
Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TBI, Its Late Consequences and the Hippocampus
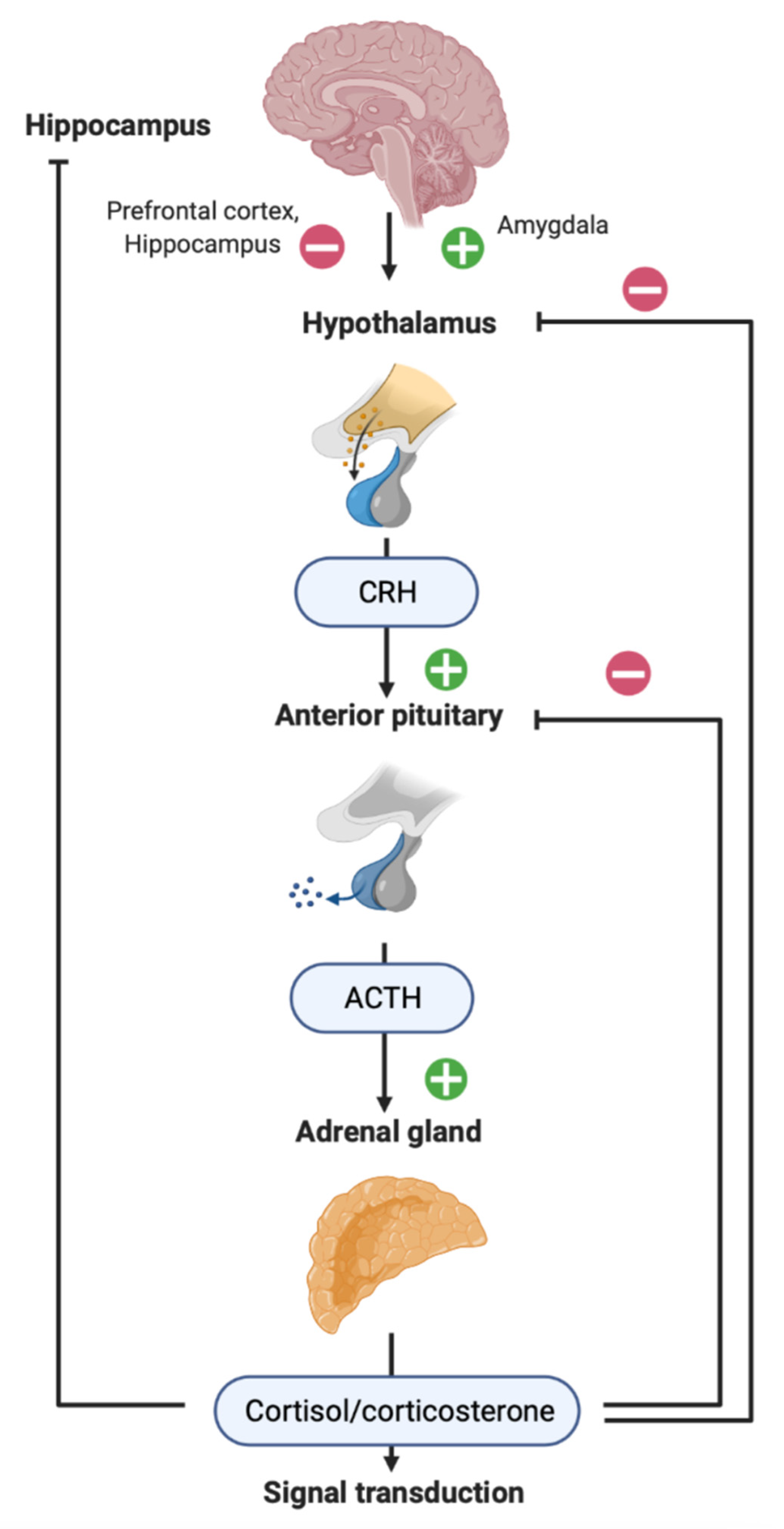
3. HPA Axis in Patients with TBI
4. Distant Hippocampal Damage in Rodent TBI Models
5. Glucocorticoid Signaling, Hippocampus and Neuronal Death

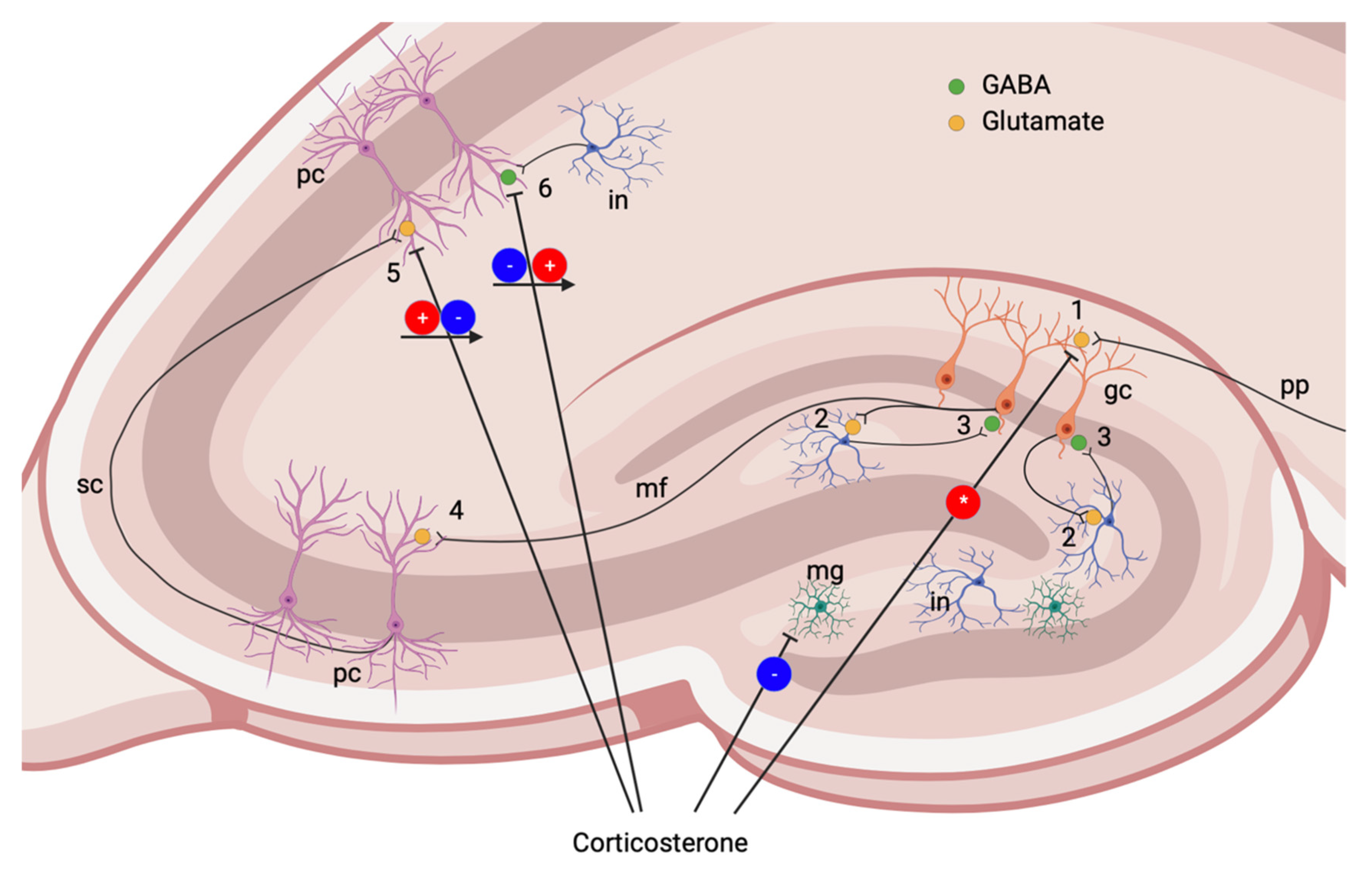
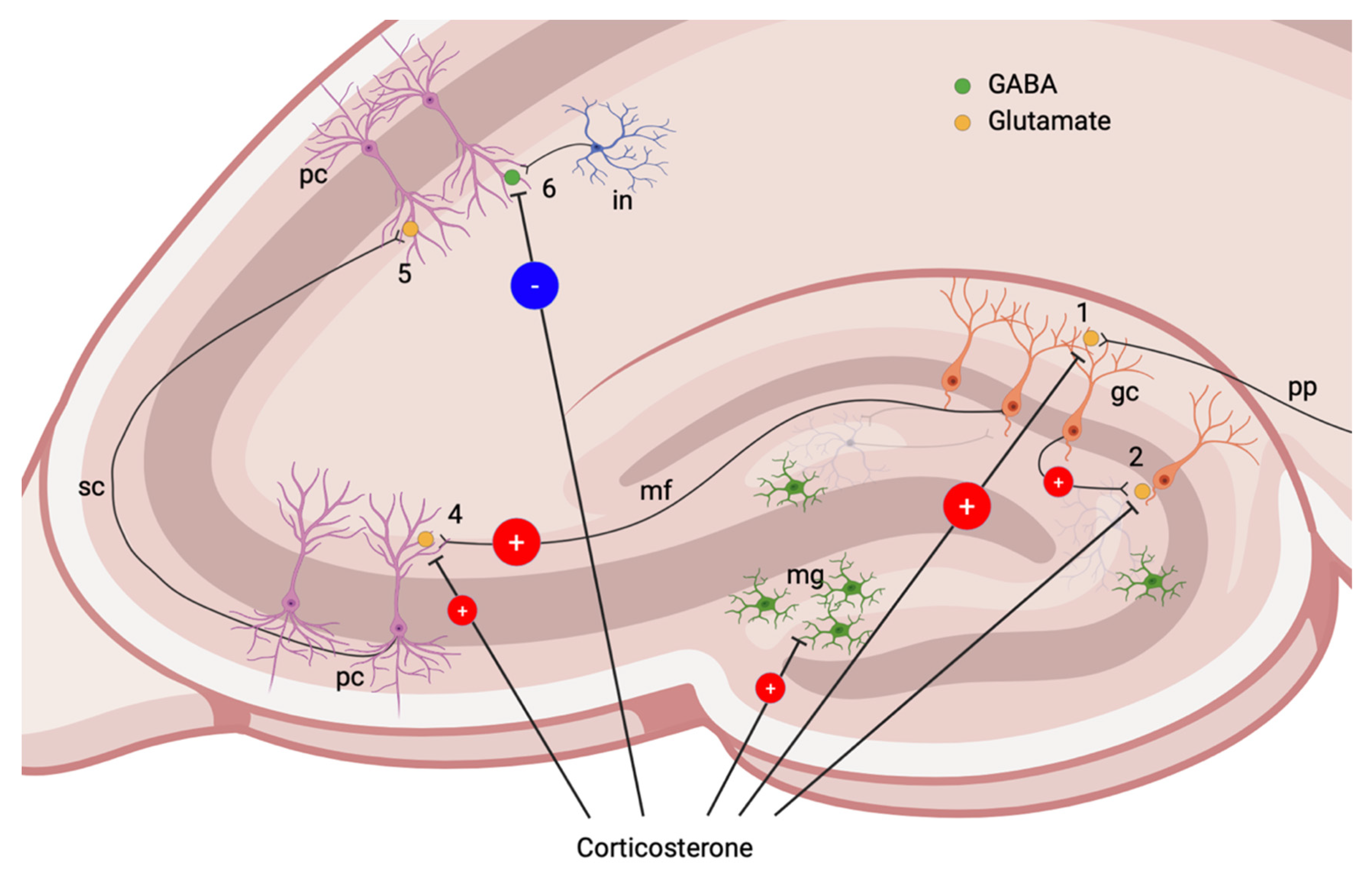
6. Neuroinflammation and TBI
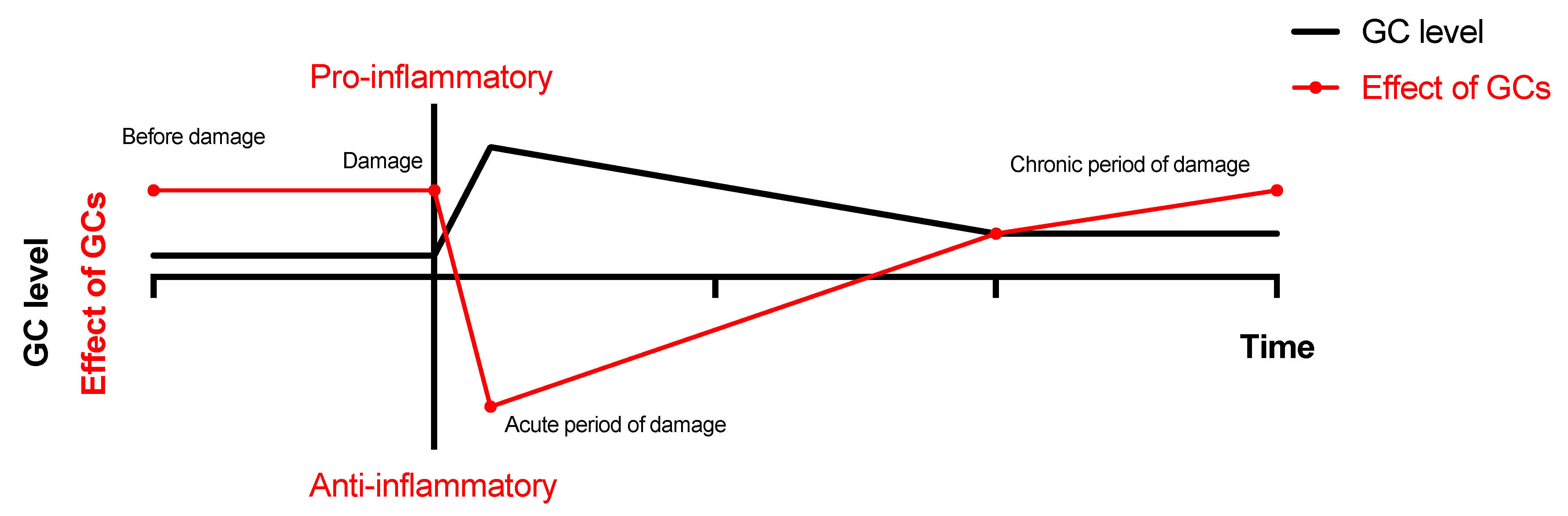
7. Neuroinflammation and GCs
8. CS Changes and Associated Events in Animal Models of TBI: Summary Table
9. Conclusions: TBI and Beyond
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotropic hormone |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| BDNF | brain-derived neurotrophic factor |
| CRH | corticotropin-releasing hormone |
| CS | corticosterone |
| DAMP | damage-associated molecular patterns |
| DG | dentate gyrus, hippocampal field |
| EPSP | excitatory postsynaptic potential |
| GABA | gamma-Aminobutyric acid |
| GCs | glucocorticoids |
| GR | glucocorticoid receptor |
| HPA | hypothalamo-pituitary axis |
| IL-1ß | interleukin 1 beta |
| IL-6 | interleukin 6 |
| iMR, iGR | intracellular cytoplasmic/nuclear receptors subtype |
| IPSC | inhibitory postsynaptic current |
| IPSP | inhibitory postsynaptic potential |
| LPS | lipopolysaccharide |
| MCAO | middle cerebral artery |
| mMR, mGR | membrane-associated receptors subtype |
| MR | mineralocorticoid receptor |
| NF-κB | nuclear factor kappa B |
| NMDA | N-methyl-D-aspartate |
| PTE | post-traumatic epilepsy |
| PV | parvalbumin |
| TBI | traumatic brain injury |
| TNFα | tumor necrosis factor alpha |
| VGCC | voltage-gated calcium channels |
References
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Roozenbeek, B.; Maas, A.I.R.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Marmarou, A.; Choi, S.; Maas, A.; Murray, G.; Steyerberg, E.W. Mortality from traumatic brain injury. In Acta Neurochirurgica, Supplementum; Springer-Verlag: Wien, Austria, 2005; Volume 95, pp. 281–285. [Google Scholar]
- Dawson, S.L.; Hirsch, C.S.; Lucas, F.V.; Sebek, B.A. The contrecoup phenomenon. Hum. Pathol. 1980, 11, 155–166. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Tudor, R.M.; Thompson, C.J. Posterior pituitary dysfunction following traumatic brain injury: Review. Pituitary 2019, 22, 296–304. [Google Scholar] [CrossRef]
- Agha, A.; Thompson, C.J. Anterior pituitary dysfunction following traumatic brain injury (TBI). Clin. Endocrinol. 2006, 64, 481–488. [Google Scholar] [CrossRef]
- Aimaretti, G.; Ghigo, E. Traumatic Brain Injury and Hypopituitarism. Sci. World J. 2005, 5, 777–781. [Google Scholar] [CrossRef]
- Webster, J.B.; Bell, K.R. Primary adrenal insufficiency following traumatic brain injury: A case report and review of the literature. Arch. Phys. Med. Rehabil. 1997, 78, 314–318. [Google Scholar] [CrossRef]
- Beez, T.; Steiger, H.-J.; Etminan, N. Pharmacological targeting of secondary brain damage following ischemic or hemorrhagic stroke, traumatic brain injury, and bacterial meningitis—A systematic review and meta-analysis. BMC Neurol. 2017, 17, 209. [Google Scholar] [CrossRef]
- Dewall, J. The ABCs of TBI. Evidence-based guidelines for adult traumatic brain injury care. JEMS 2010, 35, 54–61. [Google Scholar]
- Segatore, M. Corticosteroids and Traumatic Brain Injury: Status at the End of the Decade of the Brain. J. Neurosci. Nurs. 1999, 31, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, N.P.; Provost, A.C.; Hunter, R.G.; Guffanti, G. Noncoding RNAs: Stress, Glucocorticoids, and Posttraumatic Stress Disorder. Biol. Psychiatry 2018, 83, 849–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, C.N.; Webb, E.K.; DeRoon-Cassini, T.A.; Larson, C.L. Emotion Dysregulation Following Trauma: Shared Neurocircuitry of Traumatic Brain Injury and Trauma-Related Psychiatric Disorders. Biol. Psychiatry 2022, 91, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Vink, R.; Van Den Heuvel, C. Recent advances in the development of multifactorial therapies for the treatment of traumatic brain injury. Expert Opin. Investig. Drugs 2004, 13, 1263–1274. [Google Scholar] [CrossRef]
- Xu, C.; Fu, F.; Li, X.; Zhang, S. Mesenchymal stem cells maintain the microenvironment of central nervous system by regulating the polarization of macrophages/microglia after traumatic brain injury. Int. J. Neurosci. 2017, 127, 1124–1135. [Google Scholar] [CrossRef]
- Tapp, Z.M.; Godbout, J.P.; Kokiko-Cochran, O.N. A Tilted Axis: Maladaptive Inflammation and HPA Axis Dysfunction Contribute to Consequences of TBI. Front. Neurol. 2019, 10, 345. [Google Scholar] [CrossRef]
- Annegers, J.F.; Hauser, W.A.; Coan, S.P.; Rocca, W.A. A population-based study of seizures after traumatic brain injuries. N. Engl. J. Med. 1998, 338, 20–24. [Google Scholar] [CrossRef]
- Gupta, P.K.; Sayed, N.; Ding, K.; Agostini, M.A.; Van Ness, P.C.; Yablon, S.; Madden, C.; Mickey, B.; D’Ambrosio, R.; Diaz-Arrastia, R. Subtypes of Post-Traumatic Epilepsy: Clinical, Electrophysiological, and Imaging Features. J. Neurotrauma 2014, 31, 1439–1443. [Google Scholar] [CrossRef]
