
Shared Molecular Mechanisms among Alzheimer’s Disease, Neurovascular Unit Dysfunction and Vascular Risk Factors: A Narrative Review



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Research Strategy
3. Discussion of the Results of the Research Strategy
3.1. Type 2 Diabetes Mellitus
3.1.1. The Clinical and Epidemiological Link between AD and T2DM
3.1.2. The Role of Insulin Signalling
3.1.3. Shared Molecular Mechanisms between AD and T2DM
3.1.4. The Role of AGE/RAGE System in AD
3.2. Hypertension
3.2.1. The Clinical and Epidemiological Link between AD and Hypertension
3.2.2. Shared Molecular Mechanisms between AD and Hypertension
3.3. Dyslipidaemia
3.3.1. The Clinical and Epidemiological Link between AD and Dyslipidaemia
3.3.2. Shared Molecular Mechanisms between AD and Dyslipidaemia
3.4. Cigarette Smoking
3.4.1. The Clinical and Epidemiological Link between AD and Cigarette Smoking
3.4.2. Shared Molecular Mechanisms between AD and Cigarette Smoking
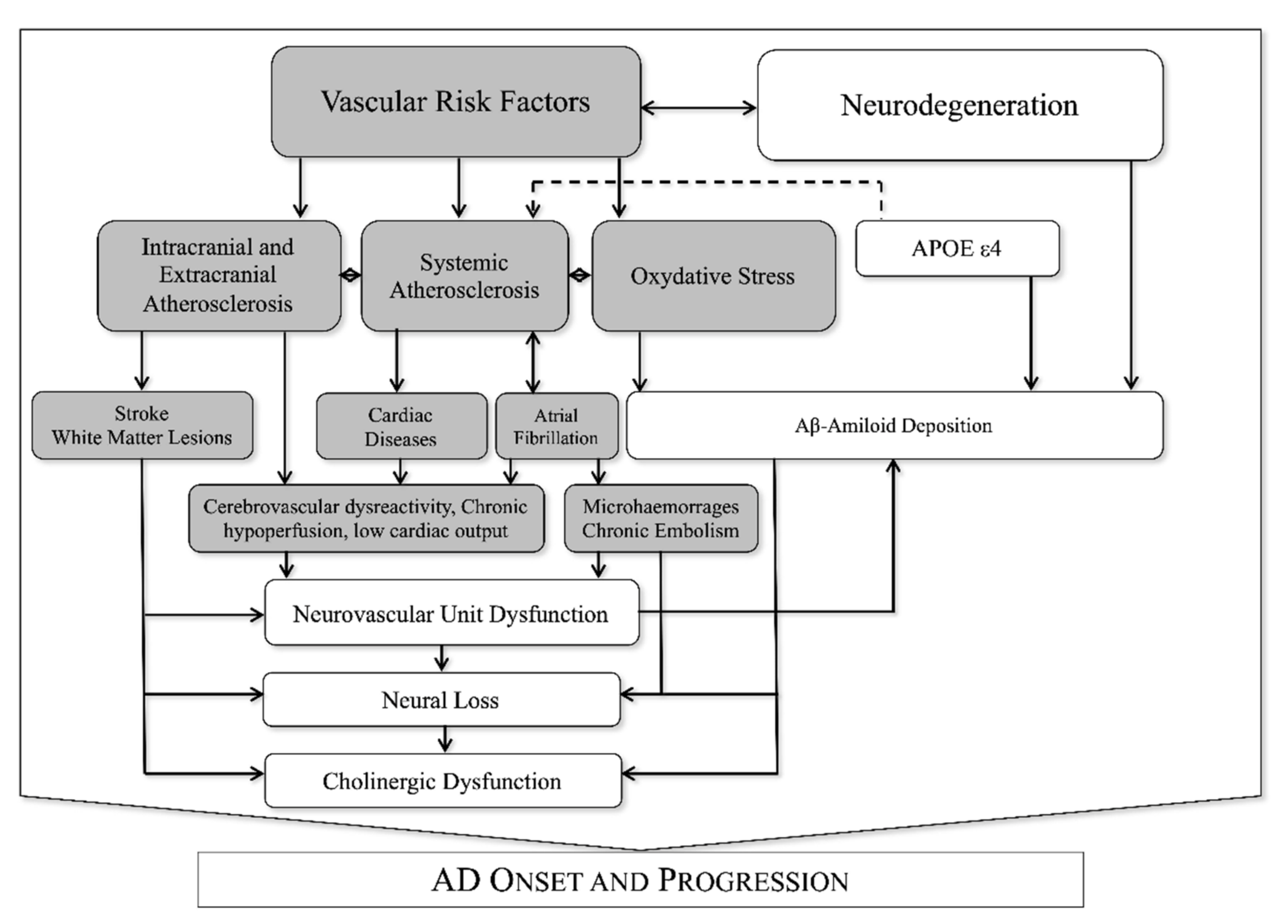
3.5. Association between VRF and NVU Dysfunction in AD
4. Conclusions
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Serio, G.; Scarpazza, C.; D’Ausilio, A.; Borgomaneri, S. Frozen in (e)motion: How reactive motor inhibition is influenced by the emotional content of stimuli in healthy and psychiatric populations. Behav. Res. Ther. 2021, 146, 103963. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the human ventromedial prefrontal cortex support fear learning, fear extinction or both? A commentary on subregional contributions. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.d.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997, 277, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Xie, S.X.; Kling, M.A.; Toledo, J.B.; Wolk, D.A.; Lee, E.B.; Van Deerlin, V.; Lee, V.M.-Y.; Trojanowski, J.Q.; Arnold, S.E. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 2012, 135, 3749–3756. [Google Scholar] [CrossRef] [PubMed]
- Silvestrini, M.; Viticchi, G.; Falsetti, L.; Balucani, C.; Vernieri, F.; Cerqua, R.; Luzzi, S.; Bartolini, M.; Provinciali, L. The role of carotid atherosclerosis in Alzheimer’s disease progression. J. Alzheimers Dis. 2011, 25, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Gabr, M. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2019, 14, 437–440. [Google Scholar] [CrossRef]
- Girouard, H.; Iadecola, C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol. 2006, 100, 328–335. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Presa, J.L.; Saravia, F.; Bagi, Z.; Filosa, J.A. Vasculo-Neuronal Coupling and Neurovascular Coupling at the Neurovascular Unit: Impact of Hypertension. Front. Physiol. 2020, 11, 584135. [Google Scholar] [CrossRef]
- Silvestrini, M.; Vernieri, F.; Pasqualetti, P.; Matteis, M.; Passarelli, F.; Troisi, E.; Caltagirone, C. Impaired Cerebral Vasoreactivity and Risk of Stroke in Patients With Asymptomatic Carotid Artery Stenosis. JAMA 2000, 283, 2122–2127. [Google Scholar] [CrossRef]
- Viticchi, G.; Falsetti, L.; Vernieri, F.; Altamura, C.; Altavilla, R.; Luzzi, S.; Bartolini, M.; Provinciali, L.; Silvestrini, M. Apolipoprotein E genotype and cerebrovascular alterations can influence conversion to dementia in patients with mild cognitive impairment. J. Alzheimers Dis. 2014, 41, 401–410. [Google Scholar] [CrossRef]
- Mogi, M.; Horiuchi, M. Neurovascular Coupling in Cognitive Impairment Associated With Diabetes Mellitus. Circ. J. 2011, 75, 1042–1048. [Google Scholar] [CrossRef] [Green Version]
- Boms, N.; Yonai, Y.; Molnar, S.; Rosengarten, B.; Bornstein, N.M.; Csiba, L.; Olah, L. Effect of Smoking Cessation on Visually Evoked Cerebral Blood Flow Response in Healthy Volunteers. J. Vasc. Res. 2010, 47, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.R.; Muldoon, M.F.; Ryan, C.; Price, J.C.; Greer, P.; Sutton-Tyrrell, K.; van der Veen, F.M.; Meltzer, C.C. Reduced cerebral blood flow response and compensation among patients with untreated hypertension. Neurology 2005, 64, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Czuba, E.; Steliga, A.; Lietzau, G.; Kowiański, P. Cholesterol as a modifying agent of the neurovascular unit structure and function under physiological and pathological conditions. Metab. Brain Dis. 2017, 32, 935–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Yan, L.-F.; Sun, Q.; Yu, Y.; Zhang, J.; Dai, Y.-J.; Yang, Y.; Hu, Y.-C.; Nan, H.-Y.; Zhang, X.; et al. Disturbed neurovascular coupling in type 2 diabetes mellitus patients: Evidence from a comprehensive fMRI analysis. NeuroImage Clin. 2019, 22, 101802. [Google Scholar] [CrossRef]
