
Functional Phenotypes of Intraplaque Macrophages and Their Distinct Roles in Atherosclerosis Development and Atheroinflammation

 , ,
, , 
Abstract
:1. Introduction
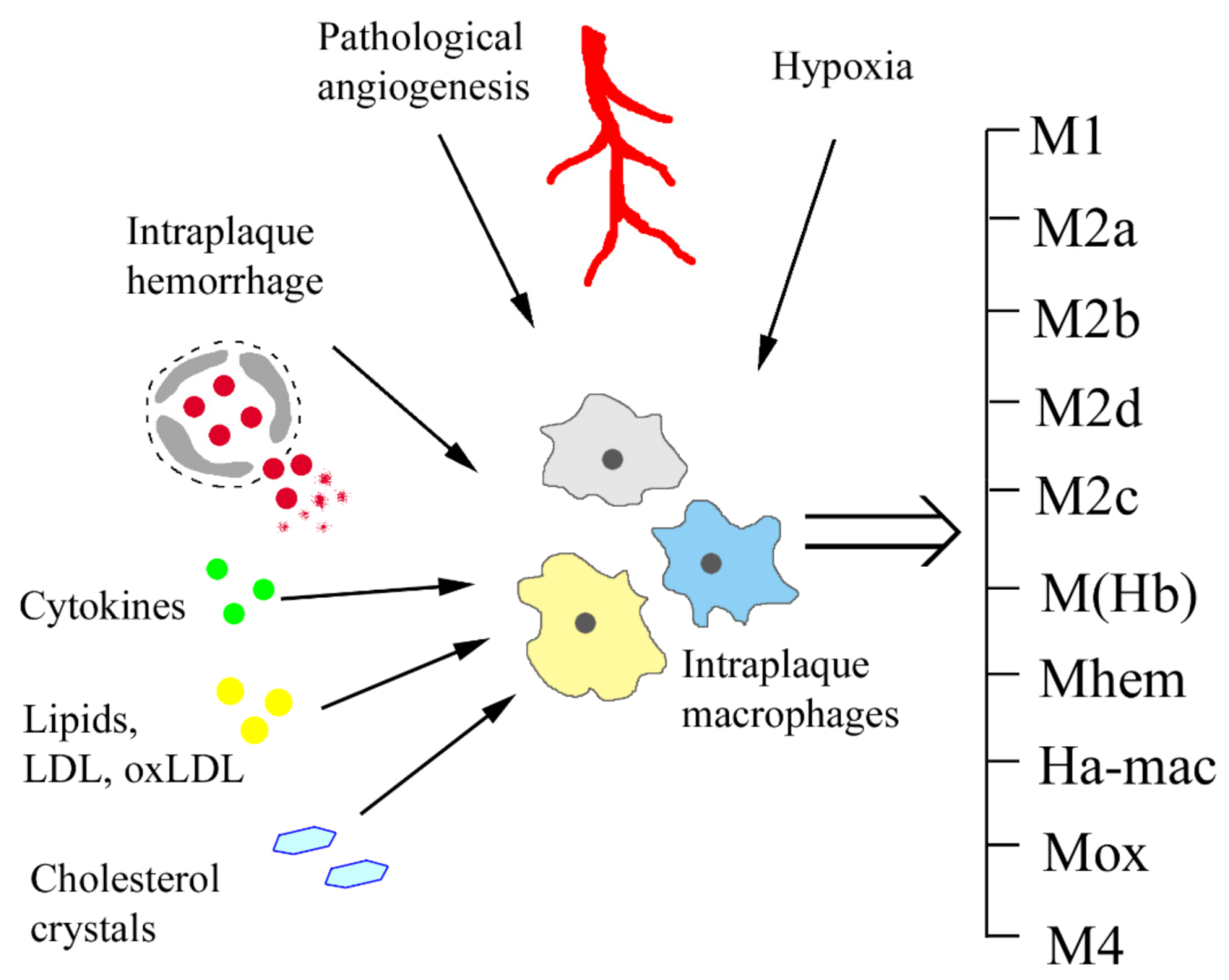
2. Atheroinflammation and Macrophage Polarization Signals
2.1. M1/M2 Polarization
2.2. Lipids as Macrophage Polarizing Signal
2.3. Hypoxia as Macrophage Polarizing Signal
2.4. Haemorrhage as Macrophage Polarizing Signal
3. Single-Cell Level of the Diversity of Intraplaque Macrophages
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Papac- Milicevic, N.; Witztum, J.L. Innate sensing of oxidation- specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Groh, L.; Keating, S.T.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Monocyte and macrophage immunometabolism in atherosclerosis. Semin. Immunopathol. 2018, 40, 203–214. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 2019, 6724903. [Google Scholar] [CrossRef] [Green Version]
- Kavurma, M.M.; Rayner, K.J.; Karunakaran, D. The walking dead: Macrophage inflammation and death in atherosclerosis. Curr. Opin. Lipidol. 2017, 28, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Koplev, S.; Fisher, E.A.; Tabas, I.; Björkegren, J.L.M.; Doran, A.C.; Kovacic, J.C. Macrophage Trafficking, Inflammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series. J. Am. Coll. Cardiol. 2018, 72, 2181–2197. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity f macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2020, 77, 1919–1932. [Google Scholar] [CrossRef]
- Han, X.; Ma, W.; Zhu, Y.; Sun, X.; Liu, N. Advanced glycation end products enhance macrophage polarization to the M1 phenotype via the HIF-1alpha/PDK4 pathway. Mol. Cell. Endocrinol. 2020, 514, 110878. [Google Scholar] [CrossRef] [PubMed]
- Borrell-Pages, M.; Romero, J.C.; Crespo, J.; Juan-Babot, O.; Badimon, L. LRP5 associates with specific subsets of macrophages: Molecular and functional effects. J. Mol. Cell. Cardiol. 2016, 90, 146–156. [Google Scholar] [CrossRef]
- Ma, Y. LRP5: A novel anti-inflammatory macrophage marker that positively regulates migration and phagocytosis. J. Mol. Cell. Cardiol. 2016, 91, 61–62. [Google Scholar] [CrossRef]
- Frodermann, V.; van Duijn, J.; van Puijvelde, G.H.; van Santbrink, P.J.; Lagraauw, H.M.; de Vries, M.R.; Quax, P.H.; Bot, I.; Foks, A.C.; de Jager, S.C.; et al. Heat-killed Staphylococcus aureus reduces atherosclerosis by inducing anti-inflammatory macrophages. J. Intern. Med. 2016, 279, 592–605. [Google Scholar] [CrossRef] [Green Version]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Pinhal-Enfield, G.; Ramanathan, M.; Hasko, G.; Vogel, S.N.; Salzman, A.L.; Boons, G.J.; Leibovich, S.J. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am. J. Pathol. 2003, 163, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Baron, M.; Bouhlel, M.A.; Vanhoutte, J.; Copin, C.; Sebti, Y.; Derudas, B.; Mayi, T.; Bories, G.; Tailleux, A.; et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the ppargamma and lxralpha pathways. Circ. Res. 2011, 108, 985–995. [Google Scholar] [CrossRef]