- Malmgren, K.; Thom, M. Hippocampal sclerosis-Origins and imaging. Epilepsia 2012, 53, 19–33. [Google Scholar] [CrossRef]
- Englander, J.; Bushnik, T.; Duong, T.T.; Cifu, D.X.; Zafonte, R.; Wright, J.; Hughes, R.; Bergman, W. Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Arch. Phys. Med. Rehabil. 2003, 84, 365–373. [Google Scholar] [CrossRef]
- Haltiner, A.M.; Temkin, N.R.; Dikmen, S.S. Risk of seizure recurrence after the first late posttraumatic seizure. Arch. Phys. Med. Rehabil. 1997, 78, 835–840. [Google Scholar] [CrossRef]
- Temkin, N.R. Risk Factors for Posttraumatic Seizures in Adults. Epilepsia 2003, 44, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann-Eden, B.; Bruckmeir, J. Predictors and dynamics of posttraumatic epilepsy. Acta Neurol. Scand. 1997, 95, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Bombardier, C.H. Rates of Major Depressive Disorder and Clinical Outcomes Following Traumatic Brain Injury. JAMA 2010, 303, 1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry 2019, 84, 1306–1328. [Google Scholar] [CrossRef] [PubMed]
- Malykhin, N.V.; Carter, R.; Seres, P.; Coupland, N.J. Structural changes in the hippocampus in major depressive disorder: Contributions of disease and treatment. J. Psychiatry Neurosci. 2010, 35, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesdorffer, D.C.; Ishihara, L.; Mynepalli, L.; Webb, D.J.; Weil, J.; Hauser, W.A. Epilepsy, suicidality, and psychiatric disorders: A bidirectional association. Ann. Neurol. 2012, 72, 184–191. [Google Scholar] [CrossRef]
- de Kloet, E.R.; Karst, H.; Joëls, M. Corticosteroid hormones in the central stress response: Quick-and-slow. Front. Neuroendocrinol. 2008, 29, 268–272. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Corticosteroid Regulation of Synaptic Plasticity in the Hippocampus. Sci. World J. 2010, 10, 462–469. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322. [Google Scholar] [CrossRef]
- Kusmenkov, T.; Braunstein, M.; Schneider, H.; Bidlingmaier, M.; Prall, W.; Flatz, W.; Boecker, W.; Bogner, V. Initial free cortisol dynamics following blunt multiple trauma and traumatic brain injury: A clinical study. J. Int. Med. Res. 2019, 47, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Kakati, A.; Devi, B.I.; Bhadrinarayan, V.; Kalra, P.; Shukla, D. Endocrine dysfunction following traumatic brain injury in acute stage. Indian J. Neurotrauma 2013, 10, 92–96. [Google Scholar] [CrossRef]
- Rao, T.P. A study of serum cortisol levels in acute head injury patients. J. Basic Clin. Physiol. Pharmacol. 2020, 32, 20190136. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Rogers, B.; Mylotte, D.; Taleb, F.; Tormey, W.; Phillips, J.; Thompson, C.J. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin. Endocrinol. 2004, 60, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Bensalah, M.; Donaldson, M.; Aribi, Y.; Iabassen, M.; Cherfi, L.; Nebbal, M.; Medjaher, M.; Haffaf, E.; Abdennebi, B.; Guenane, K.; et al. Cortisol evaluation during the acute phase of traumatic brain injury-A prospective study. Clin. Endocrinol. 2018, 88, 627–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, F.; Outtrim, J.; Lynch, A.G.; Menon, D.K.; Matta, B.F. Hemodynamic Steroid Responsiveness is Predictive of Neurological Outcome After Traumatic Brain Injury. Neurocrit. Care 2006, 5, 176–179. [Google Scholar] [CrossRef]
- Tanriverdi, F.; Schneider, H.J.; Aimaretti, G.; Masel, B.E.; Casanueva, F.F.; Kelestimur, F. Pituitary Dysfunction After Traumatic Brain Injury: A Clinical and Pathophysiological Approach. Endocr. Rev. 2015, 36, 305–342. [Google Scholar] [CrossRef] [Green Version]
- Saichan, X.; Wei, C.; Qinglong, F.; Jun, W.; Lei, X. Plasma cortisol as a noninvasive biomarker to assess severity and prognosis of patients with craniocerebral injury. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3835–3838. [Google Scholar]
- Sörbo, A.; Eiving, I.; Theodorsson, E.; Rydenhag, B.; Jonsdottir, I.H. Pre-traumatic conditions can influence cortisol levels before and after a brain injury. Acta Neurol. Scand. 2020, 141, 342–350. [Google Scholar] [CrossRef]
- Spikman, J.M.; van der Horn, H.J.; Scheenen, M.E.; de Koning, M.E.; Savas, M.; Langerak, T.; van Rossum, E.F.C.; van der Naalt, J. Coping with stress before and after mild traumatic brain injury: A pilot hair cortisol study. Brain Inj. 2021, 35, 871–879. [Google Scholar] [CrossRef]
- Bay, E.; Sikorskii, A.; Gao, F. Functional Status, Chronic Stress, and Cortisol Response After Mild-to-Moderate Traumatic Brain Injury. Biol. Res. Nurs. 2009, 10, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Bay, E.; Hagerty, B.; Williams, R.A.; Kirsch, N. Chronic Stress, Salivary Cortisol Response, Interpersonal Relatedness, and Depression Among Community-Dwelling Survivors of Traumatic Brain Injury. J. Neurosci. Nurs. 2005, 37, 4–14. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, T.K.; Noble, L.; Andrews, B.; Faden, A.I. Traumatic brain injury in the rat: Characterization of a midline fluid-percussion model. Cent. Nerv. Syst. Trauma 1987, 4, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef] [PubMed]