- Viticchi, G.; Falsetti, L.; Buratti, L.; Luzzi, S.; Bartolini, M.; Acciarri, M.C.; Provinciali, L.; Silvestrini, M. Metabolic syndrome and cerebrovascular impairment in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2015, 30, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Buratti, L.; Balucani, C.; Viticchi, G.; Falsetti, L.; Altamura, C.; Avitabile, E.; Provinciali, L.; Vernieri, F.; Silvestrini, M. Cognitive deterioration in bilateral asymptomatic severe carotid stenosis. Stroke 2014, 45, 2072–2077. [Google Scholar] [CrossRef] [Green Version]
- Buratti, L.; Viticchi, G.; Falsetti, L.; Balucani, C.; Altamura, C.; Petrelli, C.; Provinciali, L.; Vernieri, F.; Silvestrini, M. Thresholds of impaired cerebral hemodynamics that predict short-term cognitive decline in asymptomatic carotid stenosis. J. Cereb. Blood Flow Metab. 2016, 36, 1804–1812. [Google Scholar] [CrossRef]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.; Wakayama, I.; Yanagihara, R.; Garruto, R.; et al. Advanced Glycation End Products in Alzheimer’s Disease and Other Neurodegenerative Diseases. Am. J. Pathol. 1998, 153, 1149–1155. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, F.; Wang, J.; Liu, C.; Zhou, Y.; Xu, Z.; Zhang, C.; Sun, B.; Guan, Y. Pathological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: The Receptor for Advanced Glycation End Products (RAGE). Front. Aging Neurosci. 2020, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A Meta-Analysis of Cytokines in Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Fazeli, P.K.; Lee, H.; Steinhauser, M.L. Aging Is a Powerful Risk Factor for Type 2 Diabetes Mellitus Independent of Body Mass Index. Gerontology 2020, 66, 209–210. [Google Scholar] [CrossRef]
- Qiu, C.; De Ronchi, D.; Fratiglioni, L. The epidemiology of the dementias: An update. Curr. Opin. Psychiatry 2007, 20, 380–385. [Google Scholar] [CrossRef]
- Falsetti, L.; Viticchi, G.; Buratti, L.; Grigioni, F.; Capucci, A.; Silvestrini, M. Interactions between Atrial Fibrillation, Cardiovascular Risk Factors, and ApoE Genotype in Promoting Cognitive Decline in Patients with Alzheimer’s Disease: A Prospective Cohort Study. J. Alzheimers Dis. 2018, 62, 713–725. [Google Scholar] [CrossRef]
- Viticchi, G.; Falsetti, L.; Buratti, L.; Boria, C.; Luzzi, S.; Bartolini, M.; Provinciali, L.; Silvestrini, M. Framingham risk score can predict cognitive decline progression in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2940–2945. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Hofman, A.; Ott, A.; Breteler, M.M.; Bots, M.L.; Slooter, A.J.; van Harskamp, F.; van Duijn, C.N.; Van Broeckhoven, C.; Grobbee, D.E. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 1997, 349, 151–154. [Google Scholar] [CrossRef]
- Kloppenborg, R.P.; van den Berg, E.; Kappelle, L.J.; Biessels, G.J. Diabetes and other vascular risk factors for dementia: Which factor matters most? A systematic review. Eur. J. Pharmacol. 2008, 585, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Crane, P.K.; Walker, R.; Hubbard, R.A.; Li, G.; Nathan, D.M.; Zheng, H.; Haneuse, S.; Craft, S.; Montine, T.J.; Kahn, S.E.; et al. Glucose Levels and Risk of Dementia. N. Engl. J. Med. 2013, 369, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s Disease is Type 3 Diabetes—Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- de la Monte, S.M.; Tong, M.; Lester-Coll, N.; Plater, M., Jr.; Wands, J.R. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: Relevance to Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 89–109. [Google Scholar] [CrossRef]
- de la Monte, S.M. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimers Disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef]
