
Recent Advances in Biocatalysis for Drug Synthesis

 , , ,
, , ,  , , and
, , and
Abstract
:
1. Introduction
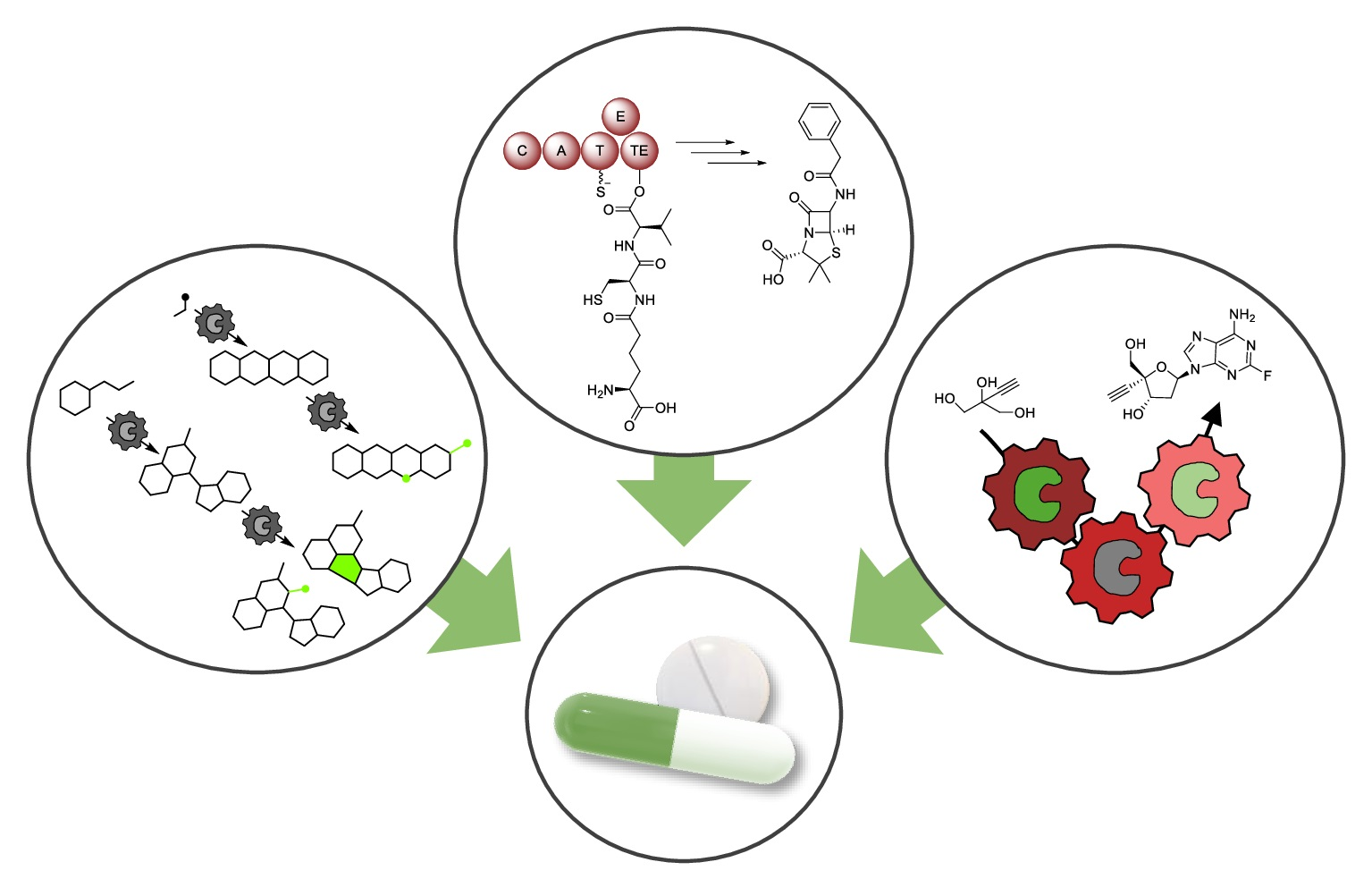
2. Biocatalysis for Drug Synthesis
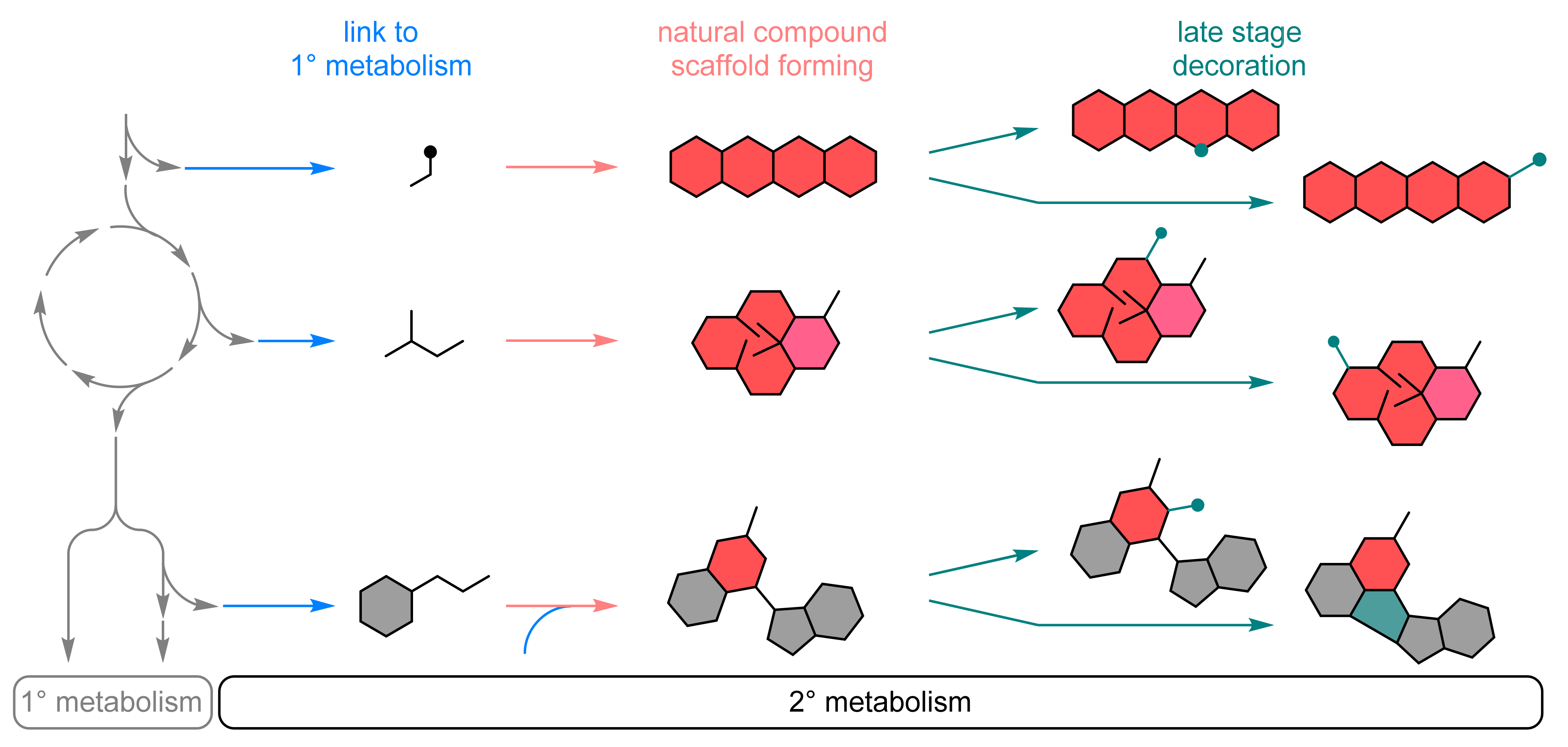
2.1. Expansion of Product Spectrum
2.1.1. Natural Metabolism as Source of Enzymes and Biochemical Pathways
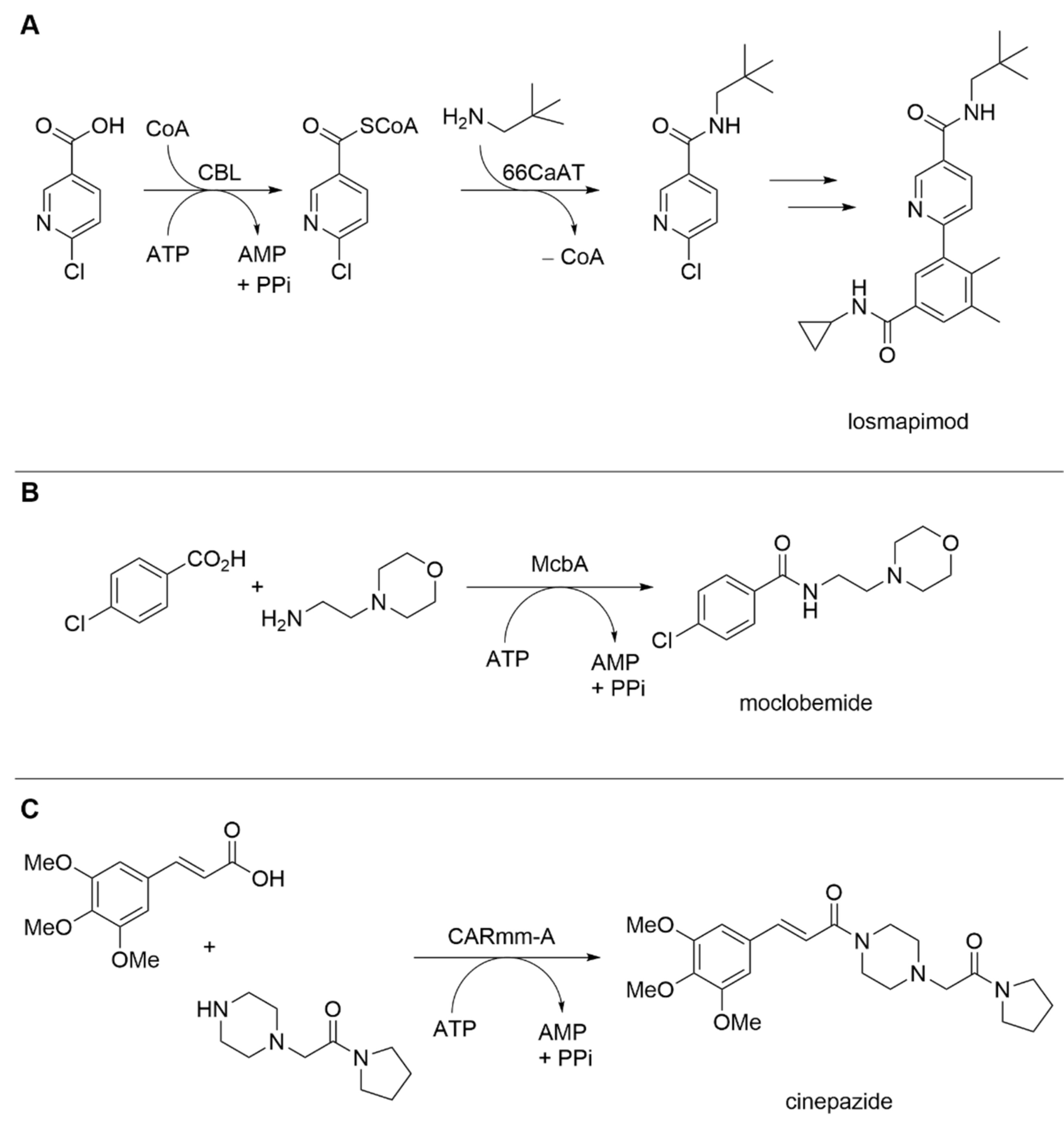
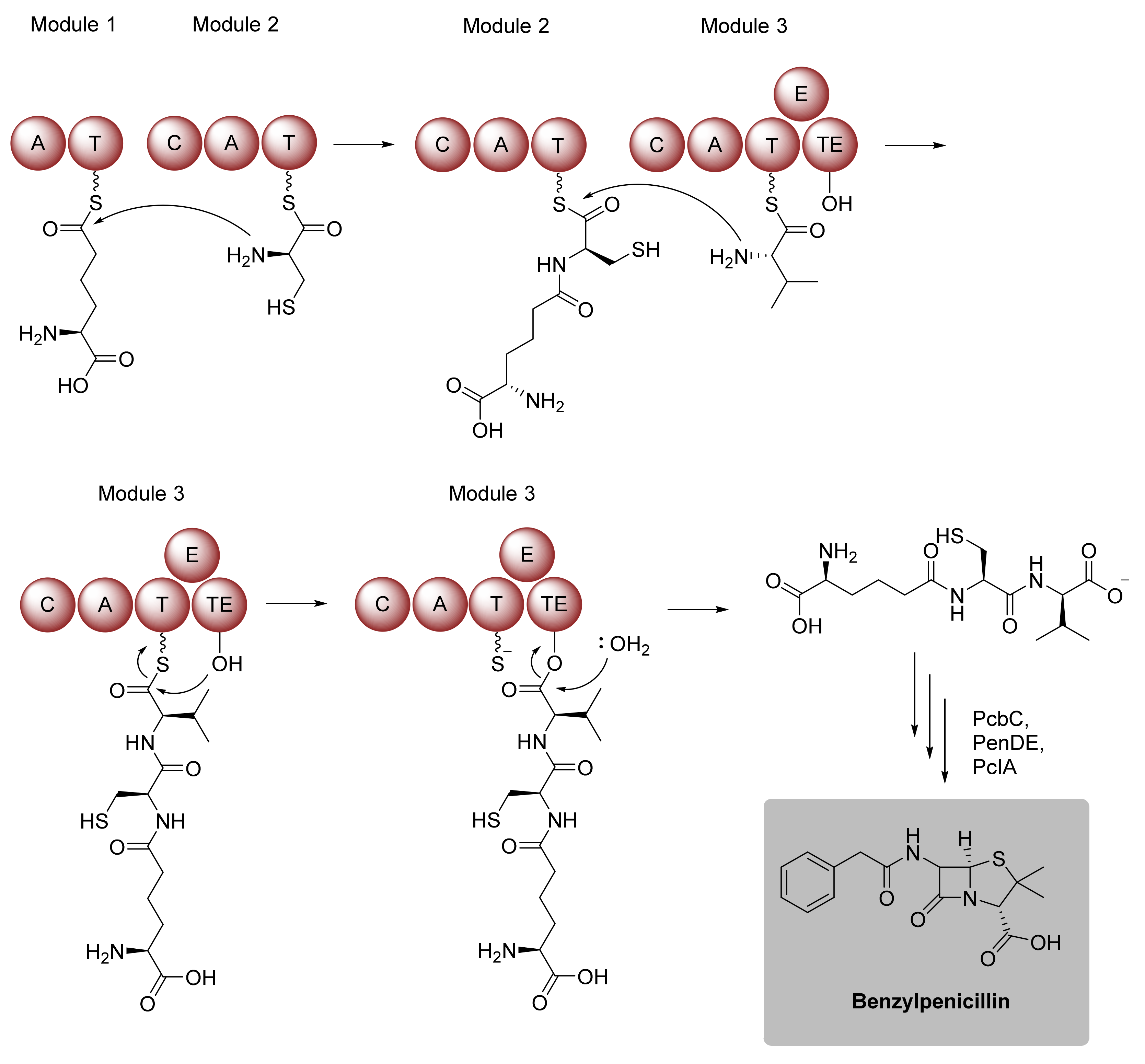
Amide Bond Formation Revisited
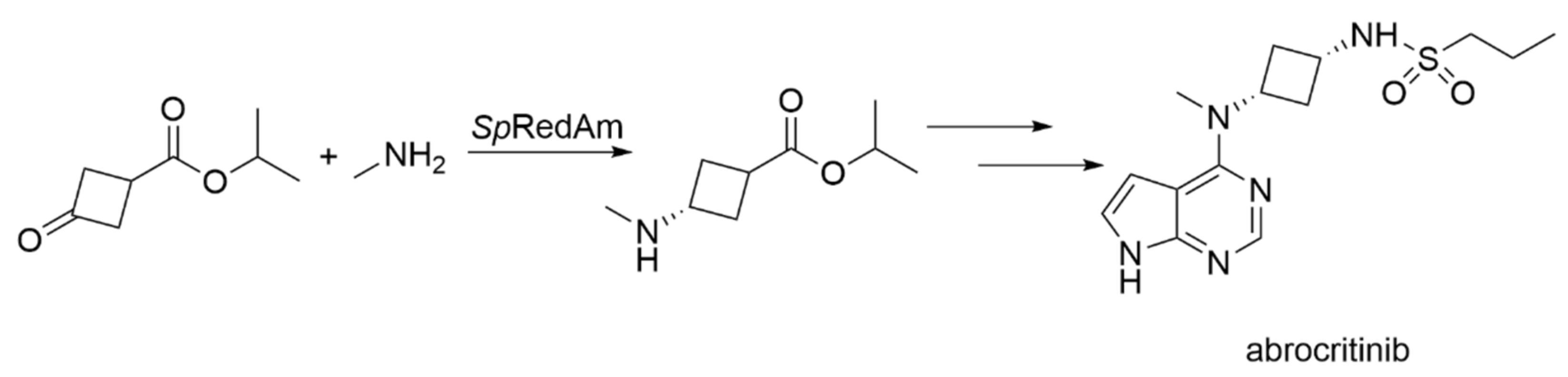
Engineered IREDs for Synthesis of Chiral Amines
Increasing the Usability of Fe2+/α-Ketoglutarate-Dependent Dioxygenases
Biocatalytic Scaffold Formation
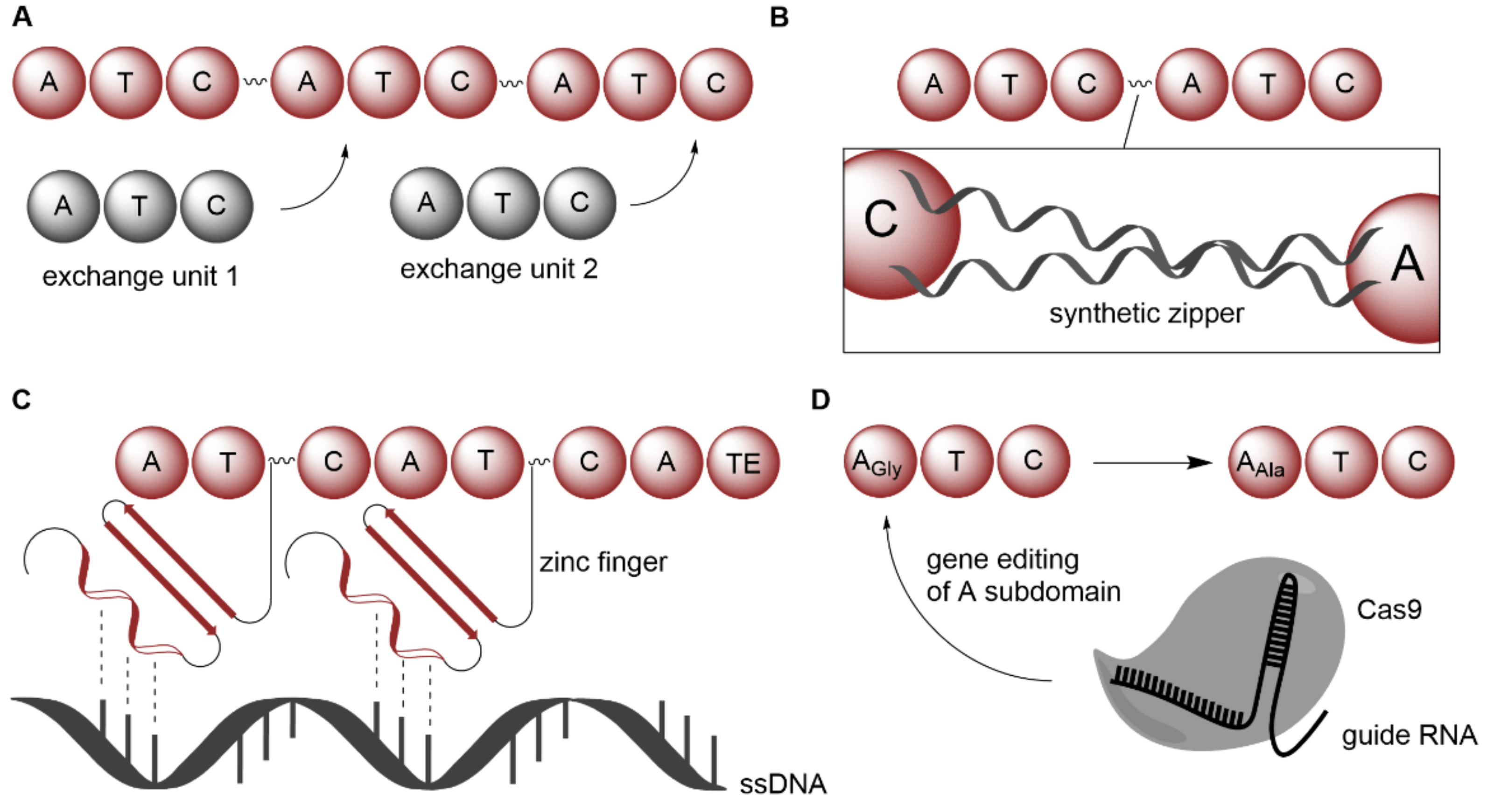
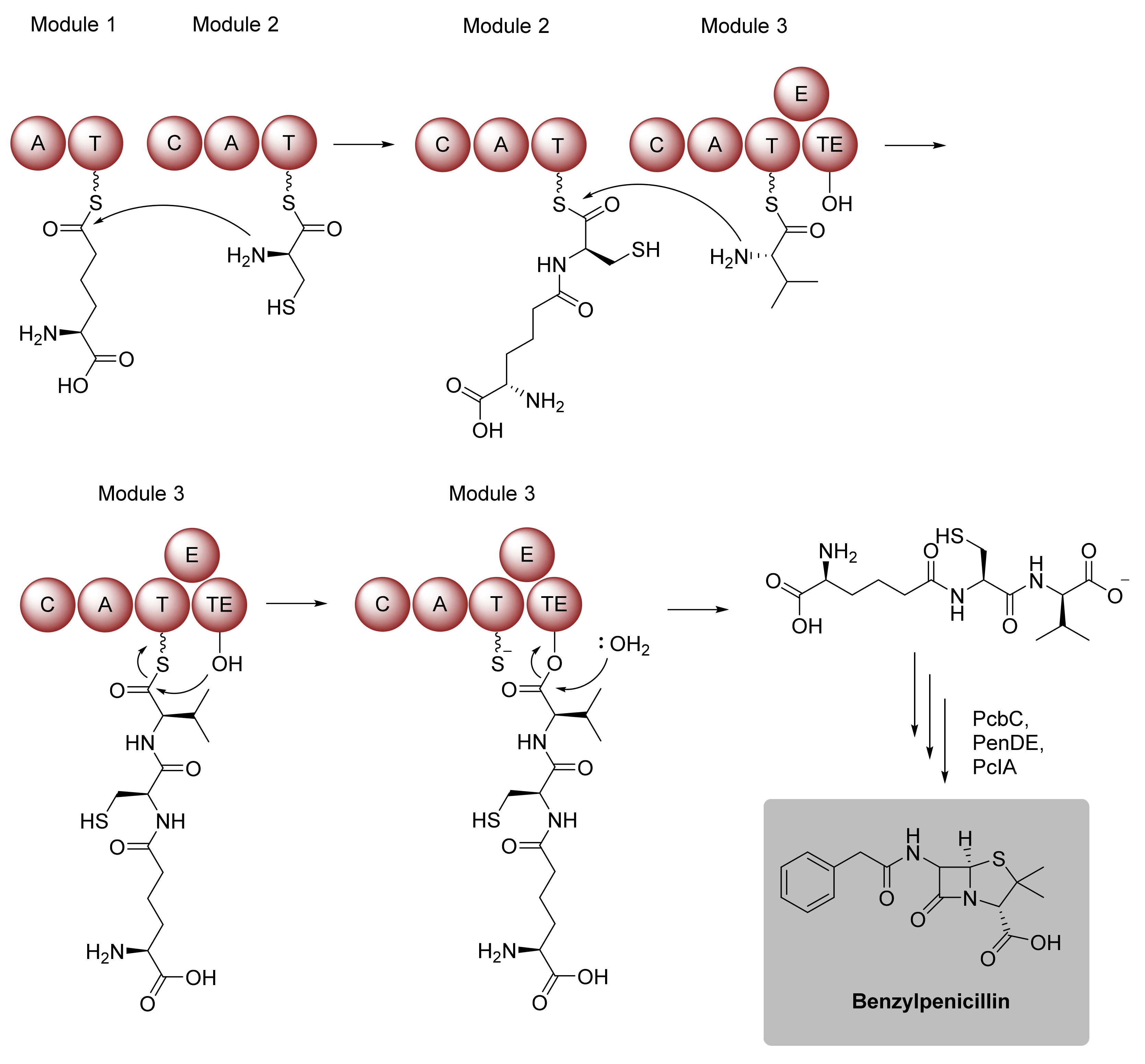
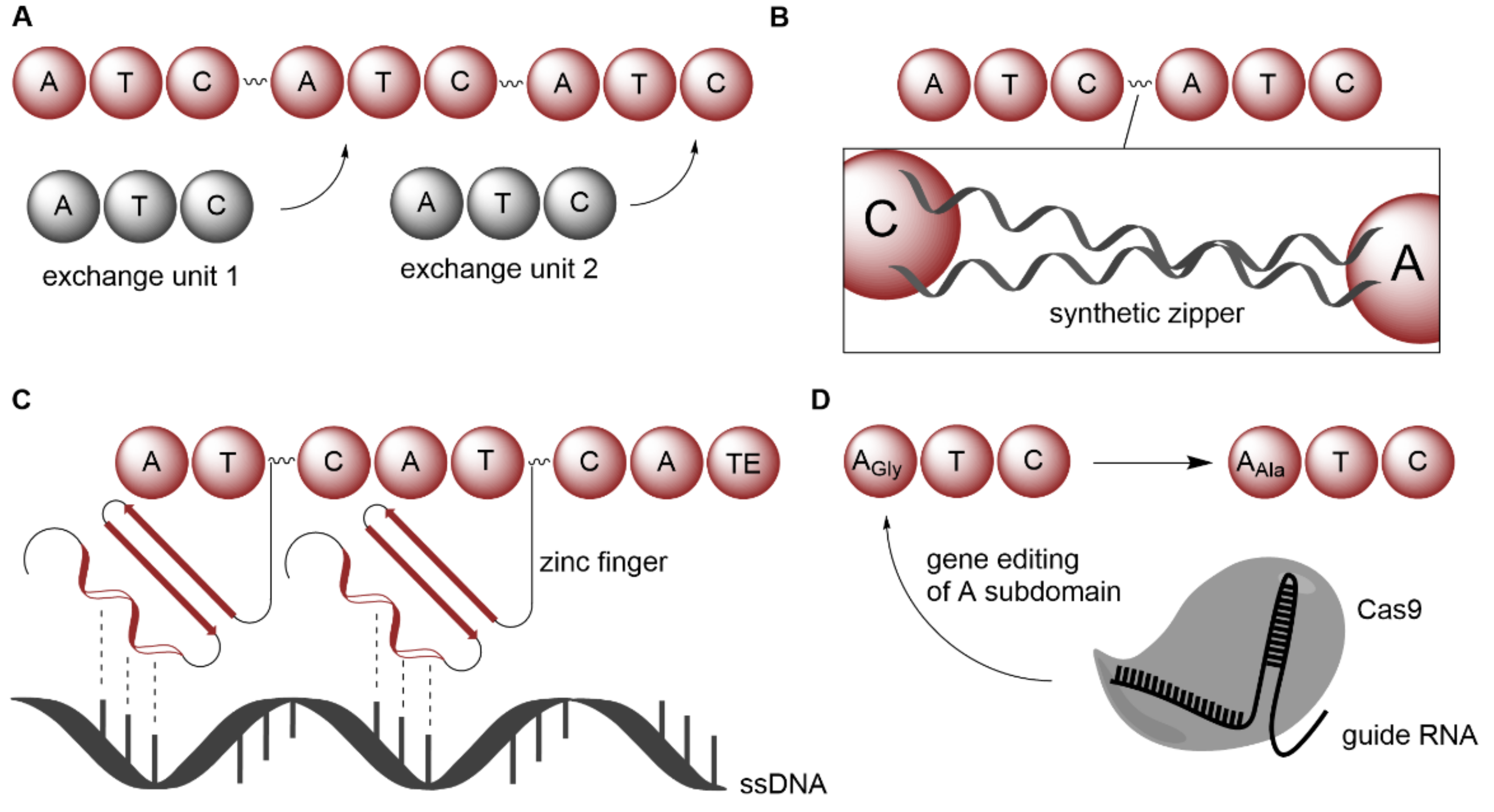
2.1.2. Expansion of Product Spectrum by Engineered Biocatalysts, as Exemplarily Illustrated for Nonribosomal Peptide Synthetases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Source Organism | Modification | Biocatalyst | Substrate | Product | Process Performance | Reference |
|---|---|---|---|---|---|---|---|
| NRPS | Xenorhabdus bovienii | Artificial splitting of NRPS by inserting natural docking domains | Whole-cell | Amino acids | Xefoampeptides derivatives | Wild-type yield | [64] |
| NRPS | Xenorhabdus; Photorhabdus | Introduction of exchange units | Whole-cell | Amino acids | Ambactin derivatives | - | [65,66] |
| NRPS | Xenorhabdus; Photorhabdus; Bacillus | Introduction of exchange unit condensation domains | Whole-cell | Amino acids | GameXPeptide derivatives | - | [67] |
| NRPS | Xenorhabdus nematophila; Photorhabdus luminescens | Introduction of synthetic zippers | Whole-cell | Amino acids | Xenotetrapeptide; GameXPeptide derivatives | Wild-type yield | [68] |
| NRPS | Brevibacillus brevis | Using zinc fingers as guidance on ssDNA | Isolated enzyme | Amino acids | Gramicidin derivatives | - | [69] |
| NRPS | Streptomycetes | Exchanging FSD that contains key active site residues within A domains | Whole-cell | Amino acids | Enduracididn derivatives | Wild-type yield | [70] |
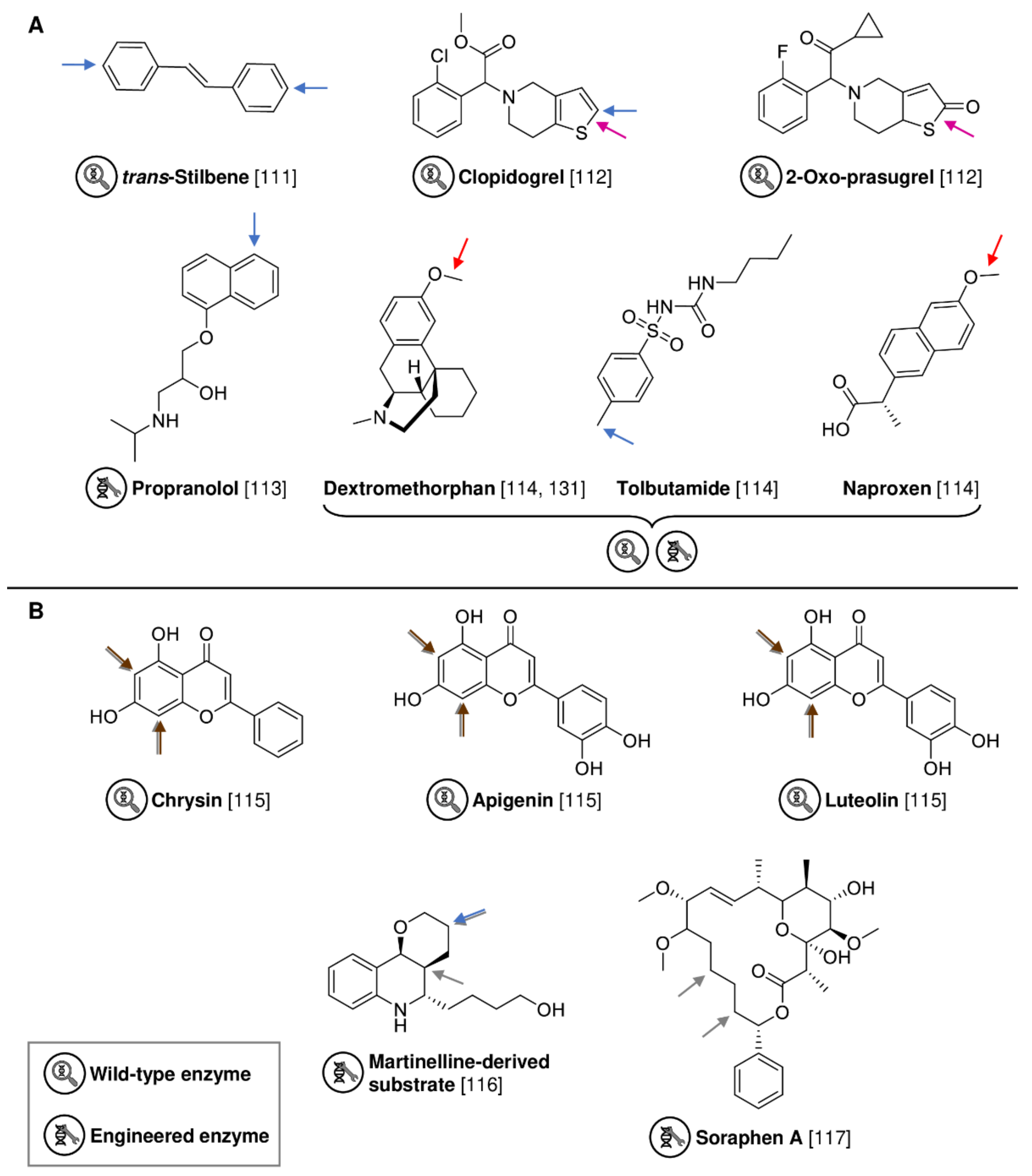
2.2. Expanding Biocatalytic Capabilities in Diversification and Late-Stage Functionalization
| Enzyme | Source Organism | Modification | Biocatalyst | Substrate | Product | Process Performance | Reference |
|---|---|---|---|---|---|---|---|
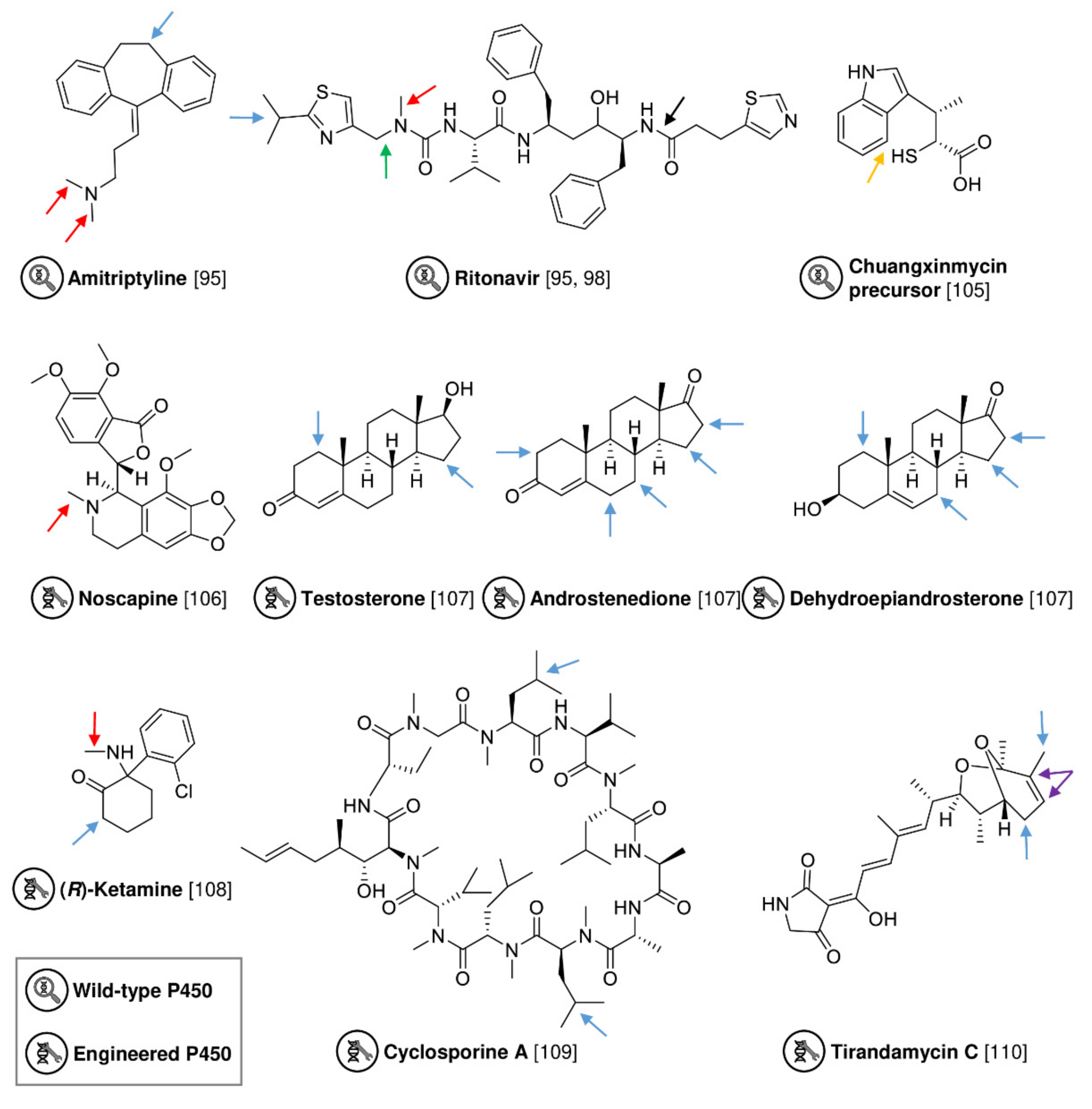
| CYP105D | Streptomyces platensis | Wild-type | E. coli cell lysate | Ritonavir | Hydroxy ritonavir | 58% conversion | [95] |
| CYP107Z | Streptomyces platensis | Wild-type | E. coli cell lysate | Amitriptyline | Demethylated/dide-methylated amitriptyline | 53%/7% conversion | [95] |
| P450 CxnD | Actinoplanes tsinanensis | Wild-type | Isolated enzyme (E. coli) | Chuangxinmycin precusor | Chuangxinmycin | - | [105] |
| P450 BM3 variant | Bacillus megaterium | Directed evolution | E. coli cell lysate | Noscapine | Demethylated noscapine | 50% conversion | [106] |
| P450 BM3 variant | Bacillus megaterium | Directed evolution | Isolated enzyme (E. coli) | Testosterone | Hydroxy testosterone | ≤76% conversion | [107] |
| CYP154E1 variant | Thermobifida fusca | Directed evolution | Whole-cell (E. coli) | (R)-Ketamine | (2R,6R)-Hydroxynorketamine | ≤85% product | [108] |
| CYP-sb21 | Sebekia benihana | Directed evolution | Isolated enzyme (E. coli) | Cyclosporin A | Hydroxy cyclosporin A | ≤94.6% substrate conversion | [109] |
| P450 TamI | Streptomyces sp. 307-9 | Directed evolution | Isolated enzyme (E. coli) | Tirandamycin | Hydroxy tirandamycin | New tirandamycin congeners | [110] |
| AaeUPO | Agrocybe aegerita | Wild-type | Isolated enzyme | trans-Stilbene | 4,4′-Dihydroxy-trans-stilbene | 94% product yield | [111] |
| MroUPO | Marasmius rotula | Wild-type | Isolated enzyme | Clopidogrel | 2-Oxo-clopidogrel | 46% product | [112] |
| MroUPO | Marasmius rotula | Wild-type | Isolated enzyme | 2-Oxo-prasugrel | Active prasugrel metabolite | 34% product | [112] |
| UPO SoLo | Agrocybe aegerita | Evolved AaeUPO variant | Isolated enzyme | Propranolol | 5′-Hydroxypropranolol | 15% isolated yield | [113] |
| UPO JaWa | Agrocybe aegerita | Evolved AaeUPO variant | Isolated enzyme | Dextromethorphan | O-Desmethylnaproxen | 82% product | [114] |
| UPO SoLo-D241G | Agrocybe aegerita | Evolved AaeUPO variant | Isolated enzyme | Naproxen | Hydroxy tolbutamide | 20% product | [114] |
| AaeUPO | Agrocybe aegerita | Wild-type | Isolated enzyme | Tolbutamide | 4-Hydroxymethyl-tolbutamide | 57% product | [114] |
| Halogenase DklH | Frankia alni | Wild-type | Whole-cell (Streptomyces albus) | Luteolin | Dichloroluteolin | 86% product | [115] |
| Halogenase WelO5* CA2 | Hapalosiphon welwitschii | Evolved WelO5* variant | E. coli cell lysate | Martinelline-derived substrate | Hydroxylated product | 30% isolated yield | [116] |
| Halogenase WelO5* VLA | Hapalosiphon welwitschii | Evolved WelO5* variant | Isolated enzyme | Soraphen A | Mono-chlorinated product | 50% conversion | [117] |
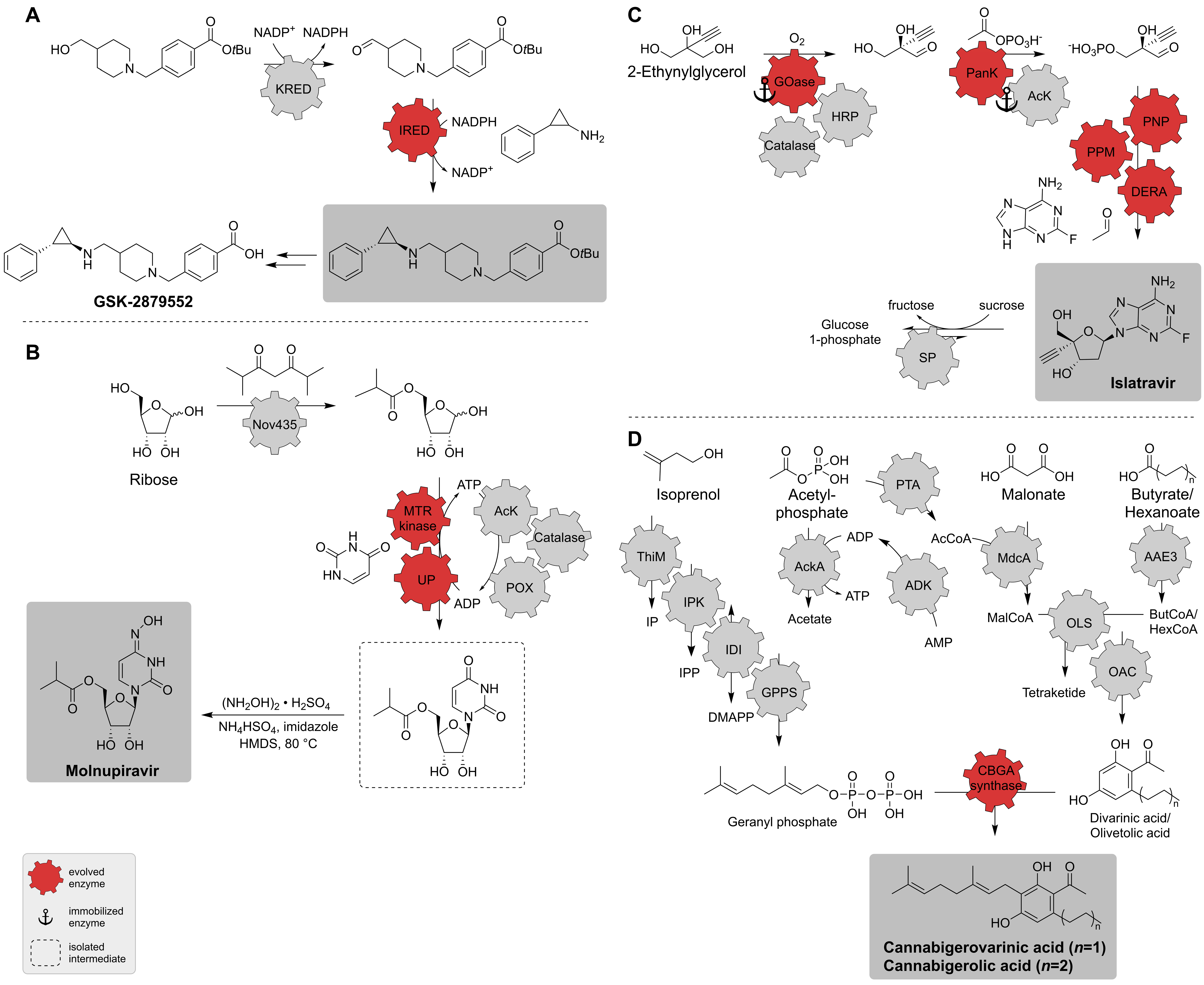
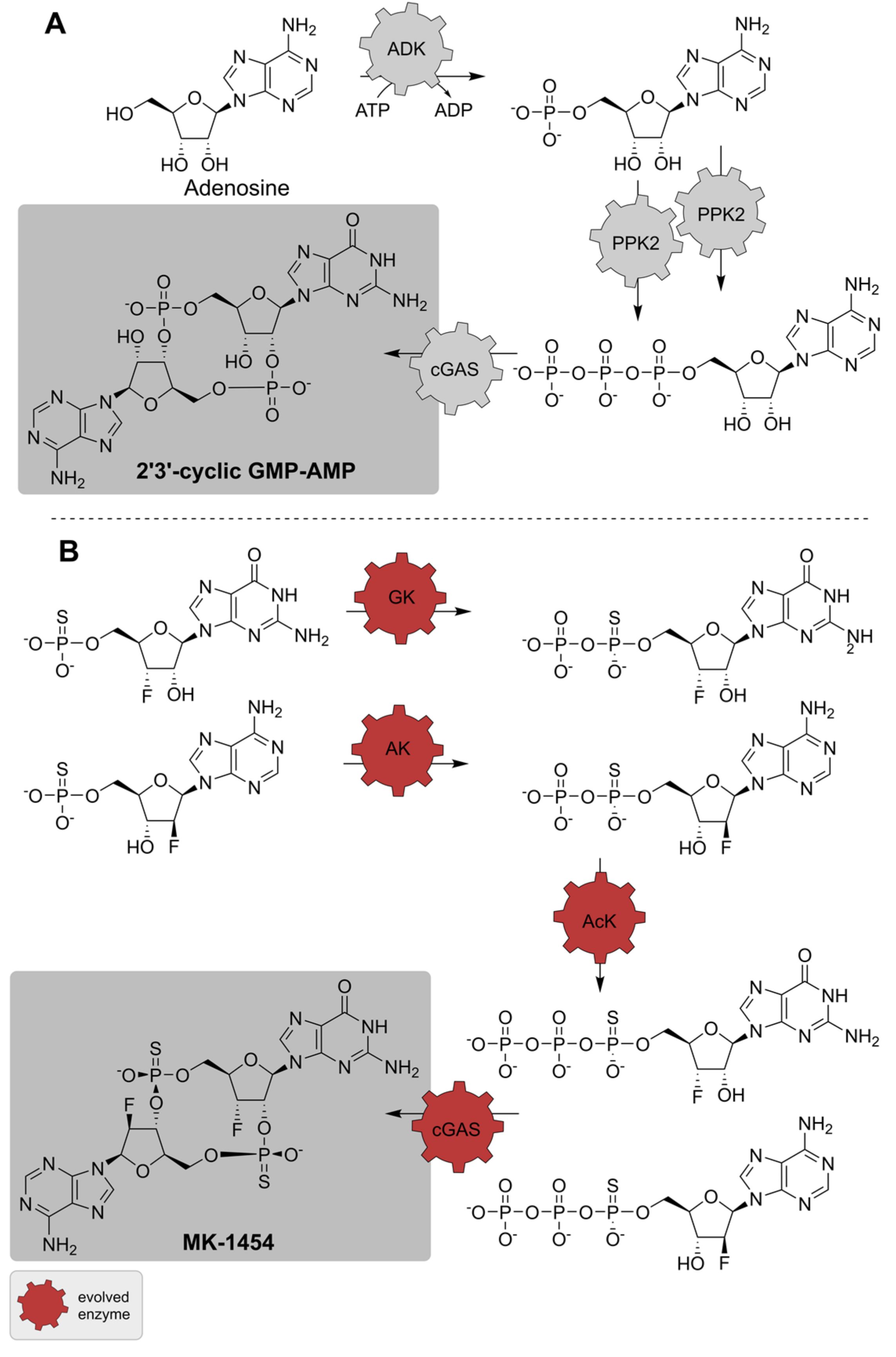