- Marklund, N. Injury Models of the Central Nervous System; Kobeissy, F.H., Dixon, C.E., Hayes, R.L., Mondello, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1462, ISBN 978-1-4939-3814-8. [Google Scholar]
- Alder, J.; Fujioka, W.; Lifshitz, J.; Crockett, D.P.; Thakker-Varia, S. Lateral Fluid Percussion: Model of Traumatic Brain Injury in Mice. J. Vis. Exp. 2011, 54, e3063. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, W.T.; Smyth, A.; Gilchrist, M.D. Animal models of traumatic brain injury: A critical evaluation. Pharmacol. Ther. 2011, 130, 106–113. [Google Scholar] [CrossRef]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting traumatic brain injury: From molecular mechanisms to therapeutic interventions. Biomedicines 2020, 8, 389. [Google Scholar] [CrossRef]
- Grady, M.S.; Charleston, J.S.; Maris, D.; Witgen, B.M.; Lifshitz, J. Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: Analysis by stereological estimation. J. Neurotrauma 2003, 20, 929–941. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Volkova, A.A.; Levshina, I.P.; Novikova, M.R.; Manolova, A.O.; Stepanichev, M.; Gulyaeva, N. The Number of IgG-Positive Neurons in the Rat Hippocampus Increases after Dosed Traumatic Brain Injury. Neurochem. J. 2018, 12, 256–261. [Google Scholar] [CrossRef]
- Tran, L.D.; Lifshitz, J.; Witgen, B.M.; Schwarzbach, E.; Cohen, A.S.; Grady, M.S. Response of the Contralateral Hippocampus to Lateral Fluid Percussion Brain Injury. J. Neurotrauma 2006, 23, 1330–1342. [Google Scholar] [CrossRef]
- Aungst, S.L.; Kabadi, S.V.; Thompson, S.M.; Stoica, B.A.; Faden, A.I. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J. Cereb. Blood Flow Metab. 2014, 34, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komoltsev, I.G.; Tret’yakova, L.V.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Butuzov, A.V.; Novikova, M.R.; Kvichansky, A.A.; Moiseeva, Y.V.; Onufriev, M.V.; et al. Neuroinflammatory Cytokine Response, Neuronal Death, and Microglial Proliferation in the Hippocampus of Rats During the Early Period After Lateral Fluid Percussion-Induced Traumatic Injury of the Neocortex. Mol. Neurobiol. 2021, 59, 1151–1167. [Google Scholar] [CrossRef] [PubMed]
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int. J. Mol. Sci. 2021, 22, 5883. [Google Scholar] [CrossRef] [PubMed]
- Ruth, R.E. Kainic-acid lesions of hippocampus produced iontophoretically: The problem of distant damage. Exp. Neurol. 1982, 76, 508–527. [Google Scholar] [CrossRef]
- Bagetta, G.; Iannone, M.; Palma, E.; Nisticò, G.; Dolly, J.O. N-methyl-D-aspartate and non-N-methyl-D-aspartate receptors mediate seizures and CA1 hippocampal damage induced by dendrotoxin-K in rats. Neuroscience 1996, 71, 613–624. [Google Scholar] [CrossRef]
- Becker, A.; Tiedge, A.; Grecksch, G.A. Diazepam—Its effects on the development of pentylenetetrazol kindling, related learning impairments, and neuronal cell loss. Pharmacol. Res. 1997, 35, 27–32. [Google Scholar] [CrossRef]
- Aniol, V.A.; Stepanichev, M.; Lazareva, N.A.; Gulyaeva, N.V. An early decrease in cell proliferation after pentylenetetrazole-induced seizures. Epilepsy Behav. 2011, 22, 433–441. [Google Scholar] [CrossRef]
- Huusko, N.; Römer, C.; Ndode-Ekane, X.E.; Lukasiuk, K.; Pitkänen, A. Loss of hippocampal interneurons and epileptogenesis: A comparison of two animal models of acquired epilepsy. Brain Struct. Funct. 2015, 220, 153–191. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Lagowska, Y.; Le Gal La Salle, G.; Tremblay, E.; Ottersen, O.P.; Naquet, R. Diazepam pretreatment reduces distant hippocampal damage induced by intra-amygdaloid injections of kainic acid. Eur. J. Pharmacol. 1978, 52, 419–420. [Google Scholar] [CrossRef]
- Bernert, H.; Turski, L. Traumatic brain damage prevented by the non-N-methyl-D-aspartate antagonist 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f] quinoxaline. Proc. Natl. Acad. Sci. USA 1996, 93, 5235–5240. [Google Scholar] [CrossRef] [Green Version]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blümcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J.; Forcelli, P.A.; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Natoli, M.; Rondouin, G.; Belaidi, M.; Baldy-Moulinier, M.; Kamenka, J.M. N-[1-(2-Thienyl)cyclohexyl]-piperidine (TCP) does not block kainic acid-induced status epilepticus but reduces secondary hippocampal damage. Neurosci. Lett. 1991, 122, 174–178. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Tremblay, E.; Ottersen, O.P.; Meldrum, B.S. The role of epileptic activity in hippocampal and “remote” cerebral lesions induced by kainic acid. Brain Res. 1980, 191, 79–97. [Google Scholar] [CrossRef]
- Dubreuil, C.I.; Marklund, N.; Deschamps, K.; McIntosh, T.K.; McKerracher, L. Activation of Rho after traumatic brain injury and seizure in rats. Exp. Neurol. 2006, 198, 361–369. [Google Scholar] [CrossRef]
- Rink, A.; Fung, K.; Trojanowski, J.Q.; Lee, V.M.; Neugebauer, E.; Mcintosh, T.K. Evidence of Apoptotic Cell Death after Experimental Traumatic Brain Injury in the Rat. Am. J. Pathol. 1995, 63, 305–309. [Google Scholar] [CrossRef]