- Lester-Coll, N.; Rivera, E.J.; Soscia, S.J.; Doiron, K.; Wands, J.R.; de la Monte, S.M. Intracerebral streptozotocin model of type 3 diabetes: Relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 13–33. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Ganju, N.; Banerjee, K.; Brown, N.V.; Luong, T.; Wands, J.R. Partial rescue of ethanol-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcohol. Clin. Exp. Res. 2000, 24, 716–726. [Google Scholar] [CrossRef]
- Myers, M.G.; Sun, X.J.; White, M.F. The IRS-1 signaling system. Trends Biochem. Sci. 1994, 19, 289–293. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.-C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef] [PubMed]
- White, M.F.; Maron, R.; Kahn, C.R. Insulin rapidly stimulates tyrosine phosphorylation of a Mr-185,000 protein in intact cells. Nature 1985, 318, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Crimmins, D.L.; Myers, M.G.; Miralpeix, M.; White, M.F. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol. Cell. Biol. 1993, 13, 7418–7428. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Z.; Wu, S.-C.; Chang, C.-M.; Lin, C.-L.; Kwan, A.-L. Arctigenin, a Potent Ingredient of Arctium lappa L., Induces Endothelial Nitric Oxide Synthase and Attenuates Subarachnoid Hemorrhage-Induced Vasospasm through PI3K/Akt Pathway in a Rat Model. BioMed Res. Int. 2015, 2015, 490209. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Basta, G.; Schmidt, A.M.; De Caterina, R. Advanced glycation end products and vascular inflammation: Implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 2004, 63, 582–592. [Google Scholar] [CrossRef]
- Burgering, B.M.T.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef]
- Delcommenne, M.; Tan, C.; Gray, V.; Rue, L.; Woodgett, J.; Dedhar, S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 11211–11216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulik, G.; Klippel, A.; Weber, M.J. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol. Cell. Biol. 1997, 17, 1595–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhang-Fallah, J.; Dikkes, P.; Warot, X.M.; Rio, C.; et al. Insulin Receptor Substrate-2 Deficiency Impairs Brain Growth and Promotes Tau Phosphorylation. J. Neurosci. 2003, 23, 7084–7092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef] [Green Version]
- Bunn, H.; Higgins, P. Reaction of monosaccharides with proteins: Possible evolutionary significance. Science 1981, 213, 222–224. [Google Scholar] [CrossRef]
- Simó, R.; Ciudin, A.; Simó-Servat, O.; Hernández, C. Cognitive impairment and dementia: A new emerging complication of type 2 diabetes—The diabetologist’s perspective. Acta Diabetol. 2017, 54, 417–424. [Google Scholar] [CrossRef]
- Miranda, H.V.; Outeiro, T.F. The sour side of neurodegenerative disorders: The effects of protein glycation. J. Pathol. 2010, 221, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, P.; Rabbani, G.; Khan, R. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mooldijk, S.S.; Licher, S.; Waqas, K.; Ikram, M.K.; Uitterlinden, A.G.; Zillikens, M.C.; Ikram, M.A. Assessment of Advanced Glycation End Products and Receptors and the Risk of Dementia. JAMA Netw. Open 2021, 4, e2033012. [Google Scholar] [CrossRef] [PubMed]
- Portegies, M.L.P.; Mirza, S.S.; Verlinden, V.J.A.; Hofman, A.; Koudstaal, P.J.; Swanson, S.A.; Ikram, M.A. Mid- to Late-Life Trajectories of Blood Pressure and the Risk of Stroke. Hypertension 2016, 67, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Kivipelto, M.; Helkala, E.-L.; Laakso, M.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef] [Green Version]
- Launer, L.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.; Foley, D.; White, L.R.; Havlik, R.J. Midlife blood pressure and dementia: The Honolulu–Asia aging study☆. Neurobiol. Aging 2000, 21, 49–55. [Google Scholar] [CrossRef]
- Posner, H.B.; Tang, M.-X.; Luchsinger, J.; Lantigua, R.; Stern, Y.; Mayeux, R. The relationship of hypertension in the elderly to AD, vascular dementia, and cognitive function. Neurology 2002, 58, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Yoshitake, T.; Kiyohara, Y.; Kato, I.; Ohmura, T.; Iwamoto, H.; Nakayama, K.; Ohmori, S.; Nomiyama, K.; Kawano, H.; Ueda, K.; et al. Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: The Hisayama Study. Neurology 1995, 45, 1161–1168. [Google Scholar] [CrossRef]