2.3. Synthetic Enzyme Cascades
| Enzyme | Source Organism | Modification | Biocatalyst | Substrate | Product | Process Performance | Reference |
|---|---|---|---|---|---|---|---|
| IRED; KRED | Diverse | Directed evolution (one enzyme) | Isolated enzymes | Amine tranylcypromine sulfate, alcohol precursor | Protected GSK-2879552 | 5 g scale with 48.3% yield, 99.5%-ee, 97.9% purity | [34] |
| Nov435; MTR kinase; UP; AcK; catalase; POX | Diverse | Directed evolution (two enzymes) | Isolated enzymes | Ribose, uracil | Molnupiravir | 69% overall yield | [149] |
| GOase; HRP; catalase; PanK; AcK; PNP; PPM; DERA; SP | Diverse | Directed evolution (five enzymes) | Isolated enzymes | 2-Ethynylglycerol | Islatravir | 51% overall yield | [141] |
| ThiM; IPK; IDI; GPPS; ADK; AcKA; PTA; MdcA; AAE3; OLS; OAC; CBGA synthase | Diverse | One engineered enzyme by Rosetta design [151] | Isolated enzymes | Isoprenol, acetylphosphate, malonate, butyrate/hexanoate | Cannabigerolic acid (CBGA); cannabigerovarinic acid (CBGVA) | 480 mg L−1 (CBGA), 580 mg L−1 (CBGVA) | [152] |
| ADK; 2 PPK2; cGAS | Diverse | - | Isolated enzymes | Adenosine, GTP | 2′3′-cGAMP | 0.08 mol per mol adenosine | [153] |
| GK; AK; AcK; cGAS | Diverse | Directed evolution (four enzymes) | Isolated enzymes | Nucleotide monothiophosphates | MK-1454 | 62% isolated yield | [154] |
3. Conclusion and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Sheldon, R.A. The E Factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Simić, S.; Zukić, E.; Schmermund, L.; Faber, K.; Winkler, C.K.; Kroutil, W. Shortening synthetic routes to small molecule active pharmaceutical ingredients employing biocatalytic methods. Chem. Rev. 2022, 122, 1052–1126. [Google Scholar] [CrossRef]
- Lütz, S.; Giver, L.; Lalonde, J. Engineered enzymes for chemical production. Biotechnol. Bioeng. 2008, 101, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, K.; Lütz, S. Recent developments and challenges of biocatalytic processes in the pharmaceutical industry. Curr. Opin. Green Sustain. Chem. 2018, 11, 58–64. [Google Scholar] [CrossRef]
- Hollmann, F.; Opperman, D.J.; Paul, C.E. Biocatalytic reduction reactions from a chemist’s perspective. Angew. Chem. Int. Ed. 2021, 60, 5644–5665. [Google Scholar] [CrossRef]
- Schwarz, J.; Rosenthal, K.; Snajdrova, R.; Kittelmann, M.; Lütz, S. The development of biocatalysis as a tool for drug discovery. Chimia (Aarau) 2020, 74, 368–377. [Google Scholar] [CrossRef]
- Arroyo, M.; de la Mata, I.; García, J.L.; Barredo, J.L. Biocatalysis for industrial production of active pharmaceutical ingredients (APIs). In Biotechnology of Microbial Enzymes: Production, Biocatalysis and Industrial Applications; Elsevier Inc.: Philadelphia, PA, USA, 2017; pp. 451–473. ISBN 9780128037461. [Google Scholar]
- Sun, H.; Zhang, H.; Ang, E.L.; Zhao, H. Biocatalysis for the synthesis of pharmaceuticals and pharmaceutical intermediates. Bioorg. Med. Chem. 2018, 26, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Classen, T.; Pietruszka, J. Complex molecules, clever solutions–Enzymatic approaches towards natural product and active agent syntheses. Bioorg. Med. Chem. 2018, 26, 1285–1303. [Google Scholar] [CrossRef]
- Walsh, C.T.; Tang, Y. Natural Product Biosynthesis; The Royal Society of Chemistry: London, UK, 2017; ISBN 978-1-78801-076-4. [Google Scholar]
- Bergman, M.E.; Davis, B.; Phillips, M.A. Medically useful plant terpenoids: Biosynthesis, occurrence, and mechanism of action. Molecules 2019, 24, 3961. [Google Scholar] [CrossRef] [Green Version]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef] [PubMed]
- Marienhagen, J.; Bott, M. Metabolic engineering of microorganisms for the synthesis of plant natural products. J. Biotechnol. 2013, 163, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Firn, R.D.; Jones, C.G. Natural products—A simple model to explain chemical diversity. Nat. Prod. Rep. 2003, 20, 382. [Google Scholar] [CrossRef] [PubMed]
- Philpott, H.K.; Thomas, P.J.; Tew, D.; Fuerst, D.E.; Lovelock, S.L. A versatile biosynthetic approach to amide bond formation. Green Chem. 2018, 20, 3426–3431. [Google Scholar] [CrossRef]
- Petchey, M.R.; Rowlinson, B.; Lloyd, R.C.; Fairlamb, I.J.S.; Grogan, G. Biocatalytic synthesis of moclobemide using the amide bond synthetase McbA coupled with an ATP recycling system. ACS Catal. 2020, 10, 4659–4663. [Google Scholar] [CrossRef]
- Lubberink, M.; Schnepel, C.; Citoler, J.; Derrington, S.R.; Finnigan, W.; Hayes, M.A.; Turner, N.J.; Flitsch, S.L. Biocatalytic monoacylation of symmetrical diamines and its application to the synthesis of pharmaceutically relevant amides. ACS Catal. 2020, 10, 10005–10009. [Google Scholar] [CrossRef]
- Kumar, R.; Karmilowicz, M.J.; Burke, D.; Burns, M.P.; Clark, L.A.; Connor, C.G.; Cordi, E.; Do, N.M.; Doyle, K.M.; Hoagland, S.; et al. Biocatalytic reductive amination from discovery to commercial manufacturing applied to abrocitinib JAK1 inhibitor. Nat. Catal. 2021, 4, 775–782. [Google Scholar] [CrossRef]
