eur. J. Inflamm. 2019, 17, 2058739219834832. [Google Scholar] [CrossRef] [Green Version]
- Bannai, T.; Mano, T.; Chen, X.; Ohtomo, G.; Ohtomo, R.; Tsuchida, T.; Koshi-Mano, K.; Hashimoto, T.; Okazawa, H.; Iwatsubo, T.; et al. Chronic cerebral hypoperfusion shifts the equilibrium of amyloid beta oligomers to aggregation-prone species with higher molecular weight. Sci. Rep. 2019, 9, 2827. [Google Scholar] [CrossRef]
- Hattori, Y.; Enmi, J.; Kitamura, A.; Yamamoto, Y.; Saito, S.; Takahashi, Y.; Iguchi, S.; Tsuji, M.; Yamahara, K.; Nagatsuka, K.; et al. A Novel Mouse Model of Subcortical Infarcts with Dementia. J. Neurosci. 2015, 35, 3915–3928. [Google Scholar] [CrossRef]
- Hattori, Y.; Kitamura, A.; Tsuji, M.; Nagatsuka, K.; Ihara, M. Motor and cognitive impairment in a mouse model of ischemic carotid artery disease. Neurosci. Lett. 2014, 581, 1–6. [Google Scholar] [CrossRef]
- Toyama, K.; Spin, J.M.; Deng, A.C.; Huang, T.T.; Wei, K.; Wagenhauser, M.U.; Yoshino, T.; Nguyen, H.; Mulorz, J.; Kundu, S.; et al. MicroRNA-Mediated Therapy Modulating Blood-Brain Barrier Disruption Improves Vascular Cognitive Impairment. Arter. Thromb. Vasc. Biol. 2018, 38, 1392–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madureira, S.; Verdelho, A.; Pantoni, L.; Scheltens, P. White matter changes: New perspectives on imaging, clinical aspects, and intervention. J. Aging Res. 2011, 2011, 841913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Yamasaki, N.; Miyakawa, T.; Kalaria, R.N.; Fujita, Y.; Ohtani, R.; Ihara, M.; Takahashi, R.; Tomimoto, H. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 2007, 38, 2826–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.M.; Maniskas, M.E.; Bix, G.J. Bilateral carotid artery stenosis causes unexpected early changes in brain extracellular matrix and blood-brain barrier integrity in mice. PLoS ONE 2018, 13, e0195765. [Google Scholar] [CrossRef]
- Pawluk, H.; Wozniak, A.; Grzesk, G.; Kolodziejska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. The Role of Selected Pro-Inflammatory Cytokines in Pathogenesis of Ischemic Stroke. Clin. Interv. Aging 2020, 15, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Amantea, D.; Nappi, G.; Bernardi, G.; Bagetta, G.; Corasaniti, M.T. Post-ischemic brain damage: Pathophysiology and role of inflammatory mediators. FEBS J. 2009, 276, 13–26. [Google Scholar] [CrossRef]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Frijns, C.J.; Kappelle, L.J. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke 2002, 33, 2115–2122. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Kunkel, S.L.; Andjelkovic, A.V. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: Signaling via Rho and Rho kinase. J. Cell Sci. 2003, 116, 4615–4628. [Google Scholar] [CrossRef] [Green Version]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, E.; Somera-Molina, K.; Van Eldik, L.J.; Watterson, D.M.; Wainwright, M.S. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J. Neuroinflammation 2008, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almutairi, M.M.; Gong, C.; Xu, Y.G.; Chang, Y.; Shi, H. Factors controlling permeability of the blood-brain barrier. Cell Mol. Life Sci. 2016, 73, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Hussain, B.; Fang, C.; Chang, J. Blood-Brain Barrier Breakdown: An Emerging Biomarker of Cognitive Impairment in Normal Aging and Dementia. Front. Neurosci. 2021, 15, 688090. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.; Zhang, H.; Lee, S.; Kim, H.; Kim, Y.; Won, M.H.; Kim, Y.M.; Kwon, Y.G. CLEC14A deficiency exacerbates neuronal loss by increasing blood-brain barrier permeability and inflammation. J. Neuroinflammation 2020, 17, 48. [Google Scholar] [CrossRef]