- Skoog, I.; Nilsson, L.; Persson, G.; Lernfelt, B.; Landahl, S.; Palmertz, B.; Andreasson, L.-A.; Odén, A.; Svanborg, A. 15-year longitudinal study of blood pressure and dementia. Lancet 1996, 347, 1141–1145. [Google Scholar] [CrossRef]
- van Dalen, J.W.; Brayne, C.; Crane, P.K.; Fratiglioni, L.; Larson, E.B.; Lobo, A.; Lobo, E.; Marcum, Z.A.; van Charante, E.P.M.; Qiu, C.; et al. Association of Systolic Blood Pressure With Dementia Risk and the Role of Age, U-Shaped Associations, and Mortality. JAMA Intern. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.J.; Coronado, P.G.; Schmitt, C.A.; Gillespie, K.M.; Chung, H.D. Blood pressure regulation in alzheimer’s disease. J. Auton. Nerv. Syst. 1994, 48, 65–71. [Google Scholar] [CrossRef]
- Skoog, I.; Andreasson, L.-A.; Landahl, S.; Lernfelt, B. A Population-Based Study on Blood Pressure and Brain Atrophy in 85-Year-Olds. Hypertension 1998, 32, 404–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Torre, J.C. Cerebral Hypoperfusion, Capillary Degeneration, and Development of Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2000, 14, S72–S81. [Google Scholar] [CrossRef]
- Carnevale, D.; Lembo, G. ‘Alzheimer-like’ pathology in a murine model of arterial hypertension. Biochem. Soc. Trans. 2011, 39, 939–944. [Google Scholar] [CrossRef]
- Carnevale, D.; Mascio, G.; D’Andrea, I.; Fardella, V.; Bell, R.D.; Branchi, I.; Pallante, F.; Zlokovic, B.; Yan, S.S.; Lembo, G. Hypertension Induces Brain β-Amyloid Accumulation, Cognitive Impairment, and Memory Deterioration Through Activation of Receptor for Advanced Glycation End Products in Brain Vasculature. Hypertension 2012, 60, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, D.; Mascio, G.; Ajmone-Cat, M.A.; D’Andrea, I.; Cifelli, G.; Madonna, M.; Cocozza, G.; Frati, A.; Carullo, P.; Carnevale, L.; et al. Role of neuroinflammation in hypertension-induced brain amyloid pathology. Neurobiol. Aging 2012, 33, 205.e19–205.e29. [Google Scholar] [CrossRef]
- Hardy, J.; Higgins, G. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Cerebrovascular transport of Alzheimer’s amyloidβ and apolipoproteins J and E: Possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 1996, 59, 1483–1497. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Chen, C.; Li, X.-H.; Tu, Y.; Sun, H.-T.; Liang, H.-Q.; Cheng, S.-X.; Zhang, S. Aβ-AGE aggravates cognitive deficit in rats via RAGE pathway. Neuroscience 2014, 257, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Lv, B.-L.; Xie, J.-Z.; Liu, J.; Zhou, X.-W.; Wang, J.-Z. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 2012, 33, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yamagishi, S.; Nakamura, Y.; Takenaka, K.; Matsui, T.; Jinnouchi, Y.; Imaizumi, T. Telmisartan inhibits expression of a receptor for advanced glycation end products (RAGE) in angiotensin-II-exposed endothelial cells and decreases serum levels of soluble RAGE in patients with essential hypertension. Microvasc. Res. 2005, 70, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.-H.; Wu, S.-Y.; Yu, M.; Huang, S.-H.; Lee, C.-W.; Jiang, M.-J.; Lin, P.-Y.; Yang, T.-T.; Kuo, Y.-M. Hypertension Accelerates Alzheimer’s Disease-Related Pathologies in Pigs and 3xTg Mice. Front. Aging Neurosci. 2018, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Deane, R.; Zetterberg, H.; Wallin, A.; Blennow, K.; Zlokovic, B.V. Impaired Lipoprotein Receptor-Mediated Peripheral Binding of Plasma Amyloid-β is an Early Biomarker for Mild Cognitive Impairment Preceding Alzheimer’s Disease. J. Alzheimers Dis. 2011, 24, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Uiterwijk, R.; Huijts, M.; Staals, J.; Rouhl, R.P.W.; De Leeuw, P.W.; Kroon, A.A.; Van Oostenbrugge, R.J. Endothelial Activation Is Associated With Cognitive Performance in Patients With Hypertension. Am. J. Hypertens. 2016, 29, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Rosei, E.A.; Rizzoni, D.; Muiesan, M.L.; Sleiman, I.; Salvetti, M.; Monteduro, C.; Porteri, E. Effects of candesartan cilexetil and enalapril on inflammatory markers of atherosclerosis in hypertensive patients with non-insulin-dependent diabetes mellitus. J. Hypertens. 2005, 23, 435–444. [Google Scholar] [CrossRef]