- Seide, S.; Arnold, L.; Wetzels, S.; Bregu, M.; Gätgens, J.; Pohl, M. From enzyme to preparative cascade reactions with immobilized enzymes: Tuning Fe(II)/α-ketoglutarate-dependent lysine hydroxylases for application in biotransformations. Catalysts 2022, 12, 354. [Google Scholar] [CrossRef]
- Meyer, F.; Frey, R.; Ligibel, M.; Sager, E.; Schroer, K.; Snajdrova, R.; Buller, R. Modulating chemoselectivity in a Fe(II)/α-ketoglutarate-dependent dioxygenase for the oxidative modification of a nonproteinogenic amino acid. ACS Catal. 2021, 11, 6261–6269. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Meierhofer, J.; Meyer, F.; Hayashi, T.; Schneider, S.; Sager, E.; Buller, R.M.U. Re-programming and optimization of a L-proline cis-4-hydroxylase for the cis-3-halogenation of its native substrate. ChemCatChem 2021, 13, 3914–3919. [Google Scholar] [CrossRef]
- Zhao, J.; Méndez-Sánchez, D.; Roddan, R.; Ward, J.M.; Hailes, H.C. Norcoclaurine synthase-mediated stereoselective synthesis of 1,1′-disubstituted, spiro- and bis-tetrahydroisoquinoline alkaloids. ACS Catal. 2020, 11, 131–138. [Google Scholar] [CrossRef]
- Schneider, P.; Henssen, B.; Paschold, B.; Chapple, B.P.; Schatton, M.; Seebeck, F.P.; Classen, T.; Pietruszka, J. Biocatalytic C3-indole methylation—A useful tool for the natural-product-inspired stereoselective synthesis of pyrroloindoles. Angew. Chem. Int. Ed. Engl. 2021, 60, 23412–23418. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Jegl, P.; Hauer, B. Stereoselective directed cationic cascades enabled by molecular anchoring in terpene cyclases. Angew. Chem. Int. Ed. Engl. 2021, 60, 13251–13256. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Chen, Q.; Ji, C.; Song, Y.; Huang, H.; Ma, J.; Tian, X.; Ju, J. Discovery of McbB, an enzyme catalyzing the β-carboline skeleton construction in the marinacarboline biosynthetic pathway. Angew. Chem. Int. Ed. 2013, 52, 9980–9984. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Chen, Q.; Li, Q.; Huang, H.; Song, Y.; Ma, J.; Ju, J. Chemoenzymatic synthesis of β-carboline derivatives using McbA, a new ATP-dependent amide synthetase. Tetrahedron Lett. 2014, 55, 4901–4904. [Google Scholar] [CrossRef]
- Petchey, M.; Cuetos, A.; Rowlinson, B.; Dannevald, S.; Frese, A.; Sutton, P.W.; Lovelock, S.; Lloyd, R.C.; Fairlamb, I.J.S.; Grogan, G. The broad aryl acid specificity of the amide bond synthetase McbA suggests potential for the biocatalytic synthesis of amides. Angew. Chem. Int. Ed. 2018, 57, 11584–11588. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.J.L.; Weise, N.J.; Frampton, J.D.; Dunstan, M.S.; Hollas, M.A.; Derrington, S.R.; Lloyd, R.C.; Quaglia, D.; Parmeggiani, F.; Leys, D.; et al. Adenylation activity of carboxylic acid reductases enables the synthesis of amides. Angew. Chem. Int. Ed. 2017, 56, 14498–14501. [Google Scholar] [CrossRef]
- Mitsukura, K.; Suzuki, M.; Shinoda, S.; Kuramoto, T.; Yoshida, T.; Nagasawa, T. Purification and characterization of a novel (R)-imine reductase from Streptomyces sp. GF3587. Biosci. Biotechnol. Biochem. 2011, 75, 1778–1782. [Google Scholar] [CrossRef] [Green Version]
- Aleku, G.A.; France, S.P.; Man, H.; Mangas-Sanchez, J.; Montgomery, S.L.; Sharma, M.; Leipold, F.; Hussain, S.; Grogan, G.; Turner, N.J. A reductive aminase from Aspergillus oryzae. Nat. Chem. 2017, 9, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.P.; Brown, M.J.B.; Diaz-Rodriguez, A.; Lloyd, R.C.; Roiban, G. Biocatalysis: A pharma perspective. Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar] [CrossRef] [Green Version]
- Schober, M.; MacDermaid, C.; Ollis, A.A.; Chang, S.; Khan, D.; Hosford, J.; Latham, J.; Ihnken, L.A.F.; Brown, M.J.B.; Fuerst, D.; et al. Chiral synthesis of LSD1 inhibitor GSK2879552 enabled by directed evolution of an imine reductase. Nat. Catal. 2019, 2, 909–915. [Google Scholar] [CrossRef]
- Herr, C.Q.; Hausinger, R.P. Amazing diversity in biochemical roles of Fe(II)/2-oxoglutarate oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Leissing, T.M.; Chowdhury, R.; Hopkinson, R.J.; Schofield, C.J. 2-Oxoglutarate-dependent oxygenases. Annu. Rev. Biochem. 2018, 87, 585–620. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Buller, R. Industrial application of 2-oxoglutarate-dependent oxygenases. Catalysts 2019, 9, 221. [Google Scholar] [CrossRef] [Green Version]
- Zwick, C.R.; Renata, H. Harnessing the biocatalytic potential of iron- and α-ketoglutarate-dependent dioxygenases in natural product total synthesis. Nat. Prod. Rep. 2020, 37, 1065–1079. [Google Scholar] [CrossRef]
- Baud, D.; Saaidi, P.L.; Monfleur, A.; Harari, M.; Cuccaro, J.; Fossey, A.; Besnard, M.; Debard, A.; Mariage, A.; Pellouin, V.; et al. Synthesis of mono- and dihydroxylated amino acids with new α-ketoglutarate-dependent dioxygenases: Biocatalytic oxidation of C-H bonds. ChemCatChem 2014, 6, 3012–3017. [Google Scholar] [CrossRef] [Green Version]
- Hara, R.; Yamagata, K.; Miyake, R.; Kawabata, H.; Uehara, H.; Kino, K. Discovery of lysine hydroxylases in the clavaminic acid synthase-like superfamily for efficient hydroxylysine bioproduction. Appl. Environ. Microbiol. 2017, 83, 585–620. [Google Scholar] [CrossRef] [Green Version]