- Huang, L.; He, Z.; Guo, L.; Wang, H. Improvement of cognitive deficit and neuronal damage in rats with chronic cerebral ischemia via relative long-term inhibition of rho-kinase. Cell Mol. Neurobiol. 2008, 28, 757–768. [Google Scholar] [CrossRef]
- An, L.; Shen, Y.; Chopp, M.; Zacharek, A.; Venkat, P.; Chen, Z.; Li, W.; Qian, Y.; Landschoot-Ward, J.; Chen, J. Deficiency of Endothelial Nitric Oxide Synthase (eNOS) Exacerbates Brain Damage and Cognitive Deficit in A Mouse Model of Vascular Dementia. Aging Dis. 2021, 12, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.C. Brain hypoperfusion: A critical factor in vascular dementia. Neurol. Res. 2004, 26, 454–458. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Y-maze (Alternation triplet) | NT | 9 | 47.22 |
| VD | 9 | 33.29 * | |
| Y-maze (Total arm entry) | NT | 9 | 48.13 |
| VD | 9 | 34.61 ** |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Barnes-maze (Time spent in target quadrant) | NT | 9 | 38.84 |
| VD | 8 | 14.74 ** | |
| Barnes-maze (Time to the find target hole) | NT | 17 | 6.060 |
| VD | 9 | 18.73 *** |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Passive avoidance | NT | 9 | 38.84 |
| VD | 8 | 14.74 ** |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Open field (Distance in periphery zone) | NT | 9 | 3192 |
| VD | 8 | 3050 | |
| Open field (Distance in center zone) | NT | 11 | 477.6 |
| VD | 10 | 504.8 | |
| Open field (Total distance) | NT | 12 | NS |
| VD | 9 | ||
| Light and dark (Transition) | NT | 13 | 22.15 |
| VD | 7 | 12.57 * |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Rock1 | NT | 9 | 1 |
| VD | 9 | 17.48 *** | |
| Rock2 | NT | 9 | 1 |
| VD | 9 | 15.01 *** |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Occludin | NT | 9 | 1 |
| VD | 9 | 0.69 | |
| Claudin-5 | NT | 9 | 1 |
| VD | 9 | 0.58 * |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Il-6 | NT | 9 | 1 |
| VD | 9 | 116.7 *** | |
| Il-1β | NT | 9 | 1 |
| VD | 9 | 20.08 ** | |
| Mcp-1 | NT | 9 | 1 |
| VD | 9 | 4.12 | |
| Ccr2 | NT | 9 | 1 |
| VD | 9 | 1.69 | |
| Vcam-1 | NT | 9 | 1 |
| VD | 9 | 2.82 **** |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| α-SMA | NT | 3 | 0 |
| VD | 1 | 0.165 | |
| TUNEL | NT | 1 | 0.187 |
| VD | 1 | 0.451 |
| Group | No. of Mice | Mean Value | |
|---|---|---|---|
| Corpus callosum | NT | 3 | 216.2 |
| VD | 2 | 163.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.C.; Hong, D.-Y.; Lee, D.-H.; Park, S.-W.; Lee, J.Y.; Jeong, J.H.; Kim, E.-Y.; Chung, H.-M.; Hong, K.-S.; Park, S.-P.; et al. Inflammation and Rho-Associated Protein Kinase-Induced Brain Changes in Vascular Dementia. Biomedicines 2022, 10, 446. https://doi.org/10.3390/biomedicines10020446
Lee EC, Hong D-Y, Lee D-H, Park S-W, Lee JY, Jeong JH, Kim E-Y, Chung H-M, Hong K-S, Park S-P, et al. Inflammation and Rho-Associated Protein Kinase-Induced Brain Changes in Vascular Dementia. Biomedicines. 2022; 10(2):446. https://doi.org/10.3390/biomedicines10020446
Chicago/Turabian StyleLee, Eun Chae, Dong-Yong Hong, Dong-Hun Lee, Sang-Won Park, Ji Young Lee, Ji Hun Jeong, Eun-Young Kim, Hyung-Min Chung, Ki-Sung Hong, Se-Pill Park, and et al. 2022. "Inflammation and Rho-Associated Protein Kinase-Induced Brain Changes in Vascular Dementia" Biomedicines 10, no. 2: 446. https://doi.org/10.3390/biomedicines10020446
APA StyleLee, E. C., Hong, D.-Y., Lee, D.-H., Park, S.-W., Lee, J. Y., Jeong, J. H., Kim, E.-Y., Chung, H.-M., Hong, K.-S., Park, S.-P., Lee, M. R., & Oh, J. S. (2022). Inflammation and Rho-Associated Protein Kinase-Induced Brain Changes in Vascular Dementia. Biomedicines, 10(2), 446. https://doi.org/10.3390/biomedicines10020446