- Akter, S.; Jesmin, S.; Iwashima, Y.; Hideaki, S.; Rahman, M.A.; Islam, M.M.; Moroi, M.; Shimojo, N.; Yamaguchi, N.; Miyauchi, T.; et al. Higher circulatory level of endothelin-1 in hypertensive subjects screened through a cross-sectional study of rural Bangladeshi women. Hypertens. Res. 2015, 38, 208–212. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Notkola, I.-L.; Sulkava, R.; Pekkanen, J.; Erkinjuntti, T.; Ehnholm, C.; Kivinen, P.; Tuomilehto, J.; Nissinen, A. Serum Total Cholesterol, Apolipoprotein E {FC12}e4 Allele, and Alzheimer’s Disease. Neuroepidemiology 1998, 17, 14–20. [Google Scholar] [CrossRef]
- Kivipelto, M.; Helkala, E.-L.; Laakso, M.P.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Iivonen, S.; Mannermaa, A.; Tuomilehto, J.; Nissinen, A.; et al. Apolipoprotein E ϵ4 Allele, Elevated Midlife Total Cholesterol Level, and High Midlife Systolic Blood Pressure Are Independent Risk Factors for Late-Life Alzheimer Disease. Ann. Intern. Med. 2002, 137, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Barrett-Connor, E.; Lin, F.; Grady, D. Serum Lipoprotein Levels, Statin Use, and Cognitive Function in Older Women. Arch. Neurol. 2002, 59, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Helzner, E.P.; Luchsinger, J.A.; Scarmeas, N.; Cosentino, S.; Brickman, A.M.; Glymour, M.M.; Stern, Y. Contribution of Vascular Risk Factors to the Progression in Alzheimer Disease. Arch. Neurol. 2009, 66, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Zandi, P.P.; Sjogren, M.; Gustafson, D.; Ostling, S.; Steen, B.; Skoog, I. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology 2005, 64, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Sidney, S.; Selby, J.; Johnston, S.C.; Yaffe, K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology 2005, 64, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Esh, C.; Kokjohn, T.A.; Kalback, W.; Luehrs, D.C.; Seward, J.D.; Sue, L.I.; Beach, T.G. Circle of Willis Atherosclerosis Is a Risk Factor for Sporadic Alzheimer’s Disease. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2055–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaurin, J.; Darabie, A.A.; Morrison, M.R. Cholesterol, a Modulator of Membrane-Associated Aβ-Fibrillogenesis. Ann. N. Y. Acad. Sci. 2002, 977, 376–383. [Google Scholar] [CrossRef]
- Sun, F.; Chen, L.; Wei, P.; Chai, M.; Ding, X.; Xu, L.; Luo, S.-Z. Dimerization and Structural Stability of Amyloid Precursor Proteins Affected by the Membrane Microenvironments. J. Chem. Inf. Model. 2017, 57, 1375–1387. [Google Scholar] [CrossRef]
- Brown, A.M.; Bevan, D.R. Influence of sequence and lipid type on membrane perturbation by human and rat amyloid β-peptide (1–42). Arch. Biochem. Biophys. 2017, 614, 1–13. [Google Scholar] [CrossRef]
- Abad-Rodriguez, J.; Ledesma, M.D.; Craessaerts, K.; Perga, S.; Medina, M.; Delacourte, A.; Dingwall, C.; De Strooper, B.; Dotti, C.G. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 2004, 167, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, G.L.; Kaye, J.A.; Quinn, J.F. Dyslipidemia and Blood-Brain Barrier Integrity in Alzheimer’s Disease. Curr. Gerontol. Geriatr. Res. 2012, 2012, 184042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, J.; Moreira, E.L.G.; dos Santos, D.B.; Piermartiri, T.C.; Dutra, R.C.; Pinton, S.; Tasca, C.I.; Farina, M.; Prediger, R.D.S.; de Bem, A.F. Increased Susceptibility to Amyloid-β-Induced Neurotoxicity in Mice Lacking the Low-Density Lipoprotein Receptor. J. Alzheimers Dis. 2014, 41, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Desikan, R.S.; Schork, A.J.; Wang, Y.; Thompson, W.K.; Dehghan, A.; Ridker, P.M.; Chasman, D.I.; McEvoy, L.K.; Holland, D.; Chen, C.-H.; et al. Polygenic Overlap Between C-Reactive Protein, Plasma Lipids, and Alzheimer Disease. Circulation 2015, 131, 2061–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A High-Density Whole-Genome Association Study Reveals That APOE Is the Major Susceptibility Gene for Sporadic Late-Onset Alzheimer’s Disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef]