- Amatuni, A.; Renata, H. Identification of a lysine 4-hydroxylase from the glidobactin biosynthesis and evaluation of its biocatalytic potential. Org. Biomol. Chem. 2019, 17, 1736–1739. [Google Scholar] [CrossRef]
- Lamarre, D.; Croteau, G.; Wardrop, E.; Bourgon, L.; Thibeault, D.; Clouette, C.; Vaillancourt, M.; Cohen, E.; Pargellis, C.; Yoakim, C.; et al. Antiviral properties of palinavir, a potent inhibitor of the human immunodeficiency virus type 1 protease. Antimicrob. Agents Chemother. 1997, 41, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhu, M.; Song, Z.; Li, C.; Wang, Y.; Zhu, Z.; Sun, D.; Lu, F.; Qin, H. Reshaping the binding pocket of lysine hydroxylase for enhanced activity. ACS Catal. 2020, 10, 13946–13956. [Google Scholar] [CrossRef]
- Zhang, X.; King-Smith, E.; Renata, H. Total synthesis of tambromycin by combining chemocatalytic and biocatalytic C−H functionalization. Angew. Chem. Int. Ed. 2018, 57, 5037–5041. [Google Scholar] [CrossRef] [PubMed]
- Amatuni, A.; Shuster, A.; Adibekian, A.; Renata, H. Concise chemoenzymatic total synthesis and identification of cellular targets of cepafungin I. Cell Chem. Biol. 2020, 27, 1318–1326.e18. [Google Scholar] [CrossRef]
- Baud, D.; Peruch, O.; Saaidi, P.-L.; Fossey, A.; Mariage, A.; Petit, J.-L.; Salanoubat, M.; Vergne-Vaxelaire, C.; de Berardinis, V.; Zaparucha, A. Biocatalytic approaches towards the synthesis of chiral amino alcohols from lysine: Cascade reactions combining alpha-keto acid oxygenase hydroxylation with pyridoxal phosphate-dependent decarboxylation. Adv. Synth. Catal. 2017, 359, 1563–1569. [Google Scholar] [CrossRef]
- Rolf, J.; Nerke, P.; Britner, A.; Krick, S.; Lütz, S.; Rosenthal, K. From cell-free protein synthesis to whole-cell biotransformation: Screening and identification of novel α-ketoglutarate-dependent dioxygenases for preparative-scale synthesis of hydroxy-L-lysine. Catalysts 2021, 11, 1038. [Google Scholar] [CrossRef]
- Rolf, J.; Rosenthal, K.; Lütz, S. Application of cell-free protein synthesis for faster biocatalyst development. Catalysts 2019, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Hetzler, B.E.; Trauner, D.; Lawrence, A.L. Natural product anticipation through synthesis. Nat. Rev. Chem. 2022, 6, 170–181. [Google Scholar] [CrossRef]
- Novak, A.J.E.; Grigglestone, C.E.; Trauner, D. A biomimetic synthesis elucidates the origin of preuisolactone A. J. Am. Chem. Soc. 2019, 141, 15515–15518. [Google Scholar] [CrossRef]
- Powers, Z.; Scharf, A.; Cheng, A.; Yang, F.; Himmelbauer, M.; Mitsuhashi, T.; Barra, L.; Taniguchi, Y.; Kikuchi, T.; Fujita, M.; et al. Biomimetic synthesis of meroterpenoids by dearomatization-driven polycyclization. Angew. Chem. Int. Ed. Engl. 2019, 58, 16141–16146. [Google Scholar] [CrossRef]
- Gu, J.H.; Wang, W.J.; Chen, J.Z.; Liu, J.S.; Li, N.P.; Cheng, M.J.; Hu, L.J.; Li, C.C.; Ye, W.C.; Wang, L. Leptosperols A and B, two cinnamoylphloroglucinol–sesquiterpenoid hybrids from Leptospermum scoparium: Structural elucidation and biomimetic synthesis. Org. Lett. 2020, 22, 1796–1800. [Google Scholar] [CrossRef]
- Kries, H.; O’Connor, S.E. Biocatalysts from alkaloid producing plants. Curr. Opin. Chem. Biol. 2016, 31, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichman, B.R. The scaffold-forming steps of plant alkaloid biosynthesis. Nat. Prod. Rep. 2021, 38, 103–129. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Seebeck, F.P. S-adenosylhomocysteine as a methyl transfer catalyst in biocatalytic methylation reactions. Nat. Catal. 2019, 2, 696–701. [Google Scholar] [CrossRef]
- Brufani, M.; Castellano, C.; Marta, M.; Oliverio, A.; Pagella, P.G.; Pavone, F.; Pomponi, M.; Rugarli, P.L. A long-lasting cholinesterase inhibitor affecting neural and behavioral processes. Pharmacol. Biochem. Behav. 1987, 26, 625–629. [Google Scholar] [CrossRef]
- Iijima, S.; Greig, N.H.; Garofalo, P.; Spangler, E.L.; Heller, B.; Brossi, A.; Ingram, D.K. Phenserine: A physostigmine derivative that is a long-acting inhibitor of cholinesterase and demonstrates a wide dose range for attenuating a scopolamine-induced learning impairment of rats in a 14-unit T-maze. Psychopharmacology 1993, 112, 415–420. [Google Scholar] [CrossRef]
- Winand, L.; Schneider, P.; Kruth, S.; Greven, N.J.; Hiller, W.; Kaiser, M.; Pietruszka, J.; Nett, M. Mutasynthesis of Physostigmines in Myxococcus xanthus. Org. Lett. 2021, 23, 6563–6567. [Google Scholar] [CrossRef]
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevels, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 2008, 5, 191–211. [Google Scholar] [CrossRef]
- Marahiel, M.A. A structural model for multimodular NRPS assembly lines. Nat. Prod. Rep. 2016, 33, 136–140. [Google Scholar] [CrossRef]
- Süssmuth, R.D.; Mainz, A. Nonribosomal peptide synthesis—Principles and prospects. Angew. Chem. Int. Ed. Engl. 2017, 56, 3770–3821. [Google Scholar] [CrossRef]