- Stoger, J.L.; Gijbels, M.J.J.; van der Velden, S.; Manca, M.; van der Loos, C.M.; Biessen, E.A.L.; Daemen, M.J.A.P.; Lutgens, E.; de Winther, M.P.J. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012, 225, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.W.; Martel, C.; Potteaux, S.; Esaulova, E.; Ingersoll, M.A.; Elvington, A.; Saunders, B.T.; Huang, L.H.; Habenicht, A.J.; Zinselmeyer, B.H.; et al. Limited Macrophage Positional Dynamics in Progressing or Regressing Murine Atherosclerotic Plaques-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1702–1710. [Google Scholar] [CrossRef] [Green Version]
- Ménégaut, L.; Thomas, C.; Jalil, A.; Julla, J.B.; Magnani, C.; Ceroi, A.; Basmaciyan, L.; Dumont, A.; Le Goff, W.; Mathew, M.J.; et al. Interplay between Liver X Receptor and Hypoxia Inducible Factor 1alpha Potentiates Interleukin-1beta Production in Human Macrophages. Cell Rep. 2020, 31, 107665. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Flores, A.M.; Hosseini-Nassab, N.; Jarr, K.U.; Ye, J.; Zhu, X.; Wirka, R.; Koh, A.L.; Tsantilas, P.; Wang, Y.; Nanda, V.; et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat. Nanotechnol. 2020, 15, 154–161. [Google Scholar] [CrossRef]
- Murata, Y.; Kotani, T.; Ohnishi, H.; Matozaki, T. The CD47-SIRPalpha signalling system: Its physiological roles and therapeutic application. J. Biochem. 2014, 155, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Fong, G.H. Potential contributions of intimal and plaque hypoxia to atherosclerosis. Curr. Atheroscler. Rep. 2015, 17, 510. [Google Scholar] [CrossRef]
- Jeziorska, M.; Woolley, D.E. Neovascularization in early atherosclerotic lesions of human carotid arteries: Its potential contribution to plaque development. Hum. Pathol. 1999, 30, 919–925. [Google Scholar] [CrossRef]
- Guo, L.; Harari, E.; Virmani, R.; Finn, A.V. Hemorrhage, Angiogenesis, Macrophages, and Iron Metabolism in Atherosclerotic Vascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e33–e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Bai, W.; Liu, Q.; Cui, J.; Zhang, W. Intermittent Hypoxia Enchances THP-1 Monocyte Adhesion and Chemotaxis and Promotes M1 Macrophage Polarization via RAGE. BioMed Res. Int. 2018, 2018, 1650456. [Google Scholar] [CrossRef] [PubMed]
- Crucet, M.; Wust, S.J.; Spielmann, P.; Luscher, T.F.; Wenger, R.H.; Matter, C.M. Hypoxia enhances lipid uptake in macrophages: Role of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis 2013, 229, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Maier, A.; Wu, H.; Cordasic, N.; Oefner, P.; Dietel, B.; Thiele, C.; Weidemann, A.; Eckardt, K.U.; Warnecke. Hypoxia-inducible protein 2 Hig2/Hilpda mediates neutral lipid accumulation in macrophages and contributes to atherosclerosis in apolipoprotein E-deficient mice. FASEB J. 2017, 31, 4971–4984. [Google Scholar] [CrossRef] [Green Version]
- Jain, T.; Nikolopoulou, E.A.; Xu, Q.; Qu, A. Hypoxia inducible factor as a therapeutic target for atherosclerosis. Pharmacol. Ther. 2018, 183, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Karshovska, E.; Wei, Y.; Subramanian, P.; Mohibullah, R.; Geißler, C.; Baatsch, I.; Popal, A.; Corbalán Campos, J.; Exner, N.; Schober, A. HIF-1alpha (Hypoxia-Inducible Factor-1alpha) Promotes Macrophage Necroptosis by Regulating miR-210 and miR-383. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 583–596. [Google Scholar] [CrossRef]