- Flowers, S.A.; Rebeck, G.W. APOE in the normal brain. Neurobiol. Dis. 2020, 136, 104724. [Google Scholar] [CrossRef]
- Bell, R.D. The Imbalance of Vascular Molecules in Alzheimer’s Disease. J. Alzheimers Dis. 2012, 32, 699–709. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Wang, Y.; Ma, Q.; Sagare, A.P.; Montagne, A.; Huuskonen, M.T.; Rege, S.V.; Kisler, K.; Dai, Z.; Körbelin, J.; et al. Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J. Exp. Med. 2021, 218, e20202207. [Google Scholar] [CrossRef]
- Nigro, P.; Satoh, K.; O’Dell, M.R.; Soe, N.N.; Cui, Z.; Mohan, A.; Abe, J.; Alexis, J.D.; Sparks, J.D.; Berk, B.C. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J. Exp. Med. 2011, 208, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Fratiglioni, L.; Wang, H.-X. Smoking and Parkinson’s and Alzheimer’s disease: Review of the epidemiological studies. Behav. Brain Res. 2000, 113, 117–120. [Google Scholar] [CrossRef]
- Anstey, K.J.; von Sanden, C.; Salim, A.; O’Kearney, R. Smoking as a Risk Factor for Dementia and Cognitive Decline: A Meta-Analysis of Prospective Studies. Am. J. Epidemiol. 2007, 166, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, J.K.; Prochaska, J.J.; Glantz, S.A. Cigarette Smoking is a Risk Factor for Alzheimer’s Disease: An Analysis Controlling for Tobacco Industry Affiliation. J. Alzheimers Dis. 2010, 19, 465–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusanen, M.; Rovio, S.; Ngandu, T.; Nissinen, A.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife Smoking, Apolipoprotein E and Risk of Dementia and Alzheimer’s Disease: A Population-Based Cardiovascular Risk Factors, Aging and Dementia Study. Dement. Geriatr. Cogn. Disord. 2010, 30, 277–284. [Google Scholar] [CrossRef]
- Merchant, C.; Tang, M.-X.; Albert, S.; Manly, J.; Stern, Y.; Mayeux, R. The influence of smoking on the risk of Alzheimer’s disease. Neurology 1999, 52, 1408. [Google Scholar] [CrossRef] [Green Version]
- Rusanen, M.; Kivipelto, M.; Quesenberry, C.P.; Zhou, J.; Whitmer, R.A. Heavy Smoking in Midlife and Long-term Risk of Alzheimer Disease and Vascular Dementia. Arch. Intern. Med. 2011, 171, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Incalzi, R.A.; Gemma, A.; Marra, C.; Muzzolon, R.; Capparella, O.; Carbonin, P. Chronic obstructive pulmonary disease: An original model of cognitive decline. Am. Rev. Respir. Dis. 1993, 148, 418–424. [Google Scholar] [CrossRef]
- Dal Negro, R.W.; Bonadiman, L.; Bricolo, F.P.; Tognella, S.; Turco, P. Cognitive dysfunction in severe chronic obstructive pulmonary disease (COPD) with or without Long-Term Oxygen Therapy (LTOT). Multidiscip. Respir. Med. 2015, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Rusanen, M.; Ngandu, T.; Laatikainen, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Chronic obstructive pulmonary disease and asthma and the risk of mild cognitive impairment and dementia: A population based CAIDE study. Curr. Alzheimer Res. 2013, 10, 549–555. [Google Scholar] [CrossRef]
- Lutsey, P.L.; Chen, N.; Mirabelli, M.C.; Lakshminarayan, K.; Knopman, D.S.; Vossel, K.A.; Gottesman, R.F.; Mosley, T.H.; Alonso, A. Impaired Lung Function, Lung Disease, and Risk of Incident Dementia. Am. J. Respir. Crit. Care Med. 2019, 199, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Akaike, A.; Takada-Takatori, Y.; Kume, T.; Izumi, Y. Mechanisms of Neuroprotective Effects of Nicotine and Acetylcholinesterase Inhibitors: Role of α4 and α7 Receptors in Neuroprotection. J. Mol. Neurosci. 2010, 40, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Teaktong, T.; Graham, A.J.; Johnson, M.; Court, J.A.; Perry, E.K. Selective changes in nicotinic acetylcholine receptor subtypes related to tobacco smoking: An immunohistochemical study. Neuropathol. Appl. Neurobiol. 2004, 30, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Egleton, R.D.; Abbruscato, T. Drug Abuse and the Neurovascular Unit. Adv. Pharmacol. 2014, 71, 451–480. [Google Scholar] [PubMed]