- Kaljunen, H.; Schiefelbein, S.H.H.; Stummer, D.; Kozak, S.; Meijers, R.; Christiansen, G.; Rentmeister, A. Structural elucidation of the bispecificity of a domains as a basis for activating non-natural amino acids. Angew. Chem. Int. Ed. Engl. 2015, 54, 8833–8836. [Google Scholar] [CrossRef]
- Winand, L.; Sester, A.; Nett, M. Bioengineering of anti-inflammatory natural products. ChemMedChem 2021, 16, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Kegler, C.; Bode, H.B. Artificial splitting of a non-ribosomal peptide synthetase by inserting natural docking domains. Angew. Chem. Int. Ed. Engl. 2020, 59, 13463–13467. [Google Scholar] [CrossRef] [PubMed]
- Bozhueyuek, K.A.J.; Fleischhacker, F.; Linck, A.; Wesche, F.; Tietze, A.; Niesert, C.-P.; Bode, H.B. De novo design and engineering of non-ribosomal peptide synthetases. Nat. Chem. 2018, 10, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Burgard, C.; Popoff, A.; Zaburannyi, N.; Zipf, G.; Maier, J.; Bernauer, H.S.; Wenzel, S.C.; Müller, R. Synthetic biology approaches and combinatorial biosynthesis towards heterologous lipopeptide production. Chem. Sci. 2018, 9, 7510–7519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozhueyuek, K.A.; Micklefield, J.; Wilkinson, B. Engineering enzymatic assembly lines to produce new antibiotics. Curr. Opin. Microbiol. 2019, 51, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Bozhueyuek, K.A.J.; Watzel, J.; Abbood, N.; Bode, H.B. Synthetic zippers as an enabling tool for engineering of non-ribosomal peptide synthetases**. Angew. Chem. 2021, 133, 17672–17679. [Google Scholar] [CrossRef]
- Huang, H.-M.; Stephan, P.; Kries, H. Engineering DNA-templated nonribosomal peptide synthesis. Cell Chem. Biol. 2021, 28, 221–227.e7. [Google Scholar] [CrossRef]
- Thong, W.L.; Zhang, Y.; Zhuo, Y.; Robins, K.J.; Fyans, J.K.; Herbert, A.J.; Law, B.J.C.; Micklefield, J. Gene editing enables rapid engineering of complex antibiotic assembly lines. Nat. Commun. 2021, 12, 6872. [Google Scholar] [CrossRef]
- Dekimpe, S.; Masschelein, J. Beyond peptide bond formation: The versatile role of condensation domains in natural product biosynthesis. Nat. Prod. Rep. 2021, 38, 1910–1937. [Google Scholar] [CrossRef]
- Calcott, M.J.; Owen, J.G.; Ackerley, D.F. Efficient rational modification of non-ribosomal peptides by adenylation domain substitution. Nat. Commun. 2020, 11, 4554. [Google Scholar] [CrossRef]
- Baunach, M.; Chowdhury, S.; Stallforth, P.; Dittmann, E. The landscape of recombination events that create nonribosomal peptide diversity. Mol. Biol. Evol. 2021, 38, 2116–2130. [Google Scholar] [CrossRef] [PubMed]
- Stanišić, A.; Hüsken, A.; Stephan, P.; Niquille, D.L.; Reinstein, J.; Kries, H. Engineered nonribosomal peptide synthetase shows opposite amino acid loading and condensation specificity. ACS Catal. 2021, 11, 8692–8700. [Google Scholar] [CrossRef]
- Hahn, M.; Stachelhaus, T. Selective interaction between nonribosomal peptide synthetases is facilitated by short communication-mediating domains. Proc. Natl. Acad. Sci. USA 2004, 101, 15585–15590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocchini, C.; Linne, U.; Stachelhaus, T. In vivo biocombinatorial synthesis of lipopeptides by COM domain-mediated reprogramming of the surfactin biosynthetic complex. Chem. Biol. 2006, 13, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, C.; Cai, X.; Kegler, C.; Zhao, L.; Weickhmann, A.K.; Wurm, J.P.; Bode, H.B.; Wöhnert, J. Structure-based redesign of docking domain interactions modulates the product spectrum of a rhabdopeptide-synthesizing NRPS. Nat. Commun. 2018, 9, 4366. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Gao, L.; Han, J.; Ma, Z.; Lu, Z.; Dai, C.; Zhang, C.; Bie, X. Biocombinatorial synthesis of novel lipopeptides by COM domain-mediated reprogramming of the plipastatin NRPS complex. Front. Microbiol. 2016, 7, 1801. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Zhao, L.; Bode, H.B. Reprogramming promiscuous nonribosomal peptide synthetases for production of specific peptides. Org. Lett. 2019, 21, 2116–2120. [Google Scholar] [CrossRef]
- Calcott, M.J.; Ackerley, D.F. Genetic manipulation of non-ribosomal peptide synthetases to generate novel bioactive peptide products. Biotechnol. Lett. 2014, 36, 2407–2416. [Google Scholar] [CrossRef]
- Niquille, D.L.; Hansen, D.A.; Hilvert, D. Reprogramming nonribosomal peptide synthesis by surgical mutation. Synlett 2019, 30, 2123–2130. [Google Scholar] [CrossRef]
- Crüsemann, M.; Kohlhaas, C.; Piel, J. Evolution-guided engineering of nonribosomal peptide synthetase adenylation domains. Chem. Sci. 2013, 4, 1041–1045. [Google Scholar] [CrossRef]
- Kries, H.; Niquille, D.L.; Hilvert, D. A subdomain swap strategy for reengineering nonribosomal peptides. Chem. Biol. 2015, 22, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Moir, M.; Danon, J.J.; Reekie, T.A.; Kassiou, M. An overview of late-stage functionalization in today’s drug discovery. Expert Opin. Drug Discov. 2019, 14, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Luo, T.; Lei, X. Late-stage diversification of natural products. ACS Cent. Sci. 2020, 6, 622–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemard, L.; Kaplaneris, N.; Ackermann, L.; Johansson, M.J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem. 2021, 5, 522–545. [Google Scholar] [CrossRef]