- Helmke, A.; Casper, J.; Nordlohne, J.; David, S.; Haller, H.; Zeisberg, E.M.; von Vietinghoff. Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. FASEB J. 2019, 33, 2278–2289. [Google Scholar] [CrossRef] [Green Version]
- Nakano, D.; Hayashi, T.; Tazawa, N.; Yamashita, C.; Inamoto, S.; Okuda, N.; Mori, T.; Sohmiya, K.; Kitaura, Y.; Okada, Y.; et al. Chronic hypoxia accelerates the progression of atherosclerosis in apolipoprotein E-knockout mice. Hypertens. Res. 2005, 28, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Rahtu-Korpela, L.; Määttä, J.; Dimova, E.Y.; Hörkkö, S.; Gylling, H.; Walkinshaw, G.; Hakkola, J.; Kivirikko, K.I.; Myllyharju, J.; Serpi, R.; et al. Hypoxia-Inducible Factor Prolyl 4-Hydroxylase-2 Inhibition Protects Against Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Christoph, M.; Ibrahim, K.; Hesse, K.; Augstein, A.; Schmeisser, A.; Braun-Dullaeus, R.C.; Simonis, G.; Wunderlich, C.; Quick, S.; Strasser, R.H.; et al. Local inhibition of hypoxia-inducible factor reduces neointima formation after arterial injury in ApoE-/- mice. Atherosclerosis 2014, 233, 641–647. [Google Scholar] [CrossRef]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 107–115. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Sandholt, B.V.; Keller, S.H.; Hansen, A.E.; Clemmensen, A.E.; Sillesen, H.; Højgaard, L.; Ripa, R.S.; Kjær, A. 64Cu-DOTATATE PET/MRI for detection of activated macrophages in carotid atherosclerotic plaques: Studies in patients undergoing endarterectomy. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained pro-inflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Que, K.T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.P.; Liu, Z.J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018, 7, 4012–4022. [Google Scholar] [CrossRef] [Green Version]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Passos Dos Santos, R.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Cai, X.; Ma, R.; Fu, W.; Zhang, C.; Du, X. Iron-load exacerbates the severity of atherosclerosis via inducing inflammation and enhancing the glycolysis in macrophages. J. Cell. Physiol. 2019, 234, 18792–18800. [Google Scholar] [CrossRef]
- Yuan, X.M.; Ward, L.J.; Forssell, C.; Siraj, N.; Li, W. Carotid Atheroma From Men Has Significantly Higher Levels of Inflammation and Iron Metabolism Enabled by Macrophages. Stroke 2018, 49, 419–425. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Gammella, E.; Invernizzi, P.; Cairo, G. Iron levels in polarized macrophages: Regulation of immunity and autoimmunity. Autoimmun. Rev. 2012, 11, 883–889. [Google Scholar] [CrossRef]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.J.; Johns, M.; Kampfer, T.; Nguyen, A.T.; Game, L.; Schaer, D.J.; Mason, J.C.; Haskard, D.O. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 2012, 110, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 174, 1097–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the hif prolyl hydroxylase by the iron chaperones pcbp1 and pcbp2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 2010, 184, 4810–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitsilos, S.; Hunt, J.; Mohler, E.R.; Prabhakar, A.M.; Poncz, M.; Dawicki, J.; Khalapyan, T.Z.; Wolfe, M.L.; Fairman, R.; Mitchell, M.; et al. Platelet factor 4 localization in carotid atherosclerotic plaques: Correlation with clinical parameters. Thromb. Haemost. 2003, 90, 1112–1120. [Google Scholar] [CrossRef]
- de Vries, M.R.; Parma, L.; Peters, H.A.B.; Schepers, A.; Hamming, J.F.; Jukema, J.W.; Goumans, M.J.T.H.; Guo, L.; Finn, A.V.; Virmani, R.; et al. Blockade of vascular endothelial growth factor receptor 2 inhibits intraplaque haemorrhage by normalization of plaque neovessels. J. Intern. Med. 2019, 285, 59–74. [Google Scholar] [CrossRef]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef]
- Xu, X.; Mao, W.; Chai, Y.; Dai, J.; Chen, Q.; Wang, L.; Zhuang, Q.; Pan, Y.; Chen, M.; Ni, G.; et al. Angiogenesis inhibitor, endostar, prevents vasa vasorum neovascularization in a swine atherosclerosis model. J. Atheroscler. Thromb. 2015, 22, 1100–1112. [Google Scholar] [CrossRef] [Green Version]
- Winnik, S.; Lohmann, C.; Siciliani, G.; von Lukowicz, T.; Kuschnerus, K.; Kraenkel, N.; Brokopp, C.E.; Enseleit, F.; Michels, S.; Ruschitzka, F.; et al. Systemic VEGF inhibition accelerates experimental atherosclerosis and disrupts endothelial homeostasis—Implications for cardiovascular safety. Int. J. Cardiol. 2013, 168, 2453–2461. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, L.; de Winther, M.P. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol. 2020, 250, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.J.; Cochain, C.; Vafadarnejad, E.; Saliba, A.E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.; Jang, M.; Jang, H.S.; Yun, T.J.; Lee, S.H.; et al. Transcriptome analysis reveals non-foamy rather than foamy plaque macrophages are pro-inflammatory in atherosclerotic murine models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Lin, J.D.; Nishi, H.; Poles, J.; Niu, X.; Mccauley, C.; Rahman, K.; Brown, E.J.; Yeung, S.T.; Vozhilla, N.; Weinstock, A.; et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 2019, 4, e124574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checkouri, E.; Blanchard, V.; Meilhac, O. Macrophages in Atherosclerosis, First or Second Row Players? Biomedicines 2021, 9, 1214. [Google Scholar] [CrossRef]
- Chen, Y.C.; Bui, A.V.; Diesch, J.; Manassch, R.; Hausding, C.; Rivera, J.; Haviv, I.; Agrotis, A.; Htun, N.M.; Jowett, J.; et al. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ. Res. 2013, 113, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Novak, M.I.; Weinheimer-Haus, E.M.; Koh, T.J. Macrophage Ativation and Skeletal Muscle Healing Following Traumatic Injury. J. Pathol. 2014, 232, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Mirza, R.E.; Fang, M.M.; Weinheimer-Haus, E.M.; Ennis, W.J.; Koh, T.J. Sustained Inflammasome Activity in Macrophages Impairs Wound Healing in Type 2 Diabetic Humans and Mice. Diabetes 2014, 63, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, S.; Moriyama, M.; Tanaka, A.; Machara, T.; Tsuboi, H.; Iizuka, M.; Hayashida, J.; Ohta, M.; Saeki, T.; Notohara, K.; et al. Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Clin. Immunol. 2015, 156, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfs, I.M.J.; Stöger, J.L.; Goossens, P.; Pöttgens, C.; Gijbels, M.J.J.; Wijnands, E.; van der Vorst-, E.P.C.; van Gorp, P.; Beckers, L.; Engel, D.; et al. Reprogramming macrophages to an anti-inflammatory phenotype by helminth antigens reduces murine atherosclerosis. FASEB J. 2014, 28, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanson, M.; Distel, E.; Fisher, E.A. HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS ONE. 2013, 8, e74676. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Rong, J.X.; Shamir, R.; Sanson, M.; Vengrenyuk, Y.; Liu, J.; Rayner, K.; Moore, K.; Garabedian, M.; Fisher, E.A. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7166–7171. [Google Scholar] [CrossRef] [Green Version]
- Härdtner, C.; Ehlert, C.A.; Hilgendorf, I. New insights in statins affecting atheromatous plaque macrophages. Curr. Opin. Lipidol. 2021, 32, 258–264. [Google Scholar] [CrossRef]
- Zhang, Q.; He, J.; Xu, F.; Huang, X.; Wang, Y.; Zhang, W.; Liu, J. Supramolecular copolymer modified statin-loaded discoidal rHDLs for atherosclerotic anti-inflammatory therapy by cholesterol efflux and M2 macrophage polarization. Biomater. Sci. 2021, 9, 6153–6168. [Google Scholar] [CrossRef]
- Cui, H.; Soga, K.; Tamehiro, N.; Adachi, R.; Hachisuka, A.; Hirose, A.; Kondo, K.; Nishimaki-Mogami, T. Statins repress needle-like carbon nanotube- or cholesterol crystal-stimulated IL-1β production by inhibiting the uptake of crystals by macrophages. Biochem. Pharmacol. 2021, 188, 114580. [Google Scholar] [CrossRef]
- Teng, C.; Lin, C.; Huang, F.; Xing, X.; Chen, S.; Ye, L.; Azevedo, H.S.; Xu, C.; Wu, Z.; Chen, Z.; et al. Intracellular codelivery of anti-inflammatory drug and anti-miR 155 to treat inflammatory disease. Acta Pharm. Sin. B 2020, 10, 1521–1533. [Google Scholar] [CrossRef]
- Heffron, S.P.; Weinstock, A.; Scolaro, B.; Chen, S.; Sansbury, B.E.; Marecki, G.; Rolling, C.C.; El Bannoudi, H.; Barrett, T.; Canary, J.W.; et al. Platelet-conditioned media induces an anti-inflammatory macrophage phenotype through EP4. J. Thromb. Haemost. 2021, 19, 562–573. [Google Scholar] [CrossRef]

{kind=link}
| Macrophage Subtype | Differentiation Stimuli | Markers (Human) | Markers (Murine) | Functional Activity in Atherosclerosis |
|---|---|---|---|---|
| M1 | Th1 cytokines, cholesterol crystals, lipopolysaccharide, oxLDL, hypoxia, AGE, iron overload | IL-1β, TNF, IL-6, IL-12, IL-23, CXCL9, CXCL10, CXCL11 | IL-1β, TNF, IL-6, IL-12, IL-23, CXCL9, CXCL10, CXCL11, arginase II | Pro-inflammatory; high ROS production; recruitment of Th1 cells; necrotic core formation; decreased plaque stability |
| M2a | Th2 cytokines (IL-4, IL-13) | MMR, IL-1Ra, factor XIIIa, CD200R, CCL18, stabilin-1, CD163 | Arginase I, resistin-like α, Ym1, Ym2, MMGL, MMR, stabilin-1, CD163, dectin-1 | Resistant to lipid accumulation; anti-inflammatory; tissue repair; pro-fibrotic |