- Abbruscato, T.J.; Lopez, S.P.; Mark, K.S.; Hawkins, B.T.; Davis, T.P. Nicotine and Cotinine Modulate Cerebral Microvascular Permeability and Protein Expression of ZO-1 through Nicotinic Acetylcholine Receptors Expressed on Brain Endothelial Cells. J. Pharm. Sci. 2002, 91, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Estrada, L.D.; Sanchez-Mejias, E.; Soto, C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat. Commun. 2013, 4, 1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W. Alzheimer’s Disease Neuroimaging Initiative. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimers Dement. 2014, 10, S122–S145. [Google Scholar] [CrossRef] [Green Version]
- Falsetti, L.; Viticchi, G.; Zaccone, V.; Tarquinio, N.; Nobili, L.; Nitti, C.; Salvi, A.; Moroncini, G.; Silvestrini, M. Chronic respiratory diseases and neurodegenerative disorders: A primer for the practicing clinician. Med. Princ. Pract. 2021, 30, 501–507. [Google Scholar] [CrossRef]
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological impacts of chronic hypoxia on alzheimer’s disease. ACS Chem. Neurosci. 2019, 10, 902–909. [Google Scholar] [CrossRef]
- Zhang, X.; Le, W. Pathological role of hypoxia in Alzheimer’s disease. Exp. Neurol. 2010, 223, 299–303. [Google Scholar] [CrossRef]
- Chow, N.; Bell, R.D.; Deane, R.; Streb, J.W.; Chen, J.; Brooks, A.; Van Nostrand, W.; Miano, J.M.; Zlokovic, B.V. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc. Natl. Acad. Sci. USA 2007, 104, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viticchi, G.; Falsetti, L.; Vernieri, F.; Altamura, C.; Bartolini, M.; Luzzi, S.; Provinciali, L.; Silvestrini, M. Vascular predictors of cognitive decline in patients with mild cognitive impairment. Neurobiol. Aging 2012, 33, 1127.e1–1127.e9. [Google Scholar] [CrossRef] [PubMed]
- Balucani, C.; Viticchi, G.; Falsetti, L.; Silvestrini, M. Cerebral hemodynamics and cognitive performance in bilateral asymptomatic carotid stenosis. Neurology 2012, 79, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta—Mol. Basis Dis. 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Halliday, M.R.; Pomara, N.; Sagare, A.P.; Mack, W.J.; Frangione, B.; Zlokovic, B.V. Relationship Between Cyclophilin A Levels and Matrix Metalloproteinase 9 Activity in Cerebrospinal Fluid of Cognitively Normal Apolipoprotein E4 Carriers and Blood-Brain Barrier Breakdown. JAMA Neurol. 2013, 70, 1198–1200. [Google Scholar] [CrossRef] [Green Version]
- Viticchi, G.; Falsetti, L.; Buratti, L.; Sajeva, G.; Luzzi, S.; Bartolini, M.; Provinciali, L.; Silvestrini, M. Framingham Risk Score and the Risk of Progression from Mild Cognitive Impairment to Dementia. J Alzheimers Dis. 2017, 59, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Suri, S.; Mackay, C.E.; Kelly, M.E.; Germuska, M.; Tunbridge, E.M.; Frisoni, G.B.; Matthews, P.M.; Ebmeier, K.P.; Bulte, D.P.; Filippini, N. Reduced cerebrovascular reactivity in young adults carrying the APOE ε4 allele. Alzheimers Dement. 2015, 11, 648–657. [Google Scholar] [CrossRef]
- Buratti, L.; Balestrini, S.; Altamura, C.; Viticchi, G.; Falsetti, L.; Luzzi, S.; Provinciali, L.; Vernieri, F.; Silvestrini, M. Markers for the risk of progression from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 883–890. [Google Scholar] [CrossRef]
- Viticchi, G.; Falsetti, L.; Burattini, M.; Zaccone, V.; Buratti, L.; Bartolini, M.; Moroncini, G.; Silvestrini, M. Atrial Fibrillation on Patients with Vascular Dementia: A Fundamental Target for Correct Management. Brain Sci. 2020, 10, 420. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Ababei, D.C.; Bild, V.; Bild, W.; Paduraru, L.; Gutu, M.M.; Tamba, B.-I. Renal Contributions in the Pathophysiology and Neuropathological Substrates Shared by Chronic Kidney Disease and Alzheimer’s Disease. Brain Sci. 2020, 10, 563. [Google Scholar] [CrossRef]