- Börgel, J.; Ritter, T. Late-stage functionalization. Chem 2020, 6, 1877–1887. [Google Scholar] [CrossRef]
- Romero, E.; Jones, B.S.; Hogg, B.N.; Rué Casamajo, A.; Hayes, M.A.; Flitsch, S.L.; Turner, N.J.; Schnepel, C. Enzymatic late-stage modifications: Better late than never. Angew. Chem. Int. Ed. 2021, 60, 16824–16855. [Google Scholar] [CrossRef]
- Martínez, A.T.; Ruiz-Dueñas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarty, S.; Wang, Y.; Perkins, J.C.; Narayan, A.R.H. Scalable biocatalytic C-H oxyfunctionalization reactions. Chem. Soc. Rev. 2020, 49, 8137–8155. [Google Scholar] [CrossRef]
- Bernhardt, R. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 2006, 124, 128–145. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, Y.; Guengerich, X.F.P.; Ma, L.; Li, S.; Zhang, W. Engineering cytochrome P450 enzyme systems for biomedical and biotechnological applications. J. Biol. Chem. 2020, 295, 833–849. [Google Scholar] [CrossRef]
- Okubo, S.; Ena, E.; Okuda, A.; Kozone, I.; Hashimoto, J.; Nishitsuji, Y.; Fujie, M.; Satoh, N.; Ikeda, H.; Shin-ya, K. Identification of functional cytochrome P450 and ferredoxin from Streptomyces sp. EAS-AB2608 by transcriptional analysis and their heterologous expression. Appl. Microbiol. Biotechnol. 2021, 105, 4177–4187. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, L.M.; Schäper, J.; Rosenthal, K.; Lütz, S. Accessing the biocatalytic potential for C-H-activation by targeted genome mining and screening. ChemCatChem 2019, 11, 5766–5777. [Google Scholar] [CrossRef] [Green Version]
- Hilberath, T.; Windeln, L.M.; Decembrino, D.; Le-Huu, P.; Bilsing, F.L.; Urlacher, V.B. Two-step screening for identification of drug-metabolizing bacterial cytochromes P450 with diversified selectivity. ChemCatChem 2020, 12, 1710–1719. [Google Scholar] [CrossRef]
- Palmer-Brown, W.; Miranda-CasoLuengo, R.; Wolfe, K.H.; Byrne, K.P.; Murphy, C.D. The CYPome of the model xenobiotic-biotransforming fungus Cunninghamella elegans. Sci. Rep. 2019, 9, 9240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Chen, K.; Cui, H.; Wan, N.; Cui, B.; Han, W.; Chen, Y. Characterization of a self-sufficient cytochrome P450 monooxygenase from Deinococcus apachensis for enantioselective benzylic hydroxylation. ChemBioChem 2020, 21, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, L.M.; Hageneier, F.; Rosenthal, K.; Busche, T.; Brandt, D.; Kalinowski, J.; Lütz, S. Recombinant expression and characterization of novel P450s from Actinosynnema mirum. Bioorg. Med. Chem. 2021, 42, 116241. [Google Scholar] [CrossRef]
- Fredenhagen, A.; Schroer, K.; Schröder, H.; Hoepfner, D.; Ligibel, M.; Porchet Zemp, L.; Radoch, C.; Freund, E.; Meishammer, A. Cladosporin derivatives obtained by biotransformation provide guidance for the focused derivatization of this antimalarial lead compound. ChemBioChem 2019, 20, 650–654. [Google Scholar] [CrossRef]
- Zhou, J.; Hu, T.; Liu, Y.; Tu, L.; Song, Y.; Lu, Y.; Zhang, Y.; Tong, Y.; Zhao, Y.; Su, P.; et al. Cytochrome P450 catalyses the 29-carboxyl group formation of celastrol. Phytochemistry 2021, 190, 112868. [Google Scholar] [CrossRef]
- Shi, Y.; Ye, F.; Song, Y.; Zhang, X.; Lu, C.; Shen, Y. Rifamycin W analogues from Amycolatopsis mediterranei S699 ∆rif-orf5 strain. Biomolecules 2021, 11, 920. [Google Scholar] [CrossRef]
- Li, X.; Fu, J.; Li, Y.; Liu, J.; Gao, R.; Shi, Y.; Li, Y.; Sun, H.; Wang, L.; Li, Y.; et al. Cytochrome P450 monooxygenase for catalyzing C-42 hydroxylation of the glycine-derived fragment in hangtaimycin biosynthesis. Org. Lett. 2022, 24, 1388–1393. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Z.; Sun, C.; Shao, M.; Ma, J.; Wei, X.; Zhang, T.; Li, W.; Ju, J. Discovery and biosynthesis of atrovimycin, an antitubercular and antifungal cyclodepsipeptide featuring vicinal-dihydroxylated cinnamic acyl chain. Org. Lett. 2019, 21, 2634–2638. [Google Scholar] [CrossRef] [PubMed]
- Go, E.B.; Kim, L.J.; Nelson, H.M.; Ohashi, M.; Tang, Y. Biosynthesis of the Fusarium mycotoxin (-)-sambutoxin. Org. Lett. 2021, 23, 7819–7823. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jiang, Z.; Hu, X.; Hu, X.; Gu, R.; Jiang, B.; Zuo, L.; Li, X.; Sun, H.; Zhang, C.; et al. The cytochrome P450 catalyzing C−S bond formation in S-heterocyclization of chuangxinmycin biosynthesis. Angew. Chem. Int. Ed. 2021, 60, 15399–15404. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.; Lutz, A.; Chalmers, D.K.; Jarrold, A.; Bowser, T.; Stevens, G.W.; Gras, S.L. Production of metabolites of the anti-cancer drug noscapine using a P450BM3 mutant library. Biotechnol. Rep. 2019, 24, e00372. [Google Scholar] [CrossRef]
- Chen, W.; Fisher, M.J.; Leung, A.; Cao, Y.; Wong, L.L. Oxidative diversification of steroids by nature-inspired scanning glycine mutagenesis of P450BM3 (CYP102A1). ACS Catal. 2020, 10, 8334–8343. [Google Scholar] [CrossRef]