- Raghupathi, R. Cell Death Mechanisms Following Traumatic Brain Injury. Brain Pathol. 2004, 14, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Newton, R. Molecular mechanisms of glucocorticoid action: What is important? Thorax 2000, 55, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Licht, T.; Sasson, E.; Bell, B.; Grunewald, M.; Kumar, S.; Kreisel, T.; Ben-Zvi, A.; Keshet, E. Hippocampal neural stem cells facilitate access from circulation via apical cytoplasmic processes. Elife 2020, 9, e52134. [Google Scholar] [CrossRef]
- Prager, E.M.; Johnson, L.R. Stress at the Synapse: Signal Transduction Mechanisms of Adrenal Steroids at Neuronal Membranes. Sci. Signal. 2009, 2, re5. [Google Scholar] [CrossRef]
- Karst, H.; Berger, S.; Turiault, M.; Tronche, F.; Schütz, G.; Joëls, M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. USA 2005, 102, 19204–19207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mifsud, K.R.; Kennedy, C.L.M.; Salatino, S.; Sharma, E.; Price, E.M.; Haque, S.N.; Gialeli, A.; Goss, H.M.; Panchenko, P.E.; Broxholme, J.; et al. Distinct regulation of hippocampal neuroplasticity and ciliary genes by corticosteroid receptors. Nat. Commun. 2021, 12, 4737. [Google Scholar] [CrossRef] [PubMed]
- Maggio, N.; Krugers, H.J.; Segal, M. Stress and steroid regulation of synaptic transmission: From physiology to pathophysiology. Front. Cell. Neurosci. 2013, 6, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnsten, A.F.T. Stress signalling pathways that impair prefrontal cortex structure and function. Nat. Rev. Neurosci. 2009, 10, 410–422. [Google Scholar] [CrossRef]
- Marin, M.-F.; Raymond, C.; Lupien, S.J. Memory and Stress. In Stress: Physiology, Biochemistry, and Pathology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 69–78. ISBN 9780128131466. [Google Scholar]
- Narla, C.; Jung, P.S.; Bautista Cruz, F.; Everest, M.; Martinez-Trujillo, J.; Poulter, M.O. CRF Mediates Stress-Induced Pathophysiological High-Frequency Oscillations in Traumatic Brain Injury. eneuro 2019, 6, ENEURO.0334-18.2019. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. Stress, Glucocorticoids, and Damage to the Nervous System: The Current State of Confusion. Stress 1996, 1, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. Stress and the brain: Individual variability and the inverted-U. Nat. Neurosci. 2015, 18, 1344–1346. [Google Scholar] [CrossRef]
- Mattson, M.P. Excitotoxicity. In Stress: Physiology, Biochemistry, and Pathology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 125–134. [Google Scholar]
- Virgin, C.E., Jr.; Ha, T.P.; Packan, D.R.; Tombaugh, G.C.; Yang, S.H.; Homer, H.C.; Sapolsky, R.M. Glucocorticoids Inhibit Glucose Transport and Glutamate Uptake in Hippocampal Astrocytes: Implications for Glucocorticoid Neurotoxicity. J. Neurochem. 1991, 57, 1422–1428. [Google Scholar] [CrossRef]
- Joëls, M. Corticosteroid effects in the brain: U-shape it. Trends Pharmacol. Sci. 2006, 27, 244–250. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Glucocorticoid regulation of the glutamatergic synapse: Mechanisms of stress-dependent neuroplasticity. Ross. Fiziol. Zhurnal Im. I. M. Sechenova Sechenov Physiol. J. 2021, 107, 518–523. [Google Scholar] [CrossRef]
- Joëls, M.; Karst, H.; Sarabdjitsingh, R.A. The stressed brain of humans and rodents. Acta Physiol. 2018, 223, e13066. [Google Scholar] [CrossRef] [PubMed]
- Karst, H.; Joëls, M. Corticosterone Slowly Enhances Miniature Excitatory Postsynaptic Current Amplitude in Mice CA1 Hippocampal Cells. J. Neurophysiol. 2005, 94, 3479–3486. [Google Scholar] [CrossRef] [PubMed]
- Kole, M.H.P.; Swan, L.; Fuchs, E. The antidepressant tianeptine persistently modulates glutamate receptor currents of the hippocampal CA3 commissural associational synapse in chronically stressed rats. Eur. J. Neurosci. 2002, 16, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Joëls, M.; Stienstra, C.; Karten, Y. Effect of Adrenalectomy on Membrane Properties and Synaptic Potentials in Rat Dentate Granule Cells. J. Neurophysiol. 2001, 85, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Karst, H.; Joëls, M. Effect of Chronic Stress on Synaptic Currents in Rat Hippocampal Dentate Gyrus Neurons. J. Neurophysiol. 2003, 89, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Joëls, M. Corticosteroid Actions in the Hippocampus. J. Neuroendocrinol. 2001, 13, 657–669. [Google Scholar] [CrossRef]
- Karst, H.; Joëls, M. Brief RU 38486 Treatment Normalizes the Effects of Chronic Stress on Calcium Currents in Rat Hippocampal CA1 Neurons. Neuropsychopharmacology 2007, 32, 1830–1839. [Google Scholar] [CrossRef]
- van Gemert, N.G.; Joëls, M. Effect of Chronic Stress and Mifepristone Treatment on Voltage-Dependent Ca2+ Currents in Rat Hippocampal Dentate Gyrus. J. Neuroendocrinol. 2006, 18, 732–741. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Differential Corticosteroid Modulation of Inhibitory Synaptic Currents in the Dorsal and Ventral Hippocampus. J. Neurosci. 2009, 29, 2857–2866. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, M.; Czéh, B.; Flügge, G.; Zhang, W. Stress Impairs GABAergic Network Function in the Hippocampus by Activating Nongenomic Glucocorticoid Receptors and Affecting the Integrity of the Parvalbumin-Expressing Neuronal Network. Neuropsychopharmacology 2010, 35, 1693–1707. [Google Scholar] [CrossRef]