- Ferrero, J.; Williams, L.; Stella, H.; Leitermann, K.; Mikulskis, A.; O’Gorman, J.; Sevigny, J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2016, 2, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, S.; Barber, P.A. Dementia risk and prevention by targeting modifiable vascular risk factors. J. Neurochem. 2018, 144, 565–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, R.; Hongisto, K.; Solomon, A.; Lönnroos, E. Physical Activity and Alzheimer’s Disease: A Systematic Review. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 733–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.Y.; Byun, M.S.; Yi, D.; Lee, J.-H.; Ko, K.; Sohn, B.K.; Lee, J.-Y.; Ryu, S.-H.; Lee, D.W.; Shin, S.A.; et al. Midlife Lifestyle Activities Moderate APOE ε4 Effect on in vivo Alzheimer’s Disease Pathologies. Front. Aging Neurosci. 2020, 12, 42. [Google Scholar] [CrossRef] [Green Version]
- Nagai, M.; Hoshide, S.; Kario, K. Hypertension and Dementia. Am. J. Hypertens. 2010, 23, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Alkasabera, A.; Onyali, C.B.; Anim-Koranteng, C.; Shah, H.E.; Ethirajulu, A.; Bhawnani, N.; Mostafa, J.A. The Effect of Type-2 Diabetes on Cognitive Status and the Role of Anti-diabetes Medications. Cureus 2021, 13, e19176. [Google Scholar] [CrossRef]
- Olmastroni, E.; Molari, G.; De Beni, N.; Colpani, O.; Galimberti, F.; Gazzotti, M.; Zambon, A.; Catapano, A.L.; Casula, M. Statin use and risk of dementia or Alzheimer’s disease: A systematic review and meta-analysis of observational studies. Eur. J. Prev. Cardiol. 2021. [Google Scholar] [CrossRef]
- Sheng, M.; Lu, H.; Liu, P.; Li, Y.; Ravi, H.; Peng, S.-L.; Diaz-Arrastia, R.; Devous, M.D.; Womack, K.B. Sildenafil Improves Vascular and Metabolic Function in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Sanders, O. Sildenafil for the Treatment of Alzheimer’s Disease: A Systematic Review. J. Alzheimers Dis. Rep. 2020, 4, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Zuccarello, E.; Acquarone, E.; Calcagno, E.; Argyrousi, E.K.; Deng, S.-X.; Landry, D.W.; Arancio, O.; Fiorito, J. Development of novel phosphodiesterase 5 inhibitors for the therapy of Alzheimer’s disease. Biochem. Pharmacol. 2020, 176, 113818. [Google Scholar] [CrossRef]
- Lattanzi, S.; Carbonari, L.; Pagliariccio, G.; Bartolini, M.; Cagnetti, C.; Viticchi, G.; Buratti, L.; Provinciali, L.; Silvestrini, M. Neurocognitive functioning and cerebrovascular reactivity after carotid endarterectomy. Neurology 2018, 90, e307–e315. [Google Scholar] [CrossRef] [PubMed]
- Buss, S.S.; Fried, P.J.; Pascual-Leone, A. Therapeutic noninvasive brain stimulation in Alzheimer’s disease and related dementias. Curr. Opin. Neurol. 2019, 32, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Borgomaneri, S.; Battaglia, S.; Avenanti, A.; di Pellegrino, G. Don’t Hurt Me No More: State-dependent Transcranial Magnetic Stimulation for the treatment of specific phobia. J. Affect. Disord. 2021, 286, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Borgomaneri, S.; Battaglia, S.; Sciamanna, G.; Tortora, F.; Laricchiuta, D. Memories are not written in stone: Re-writing fear memories by means of non-invasive brain stimulation and optogenetic manipulations. Neurosci. Biobehav. Rev. 2021, 127, 334–352. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falsetti, L.; Viticchi, G.; Zaccone, V.; Guerrieri, E.; Moroncini, G.; Luzzi, S.; Silvestrini, M. Shared Molecular Mechanisms among Alzheimer’s Disease, Neurovascular Unit Dysfunction and Vascular Risk Factors: A Narrative Review. Biomedicines 2022, 10, 439. https://doi.org/10.3390/biomedicines10020439
Falsetti L, Viticchi G, Zaccone V, Guerrieri E, Moroncini G, Luzzi S, Silvestrini M. Shared Molecular Mechanisms among Alzheimer’s Disease, Neurovascular Unit Dysfunction and Vascular Risk Factors: A Narrative Review. Biomedicines. 2022; 10(2):439. https://doi.org/10.3390/biomedicines10020439
Chicago/Turabian StyleFalsetti, Lorenzo, Giovanna Viticchi, Vincenzo Zaccone, Emanuele Guerrieri, Gianluca Moroncini, Simona Luzzi, and Mauro Silvestrini. 2022. "Shared Molecular Mechanisms among Alzheimer’s Disease, Neurovascular Unit Dysfunction and Vascular Risk Factors: A Narrative Review" Biomedicines 10, no. 2: 439. https://doi.org/10.3390/biomedicines10020439
APA StyleFalsetti, L., Viticchi, G., Zaccone, V., Guerrieri, E., Moroncini, G., Luzzi, S., & Silvestrini, M. (2022). Shared Molecular Mechanisms among Alzheimer’s Disease, Neurovascular Unit Dysfunction and Vascular Risk Factors: A Narrative Review. Biomedicines, 10(2), 439. https://doi.org/10.3390/biomedicines10020439