- Bokel, A.; Hutter, M.C.; Urlacher, V.B. Molecular evolution of a cytochrome P450 for the synthesis of potential antidepressant (2R,6R)-hydroxynorketamine. Chem. Commun. 2021, 57, 520–523. [Google Scholar] [CrossRef]
- Li, F.; Ma, L.; Zhang, X.; Chen, J.; Qi, F.; Huang, Y.; Qu, Z.; Yao, L.; Zhang, W.; Kim, E.S.; et al. Structure-guided manipulation of the regioselectivity of the cyclosporine A hydroxylase CYP-sb21 from Sebekia benihana. Synth. Syst. Biotechnol. 2020, 5, 236–243. [Google Scholar] [CrossRef]
- Espinoza, R.V.; Haatveit, K.C.; Grossman, S.W.; Tan, J.Y.; McGlade, C.A.; Khatri, Y.; Newmister, S.A.; Schmidt, J.J.; Garcia-Borràs, M.; Montgomery, J.; et al. Engineering P450 TamI as an iterative biocatalyst for selective late-stage C-H functionalization and epoxidation of tirandamycin antibiotics. ACS Catal. 2021, 11, 8304–8316. [Google Scholar] [CrossRef]
- Aranda, C.; Ullrich, R.; Kiebist, J.; Scheibner, K.; Del Río, J.C.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Selective synthesis of the resveratrol analogue 4,4′-dihydroxy-trans-stilbene and stilbenoids modification by fungal peroxygenases. Catal. Sci. Technol. 2018, 8, 2394–2401. [Google Scholar] [CrossRef] [Green Version]
- Kiebist, J.; Schmidtke, K.U.; Schramm, M.; König, R.; Quint, S.; Kohlmann, J.; Zuhse, R.; Ullrich, R.; Hofrichter, M.; Scheibner, K. Biocatalytic syntheses of antiplatelet metabolites of the thienopyridines clopidogrel and prasugrel using fungal peroxygenases. J. Fungi 2021, 7, 752. [Google Scholar] [CrossRef]
- Gómez de Santos, P.; Cañellas, M.; Tieves, F.; Younes, S.H.H.; Molina-Espeja, P.; Hofrichter, M.; Hollmann, F.; Guallar, V.; Alcalde, M. Selective synthesis of the human drug metabolite 5′-hydroxypropranolol by an evolved self-sufficient peroxygenase. ACS Catal. 2018, 8, 4789–4799. [Google Scholar] [CrossRef] [Green Version]
- Gómez de Santos, P.; Cervantes, F.V.; Tieves, F.; Plou, F.J.; Hollmann, F.; Alcalde, M. Benchmarking of laboratory evolved unspecific peroxygenases for the synthesis of human drug metabolites. Tetrahedron 2019, 75, 1827–1831. [Google Scholar] [CrossRef] [Green Version]
- Kolling, D.; Stierhof, M.; Lasch, C.; Myronovskyi, M.; Luzhetskyy, A. A promiscuous halogenase for the derivatization of flavonoids. Molecules 2021, 26, 6220. [Google Scholar] [CrossRef]
- Hayashi, T.; Ligibel, M.; Sager, E.; Voss, M.; Hunziker, J.; Schroer, K.; Snajdrova, R.; Buller, R. Evolved aliphatic halogenases enable regiocomplementary C−H functionalization of a pharmaceutically relevant compound. Angew. Chem. Int. Ed. 2019, 58, 18535–18539. [Google Scholar] [CrossRef]
- Büchler, J.; Malca, S.H.; Patsch, D.; Voss, M.; Turner, N.J.; Bornscheuer, U.T.; Allemann, O.; Le Chapelain, C.; Lumbroso, A.; Loiseleur, O.; et al. Algorithm-aided engineering of aliphatic halogenase WelO5* for the asymmetric late-stage functionalization of soraphens. Nat. Commun. 2022, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Sawayama, A.M.; Chen, M.M.Y.; Kulanthaivel, P.; Kuo, M.S.; Hemmerle, H.; Arnold, F.H. A panel of cytochrome P450 BM3 variants to produce drug metabolites and diversify lead compounds. Chem. Eur. J. 2009, 15, 11723–11729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmichael, A.B.; Wong, L.L. Protein engineering of Bacillus megaterium CYP102: The oxidation of polycyclic aromatic hydrocarbons. Eur. J. Biochem. 2001, 268, 3117–3125. [Google Scholar] [CrossRef]
- Ost, T.W.B.; Miles, C.S.; Murdoch, J.; Cheung, Y.F.; Reid, G.A.; Chapman, S.K.; Munro, A.W. Rational re-design of the substrate binding site of flavocytochrome P450 BM3. FEBS Lett. 2000, 486, 173–177. [Google Scholar] [CrossRef]
- Nguyen, N.A.; Jang, J.; Le, T.K.; Nguyen, T.H.H.; Woo, S.M.; Yoo, S.K.; Lee, Y.J.; Park, K.D.; Yeom, S.J.; Kim, G.J.; et al. Biocatalytic production of a potent inhibitor of adipocyte differentiation from phloretin using engineered CYP102A1. J. Agric. Food Chem. 2020, 68, 6683–6691. [Google Scholar] [CrossRef]
- Coelho, P.S.; Wang, Z.J.; Ener, M.E.; Baril, S.A.; Kannan, A.; Arnold, F.H.; Brustad, E.M. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol. 2013, 9, 485–487. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.J.; Gao, S.; Arnold, F.H. Enzymatic primary amination of benzylic and allylic C(sp3)-H bonds. J. Am. Chem. Soc. 2020, 142, 10279–10283. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F. Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges? Curr. Opin. Chem. Biol. 2017, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hofrichter, M.; Kellner, H.; Pecyna, M.J.; Ullrich, R. Fungal unspecific peroxygenases: Heme-thiolate proteins that combine peroxidase and cytochrome P450 properties. In Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450; Hrycay, E.G., Bandiera, S.M., Eds.; Springer: New York, NY, USA, 2015; pp. 341–368. [Google Scholar]
- Kinner, A.; Rosenthal, K.; Lütz, S. Identification and expression of new unspecific peroxygenases–Recent advances, challenges and opportunities. Front. Bioeng. Biotechnol. 2021, 9, 705630. [Google Scholar] [CrossRef] [PubMed]