- de Kloet, E.R.; Meijer, O.C.; de Nicola, A.F.; de Rijk, R.H.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 2018, 49, 124–145. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, S.F.; Munhoz, C.D.; Manley, N.C.; Yen, S.; Sapolsky, R.M. Glucocorticoids Increase Excitotoxic Injury and Inflammation in the Hippocampus of Adult Male Rats. Neuroendocrinology 2014, 100, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, R.F.; Haselhorst, L.A.; Schoch, K.M.; Bach, E.C.; Rios-Pilier, J.; Scheff, S.W.; Saatman, K.E.; Smith, B.N. Posttraumatic mossy fiber sprouting is related to the degree of cortical damage in three mouse strains. Epilepsy Res. 2012, 99, 167–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowenstein, D.H.; Thomas, M.J.; Smith, D.H.; McIntosh, T.K. Selective vulnerability of dentate hilar neurons following traumatic brain injury: A potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 1992, 12, 4846–4853. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Auvin, S.; Ravizza, T.; Aronica, E. Glia-Neuronal Interactions in Ictogenesis and Epileptogenesis: Role of Inflammatory Mediators. In Jasper’s Basic Mechanisms of the Epilepsies [Internet], 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M., Olsen, R., Delgado-Escueta, A., Eds.; Oxford University Press: Oxford, UK, 2012; ISBN 9780199746545. [Google Scholar]
- Vezzani, A.; Balosso, S.; Ravizza, T. Inflammation and epilepsy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 107, pp. 163–175. ISBN 9780444528988. [Google Scholar]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Tobin, R.P.; Mukherjee, S.; Kain, J.M.; Rogers, S.K.; Henderson, S.K.; Motal, H.L.; Rogers, M.K.N.; Shapiro, L.A. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol. Commun. 2014, 2, 143. [Google Scholar] [CrossRef] [Green Version]
- Hicks, R.; Soares, H.; Smith, D.; McIntosh, T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996, 91, 236–246. [Google Scholar] [CrossRef]
- Cortez, S.C.; McIntosh, T.K.; Noble, L.J. Experimental fluid percussion brain injury: Vascular disruption and neuronal and glial alterations. Brain Res. 1989, 482, 271–282. [Google Scholar] [CrossRef]
- Lescot, T.; Fulla-Oller, L.; Po, C.; Chen, X.R.; Puybasset, L.; Gillet, B.; Plotkine, M.; Meric, P.; Marchand-Leroux, C. Temporal and Regional Changes after Focal Traumatic Brain Injury. J. Neurotrauma 2010, 27, 85–94. [Google Scholar] [CrossRef]
- Xu, S.; Sun, Q.; Fan, J.; Jiang, Y.; Yang, W.; Cui, Y.; Yu, Z.; Jiang, H.; Li, B. Role of Astrocytes in Post-traumatic Epilepsy. Front. Neurol. 2019, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- Thrane, A.S.; Rangroo Thrane, V.; Nedergaard, M. Drowning stars: Reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014, 37, 620–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’amico, R.; Salinaro, A.T.; Fusco, R.; Cordaro, M.; Impellizzeri, D.; Scuto, M.; Ontario, M.L.; Dico, G.L.; Cuzzocrea, S.; Di Paola, R.; et al. Hericium erinaceus and coriolus versicolor modulate molecular and biochemical changes after traumatic brain injury. Antioxidants 2021, 10, 898. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-C.; Liao, Y.-E.; Yang, L.-Y.; Wang, J.-Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.-Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Miguel, Z.D.; Watkins, L.R.; Maier, S.F. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain. Behav. Immun. 2010, 24, 19–30. [Google Scholar] [CrossRef]
- Espinosa-Oliva, A.M.; de Pablos, R.M.; Villarán, R.F.; Argüelles, S.; Venero, J.L.; Machado, A.; Cano, J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol. Aging 2011, 32, 85–102. [Google Scholar] [CrossRef]
- Munhoz, C.D. Chronic Unpredictable Stress Exacerbates Lipopolysaccharide-Induced Activation of Nuclear Factor- B in the Frontal Cortex and Hippocampus via Glucocorticoid Secretion. J. Neurosci. 2006, 26, 3813–3820. [Google Scholar] [CrossRef]
- Munhoz, C.D.; Sorrells, S.F.; Caso, J.R.; Scavone, C.; Sapolsky, R.M. Glucocorticoids Exacerbate Lipopolysaccharide-Induced Signaling in the Frontal Cortex and Hippocampus in a Dose-Dependent Manner. J. Neurosci. 2010, 30, 13690–13698. [Google Scholar] [CrossRef] [Green Version]
- Tretyakova, L.V.; Kvichansky, A.A.; Bolshakov, A.P.; Gulyaeva, N.V. Dexamethasone Modulates Lipopolysaccharide-Induced Expression of Proinflammatory Cytokines in Rat Hippocampus. Neurochem. J. 2021, 15, 302–307. [Google Scholar] [CrossRef]