| M2b | Immune complexes and IL-1β or lipopolysaccharide | IL-10high, IL-12low | IL-10high, IL-12low | Resistant to lipid accumulation; anti-inflammatory |
| M2c | IL-1, TGF-β, glucocorticoids | MMR, MerTK | Arginase I | Resistant to lipid accumulation; effective efferocytosis; anti-inflammatory |
| M2d | Adenosine A2 receptor agonists, TLR agonists | Not identified | IL-10, iNOS, VEGF | Pro-angoigenic |
| M(Hb) | Haemoglobin-haptoglobin complexes | CD163, MMR | CD163, MMR | Resistant to lipid accumulation; pro-angoigenic |
| Mhem | Haem | CD163, ATF-1 | CD163, ATF-1 | Resistant to lipid accumulation; pro-angoigenic |
| HA-mac | Haemoglobin-haptoglobin complexes | CD163, HLA-DRlow | CD163, HLA-DRlow | Pro-angiogenic |
| Mox | Oxidized phospholipids | Antioxidant; reduced phagocytic activity | ||
| M4 | CXCL4 | MMP-7, S100-A8, MMR | IL-6, TNF, MMP-7, S100-A8, MMR | Low phagocytic activity; pro-inflammatory; resistant to foam cell formation |
| Type | Resident-Like Macrophages | Inflammatory Macrophages | TREM2hi Macrophages |
|---|---|---|---|
| Markers | Lyve1, Cxcr1, Folr2, Cd206, F13a1, Cbr2, Sepp1, Cxcl4, Gas6, Mafb | Tnf, Nlrp3, Il1b, Egr1, Cepbp, Cxcl1, Ccl2–5, Nfkbia | Trem2, Cd9, Lgals3, Ctsb, Spp1 |
| Functional pathways | Endocytosis, proliferation, anti-inflammatory | Inflammatory response | Cholesterol metabolism, oxidative phosphorylation, lipid accumulation, anti-inflammatory |
| Localization | Adventitia | Intima, plaque shoulder | Intima, necrotic core |
| Markers of corresponding population in human atherosclerosis | Cd206, Cd163 | Hla-dra, Cd74, Cyba, Lyz2, Aif1, S100A8/A9, Malat1, JunB, Nfkbia | Apoc1, ApoE, Ctsb, Fabp5, Plin2, Lgals3, Trem2, Cd9, Lxr, Stat6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mushenkova, N.V.; Nikiforov, N.G.; Melnichenko, A.A.; Kalmykov, V.; Shakhpazyan, N.K.; Orekhova, V.A.; Orekhov, A.N. Functional Phenotypes of Intraplaque Macrophages and Their Distinct Roles in Atherosclerosis Development and Atheroinflammation. Biomedicines 2022, 10, 452. https://doi.org/10.3390/biomedicines10020452
Mushenkova NV, Nikiforov NG, Melnichenko AA, Kalmykov V, Shakhpazyan NK, Orekhova VA, Orekhov AN. Functional Phenotypes of Intraplaque Macrophages and Their Distinct Roles in Atherosclerosis Development and Atheroinflammation. Biomedicines. 2022; 10(2):452. https://doi.org/10.3390/biomedicines10020452
Chicago/Turabian StyleMushenkova, Nataliya V., Nikita G. Nikiforov, Alexandra A. Melnichenko, Vladislav Kalmykov, Nikolay K. Shakhpazyan, Varvara A. Orekhova, and Alexander N. Orekhov. 2022. "Functional Phenotypes of Intraplaque Macrophages and Their Distinct Roles in Atherosclerosis Development and Atheroinflammation" Biomedicines 10, no. 2: 452. https://doi.org/10.3390/biomedicines10020452
APA StyleMushenkova, N. V., Nikiforov, N. G., Melnichenko, A. A., Kalmykov, V., Shakhpazyan, N. K., Orekhova, V. A., & Orekhov, A. N. (2022). Functional Phenotypes of Intraplaque Macrophages and Their Distinct Roles in Atherosclerosis Development and Atheroinflammation. Biomedicines, 10(2), 452. https://doi.org/10.3390/biomedicines10020452