- Skupio, U.; Tertil, M.; Sikora, M.; Golda, S.; Wawrzczak-Bargiela, A.; Przewlocki, R. Behavioral and molecular alterations in mice resulting from chronic treatment with dexamethasone: Relevance to depression. Neuroscience 2015, 286, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Hershman, S.A.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 2014, 40, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Fenn, A.M.; Gensel, J.C.; Huang, Y.; Popovich, P.G.; Lifshitz, J.; Godbout, J.P. Immune Activation Promotes Depression 1 Month After Diffuse Brain Injury: A Role for Primed Microglia. Biol. Psychiatry 2014, 76, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, T.; Zhi, L.; Bhayana, B.; Wu, M.X. Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury. J. Neuroinflamm. 2016, 13, 197. [Google Scholar] [CrossRef] [Green Version]
- Raefsky, S.M.; Mattson, M.P. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 2017, 102, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulyaeva, N.V.; Onufriev, M.V.; Moiseeva, Y.V. Ischemic Stroke, Glucocorticoids, and Remote Hippocampal Damage: A Translational Outlook and Implications for Modeling. Front. Neurosci. 2021, 15, 1–10. [Google Scholar] [CrossRef]
- Onufriev, M.V.; Moiseeva, Y.V.; Zhanina, M.Y.; Lazareva, N.A.; Gulyaeva, N.V. A Comparative Study of Koizumi and Longa Methods of Intraluminal Filament Middle Cerebral Artery Occlusion in Rats: Early Corticosterone and Inflammatory Response in the Hippocampus and Frontal Cortex. Int. J. Mol. Sci. 2021, 22, 13544. [Google Scholar] [CrossRef]
- Ben Assayag, E.; Tene, O.; Korczyn, A.D.; Shopin, L.; Auriel, E.; Molad, J.; Hallevi, H.; Kirschbaum, C.; Bornstein, N.M.; Shenhar-Tsarfaty, S.; et al. High hair cortisol concentrations predict worse cognitive outcome after stroke: Results from the TABASCO prospective cohort study. Psychoneuroendocrinology 2017, 82, 133–139. [Google Scholar] [CrossRef]
- Kwon, S.K.C.; Kovesdi, E.; Gyorgy, A.B.; Wingo, D.; Kamnaksh, A.; Walker, J.; Long, J.B.; Agoston, D.V. Stress and traumatic brain injury: A behavioral, proteomics, and histological study. Front. Neurol. 2011, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Kamnaksh, A.; Kovesdi, E.; Kwon, S.-K.; Wingo, D.; Ahmed, F.; Grunberg, N.E.; Long, J.; Agoston, D.V. Factors Affecting Blast Traumatic Brain Injury. J. Neurotrauma 2011, 28, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.L.; Richardson, M.R.; Bauman, B.M.; Hernandez, I.M.; Saperstein, S.; Handa, R.J.; Wu, T.J. Differential Responses of the HPA Axis to Mild Blast Traumatic Brain Injury in Male and Female Mice. Endocrinology 2018, 159, 2363–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Xu, X.; Niu, F.; Mao, X.; Dong, J.; Yang, M.; Gao, F.; Liu, B. Corticosterone Replacement Alleviates Hippocampal Neuronal Apoptosis and Spatial Memory Impairment Induced by Dexamethasone via Promoting Brain Corticosteroid Receptor Rebalance after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 262–272. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, M.; Yan, Q.; Xu, X.; Niu, F.; Dong, J.; Zhuang, Y.; Lu, S.; Ge, Q.; Liu, B. The Dual Dose-Dependent Effects of Corticosterone on Hippocampal Cell Apoptosis After Traumatic Brain Injury Depend on the Activation Ratio of Mineralocorticoid Receptors to Glucocorticoid Receptors. Front. Pharmacol. 2021, 12, 713715. [Google Scholar] [CrossRef]
- Geddes, R.I.; Kapoor, A.; Hayashi, K.; Rauh, R.; Wehber, M.; Bongers, Q.; Jansen, A.D.; Anderson, I.M.; Farquhar, G.; Vadakkadath-Meethal, S.; et al. Hypogonadism induced by surgical stress and brain trauma is reversed by human chorionic gonadotropin in male rats: A potential therapy for surgical and TBI-induced hypogonadism? Endocrinol. Diabetes Metab. 2021, 4, e00239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhu, X.; Wang, L.; Hou, Z.; Hao, S.; Yang, M.; Gao, F.; Liu, B. Inadequate Expression and Activation of Mineralocorticoid Receptor Aggravates Spatial Memory Impairment after Traumatic Brain Injury. Neuroscience 2020, 424, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Bai, M.; Xu, X.; Yang, M.; Niu, F.; Gao, F.; Liu, B. Corticosteroid receptor rebalancing alleviates critical illness-related corticosteroid insufficiency after traumatic brain injury by promoting paraventricular nuclear cell survival via Akt/CREB/BDNF signaling. J. Neuroinflammat. 2020, 17, 318. [Google Scholar] [CrossRef]
- Gottesfeld, Z.; Moore, A.N.; Dash, P.K. Acute Ethanol Intake Attenuates Inflammatory Cytokines after Brain Injury in Rats: A Possible Role for Corticosterone. J. Neurotrauma 2002, 19, 317–326. [Google Scholar] [CrossRef]
- Taylor, A.N.; Rahman, S.U.; Tio, D.L.; Sanders, M.J.; Bando, J.K.; Truong, A.H.; Prolo, P. Lasting Neuroendocrine-Immune Effects of Traumatic Brain Injury in Rats. J. Neurotrauma 2006, 23, 1802–1813. [Google Scholar] [CrossRef]
- Niesman, P.J.; Wei, J.; LaPorte, M.J.; Carlson, L.J.; Nassau, K.L.; Bao, G.C.; Cheng, J.P.; de la Tremblaye, P.; Lajud, N.; Bondi, C.O.; et al. Albeit nocturnal, rats subjected to traumatic brain injury do not differ in neurobehavioral performance whether tested during the day or night. Neurosci. Lett. 2018, 665, 212–216. [Google Scholar] [CrossRef]
- Meffre, D.; Pianos, A.; Liere, P.; Eychenne, B.; Cambourg, A.; Schumacher, M.; Stein, D.G.; Guennoun, R. Steroid Profiling in Brain and Plasma of Male and Pseudopregnant Female Rats after Traumatic Brain Injury: Analysis by Gas Chromatography/Mass Spectrometry. Endocrinology 2007, 148, 2505–2517. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.N.; Tio, D.L.; Paydar, A.; Sutton, R.L. Sex Differences in Thermal, Stress, and Inflammatory Responses to Minocycline Administration in Rats with Traumatic Brain Injury. J. Neurotrauma 2018, 35, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Geddes, R.I.; Hayashi, K.; Bongers, Q.; Wehber, M.; Anderson, I.M.; Jansen, A.D.; Nier, C.; Fares, E.; Farquhar, G.; Kapoor, A.; et al. Conjugated Linoleic Acid Administration Induces Amnesia in Male Sprague Dawley Rats and Exacerbates Recovery from Functional Deficits Induced by a Controlled Cortical Impact Injury. PLoS ONE 2017, 12, e0169494. [Google Scholar] [CrossRef] [PubMed]
- McCullers, D.L.; Sullivan, P.G.; Scheff, S.W.; Herman, J.P. Traumatic brain injury regulates adrenocorticosteroid receptor mRNA levels in rat hippocampus. Brain Res. 2002, 947, 41–49. [Google Scholar] [CrossRef]
- Taylor, A.N.; Rahman, S.U.; Sanders, N.C.; Tio, D.L.; Prolo, P.; Sutton, R.L. Injury Severity Differentially Affects Short- and Long-Term Neuroendocrine Outcomes of Traumatic Brain Injury. J. Neurotrauma 2008, 25, 311–323. [Google Scholar] [CrossRef]
- Taylor, A.N.; Tio, D.L.; Sutton, R.L. Restoration of Neuroendocrine Stress Response by Glucocorticoid Receptor or GABA A Receptor Antagonists after Experimental Traumatic Brain Injury. J. Neurotrauma 2013, 30, 1250–1256. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Ko, I.-G.; Kim, B.-K.; Kim, T.-W.; Kim, S.-E.; Shin, M.-S.; Kim, C.-J.; Kim, H.; Kim, K.-M.; Baek, S.-S. Treadmill exercise inhibits traumatic brain injury-induced hippocampal apoptosis. Physiol. Behav. 2010, 101, 660–665. [Google Scholar] [CrossRef]
- Lajud, N.; Roque, A.; Cheng, J.P.; Bondi, C.O.; Kline, A.E. Early Life Stress Preceding Mild Pediatric Traumatic Brain Injury Increases Neuroinflammation but Does Not Exacerbate Impairment of Cognitive Flexibility during Adolescence. J. Neurotrauma 2021, 38, 411–421. [Google Scholar] [CrossRef]
- Taylor, A.N.; Rahman, S.U.; Tio, D.L.; Gardner, S.M.; Kim, C.J.; Sutton, R.L. Injury Severity Differentially Alters Sensitivity to Dexamethasone after Traumatic Brain Injury. J. Neurotrauma 2010, 27, 1081–1089. [Google Scholar] [CrossRef]
- Baykara, B.; Aksu, I.; Buyuk, E.; Kiray, M.; Sisman, A.; Baykara, B.; Dayi, A.; Tas, A.; Ozdemir, D.; Arda, M.; et al. Progesterone treatment decreases traumatic brain injury induced anxiety and is correlated with increased serum IGF-1 levels; prefrontal cortex, amygdala, hippocampus neuron density; and reduced serum corticosterone levels in immature rats. Biotech. Histochem. 2013, 88, 250–257. [Google Scholar] [CrossRef]
- Baykara, B.; Cetin, F.; Baykara, B.; Aksu, I.; Dayi, A.; Kiray, M.; Sisman, A.R.; Ozdemir, D.; Arda, M.N.; Uysal, N. Anxiety caused by traumatic brain injury correlates to decreased prefrontal cortex vegf immunoreactivity and neuron density in immature rats. Turk. Neurosurg. 2012, 22, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Stetter, C.; Lopez-Caperuchipi, S.; Hopp-Krämer, S.; Bieber, M.; Kleinschnitz, C.; Sirén, A.-L.; Albert-Weißenberger, C. Amelioration of Cognitive and Behavioral Deficits after Traumatic Brain Injury in Coagulation Factor XII Deficient Mice. Int. J. Mol. Sci. 2021, 22, 4855. [Google Scholar] [CrossRef] [PubMed]
- Macolino, C.M.; Daiutolo, B.V.; Albertson, B.K.; Elliott, M.B. Mechanical allodynia induced by traumatic brain injury is independent of restraint stress. J. Neurosci. Methods 2014, 226, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kosari-Nasab, M.; Sadeghi, T.; Bashiri, H.; Shokouhi, G.; Salari, A.-A. The blockade of corticotropin-releasing factor 1 receptor attenuates anxiety-related symptoms and hypothalamus–pituitary–adrenal axis reactivity in mice with mild traumatic brain injury. Behav. Pharmacol. 2019, 30, 220–228. [Google Scholar] [CrossRef]
- Sherman, M.; Liu, M.-M.; Birnbaum, S.; Wolf, S.E.; Minei, J.P.; Gatson, J.W. Adult obese mice suffer from chronic secondary brain injury after mild TBI. J. Neuroinflammat. 2016, 13, 171. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.M.; Titus, D.J.; Wilson, N.M.; Freund, J.E.; Atkins, C.M. Early Life Stress Exacerbates Outcome after Traumatic Brain Injury. J. Neurotrauma 2021, 38, 555–565. [Google Scholar] [CrossRef]
- WHITE-GBADEBO, D.; HAMM, R.J. Chronic Corticosterone Treatment Potentiates Deficits Following Traumatic Brain Injury in Rats: Implications for Aging. J. Neurotrauma 1993, 10, 297–306. [Google Scholar] [CrossRef]
- Grundy, P.L.; Patel, N.; Harbuz, M.S.; Lightman, S.L.; Sharples, P.M. Adrenalectomy further suppresses the NT-3 mRNA response to traumatic brain injury but this effect is not reversed with corticosterone. Mol. Brain Res. 2004, 120, 188–192. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Z.; Chai, Y.; Luo, L.; Jiang, R.; Zhang, J. The incidence of critical-illness-related-corticosteroid-insufficiency is associated with severity of traumatic brain injury in adult rats. J. Neurol. Sci. 2014, 342, 93–100. [Google Scholar] [CrossRef]
- Grundy, P.L.; Patel, N.; Harbuz, M.S.; Lightman, S.L.; Sharples, P.M. Glucocorticoids modulate BDNF mRNA expression in the rat hippocampus after traumatic brain injury. Neuroreport 2000, 11, 3381–3384. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, B.; Chai, Y.; Dong, B.; Lei, P.; Jiang, R.; Zhang, J. Methylprednisolone exacerbates acute critical illness-related corticosteroid insufficiency associated with traumatic brain injury in rats. Brain Res. 2011, 1382, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Griesbach, G.S.; Tio, D.L.; Nair, S.; Hovda, D.A. Recovery of Stress Response Coincides with Responsiveness to Voluntary Exercise after Traumatic Brain Injury. J. Neurotrauma 2014, 31, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesbach, G.S.; Tio, D.L.; Vincelli, J.; McArthur, D.L.; Taylor, A.N. Differential Effects of Voluntary and Forced Exercise on Stress Responses after Traumatic Brain Injury. J. Neurotrauma 2012, 29, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Zhao, Z.; Zhou, Y.; Chen, X.; Li, Y.; Liu, X.; Lu, H.; Zhang, Y.; Zhang, J. High-dose glucocorticoid aggravates TBI-associated corticosteroid insufficiency by inducing hypothalamic neuronal apoptosis. Brain Res. 2013, 1541, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Tapp, Z.M.; Kumar, J.E.; Witcher, K.G.; Atluri, R.R.; Velasquez, J.A.; O’Neil, S.M.; Dziabis, J.E.; Bray, C.E.; Sheridan, J.F.; Godbout, J.P.; et al. Sleep Disruption Exacerbates and Prolongs the Inflammatory Response to Traumatic Brain Injury. J. Neurotrauma 2020, 37, 1829–1843. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.K.; Ortiz, J.B.; Thomas, T.C. Mild and Moderate Traumatic Brain Injury and Repeated Stress Affect Corticosterone in the Rat. Neurotrauma Rep. 2020, 1, 113–124. [Google Scholar] [CrossRef]
- Rowe, R.K.; Rumney, B.M.; May, H.G.; Permana, P.; Adelson, P.D.; Harman, S.M.; Lifshitz, J.; Thomas, T.C. Diffuse traumatic brain injury affects chronic corticosterone function in the rat. Endocr. Connect. 2016, 5, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.C.; Stockhausen, E.M.; Law, L.M.; Khodadad, A.; Lifshitz, J. Rehabilitation modality and onset differentially influence whisker sensory hypersensitivity after diffuse traumatic brain injury in the rat. Restor. Neurol. Neurosci. 2017, 35, 611–629. [Google Scholar] [CrossRef] [Green Version]
- McNamara, K.C.S.; Lisembee, A.M.; Lifshitz, J. The Whisker Nuisance Task Identifies a Late-Onset, Persistent Sensory Sensitivity in Diffuse Brain-Injured Rats. J. Neurotrauma 2010, 27, 695–706. [Google Scholar] [CrossRef]
- Rowe, R.K.; Harrison, J.L.; O’Hara, B.F.; Lifshitz, J. Recovery of Neurological Function Despite Immediate Sleep Disruption Following Diffuse Brain Injury in the Mouse: Clinical Relevance to Medically Untreated Concussion. Sleep 2014, 37, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, F.; Pappas, G.; Chen, Y.; Maudlin-Jeronimo, E.; McCarron, R. Effect of acute restraint stress in a polytrauma rat model. Neurosci. Lett. 2018, 684, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Choudhury, M.E.; Miyanishi, K.; Nakanishi, Y.; Kameda, K.; Abe, N.; Yano, H.; Yorozuya, T.; Tanaka, J. Aggravating effects of treadmill exercises during the early-onset period in a rat traumatic brain injury model: When should rehabilitation exercises be initiated? IBRO Rep. 2019, 7, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, N.; Hammamieh, R.; Gautam, A.; Miller, S.-A.; Condlin, M.L.; Jett, M.; Scrimgeour, A.G. TBI weight-drop model with variable impact heights differentially perturbs hippocampus-cerebellum specific transcriptomic profile. Exp. Neurol. 2021, 335, 113516. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Mahesh, R.; Bhatt, S.; Pandey, D. Molecular modifications by regulating cAMP signaling and oxidant-antioxidant defence mechanisms, produce antidepressant-like effect: A possible mechanism of etazolate aftermaths of impact accelerated traumatic brain injury in rat model. Neurochem. Int. 2017, 111, 3–11. [Google Scholar] [CrossRef]
- Doulames, V.M.; Vilcans, M.; Lee, S.; Shea, T.B. Social interaction attenuates the extent of secondary neuronal damage following closed head injury in mice. Front. Behav. Neurosci. 2015, 9, 275. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoltsev, I.G.; Gulyaeva, N.V. Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus. Biomedicines 2022, 10, 1139. https://doi.org/10.3390/biomedicines10051139
Komoltsev IG, Gulyaeva NV. Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus. Biomedicines. 2022; 10(5):1139. https://doi.org/10.3390/biomedicines10051139
Chicago/Turabian StyleKomoltsev, Ilia G., and Natalia V. Gulyaeva. 2022. "Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus" Biomedicines 10, no. 5: 1139. https://doi.org/10.3390/biomedicines10051139
APA StyleKomoltsev, I. G., & Gulyaeva, N. V. (2022). Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus. Biomedicines, 10(5), 1139. https://doi.org/10.3390/biomedicines10051139