- Hofrichter, M.; Kellner, H.; Herzog, R.; Karich, A.; Liers, C.; Scheibner, K.; Wambui Kimani, V.; Ullrich, R. Fungal peroxygenases: A phylogenetically old superfamily of heme enzymes with promiscuity for oxygen transfer reactions. In Grand Challenges in Fungal Biotechnology; Nevalainen, H., Ed.; Springer: Cham, Switzerland, 2020; pp. 369–403. ISBN 978-3-030-29541-7. [Google Scholar]
- González-Benjumea, A.; Linde, D.; Carro, J.; Ullrich, R.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Regioselective and stereoselective epoxidation of n-3 and n-6 fatty acids by fungal peroxygenases. Antioxidants 2021, 10, 1888. [Google Scholar] [CrossRef]
- Aranda, C.; Municoy, M.; Guallar, V.; Kiebist, J.; Scheibner, K.; Ullrich, R.; Del Río, J.C.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Selective synthesis of 4-hydroxyisophorone and 4-ketoisophorone by fungal peroxygenases. Catal. Sci. Technol. 2019, 9, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Knorrscheidt, A.; Soler, J.; Hünecke, N.; Püllmann, P.; Garcia-Borràs, M.; Weissenborn, M.J. Simultaneous screening of multiple substrates with an unspecific peroxygenase enabled modified alkane and alkene oxyfunctionalisations. Catal. Sci. Technol. 2021, 11, 6058–6064. [Google Scholar] [CrossRef]
- Gómez de Santos, P.; Lazaro, S.; Vinã-Gonzalez, J.; Hoang, M.D.; Sánchez-Moreno, I.; Glieder, A.; Hollmann, F.; Alcalde, M. Evolved peroxygenase-aryl alcohol oxidase fusions for self-sufficient oxyfunctionalization reactions. ACS Catal. 2020, 10, 13524–13534. [Google Scholar] [CrossRef]
- Beltrán-Nogal, A.; Sánchez-Moreno, I.; Méndez-Sánchez, D.; Gómez de Santos, P.; Hollmann, F.; Alcalde, M. Surfing the wave of oxyfunctionalization chemistry by engineering fungal unspecific peroxygenases. Curr. Opin. Struct. Biol. 2022, 73, 102342. [Google Scholar] [CrossRef]
- Karich, A.; Scheibner, K.; Ullrich, R.; Hofrichter, M. Exploring the catalase activity of unspecific peroxygenases and the mechanism of peroxide-dependent heme destruction. J. Mol. Catal. B Enzym. 2016, 134, 238–246. [Google Scholar] [CrossRef]
- Myronovskyi, M.; Rosenkränzer, B.; Nadmid, S.; Pujic, P.; Normand, P.; Luzhetskyy, A. Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab. Eng. 2018, 49, 316–324. [Google Scholar] [CrossRef]
- Schmitz, L.M.; Kinner, A.; Althoff, K.; Lütz, S. Investigation of vitamin D2 and vitamin D3 hydroxylation by Kutzneria albida. ChemBioChem 2021, 22, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Burek, B.O.; Fernández-Fueyo, E.; Alcalde, M.; Bloh, J.Z.; Hollmann, F. Selective activation of C−H bonds in a cascade process combining photochemistry and biocatalysis. Angew. Chem. Int. Ed. 2017, 56, 15451–15455. [Google Scholar] [CrossRef] [PubMed]
- Dangi, B.; Park, H.; Oh, T.J. Effects of alternative redox partners and oxidizing agents on CYP154C8 catalytic activity and product distribution. ChemBioChem 2018, 19, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Aranda, C.; Carro, J.; González-Benjumea, A.; Babot, E.D.; Olmedo, A.; Linde, D.; Martínez, A.T.; Gutiérrez, A. Advances in enzymatic oxyfunctionalization of aliphatic compounds. Biotechnol. Adv. 2021, 51, 107703. [Google Scholar] [CrossRef]
- Kara, S.; Rudroff, F. Introduction. In Enzyme Cascade Design and Modelling; Springer: Cham, Switzerland, 2021; pp. 1–6. [Google Scholar]
- Siedentop, R.; Claaßen, C.; Rother, D.; Lütz, S.; Rosenthal, K. Getting the most out of enzyme cascades: Strategies to optimize in vitro multi-enzymatic reactions. Catalysts 2021, 11, 1183. [Google Scholar] [CrossRef]
- Huffman, M.A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K.R.; Canada, K.A.; Devine, P.N.; Duan, D.; Forstater, J.H.; Grosser, S.T.; et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2020, 368, 1255–1259. [Google Scholar] [CrossRef]
- Marx, L.; Ríos-Lombardía, N.; Süss, P.; Höhne, M.; Morís, F.; González-Sabín, J.; Berglund, P. Chemoenzymatic synthesis of sertraline. Eur. J. Org. Chem. 2020, 2020, 510–513. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.M.; Besse, B.; Martinez-Marti, A.; Trigo, J.M.; Moreno, V.; Garrido, P.; Ferron-Brady, G.; Wu, Y.; Park, J.; Collingwood, T.; et al. Phase I, open-label, dose-escalation study of the safety, pharmacokinetics, pharmacodynamics, and efficacy of GSK2879552 in relapsed/refractory SCLC. J. Thorac. Oncol. 2019, 14, 1828–1838. [Google Scholar] [CrossRef] [Green Version]
- Roboz, G.J.; Yee, K.; Verma, A.; Borthakur, G.; de la Fuente Burguera, A.; Sanz, G.; Mohammad, H.P.; Kruger, R.G.; Karpinich, N.O.; Ferron-Brady, G.; et al. Phase I trials of the lysine-specific demethylase 1 inhibitor, GSK2879552, as mono- and combination-therapy in relapsed/refractory acute myeloid leukemia or high-risk myelodysplastic syndromes. Leuk. Lymphoma 2022, 63, 463–467. [Google Scholar] [CrossRef]
- Kinsella, S.M.; Carvalho, B.; Dyer, R.A.; Fernando, R.; McDonnell, N.; Mercier, F.J.; Palanisamy, A.; Sia, A.T.H.; Van de Velde, M.; Vercueil, A. International consensus statement on the management of hypotension with vasopressors during caesarean section under spinal anaesthesia. Anaesthesia 2018, 73, 71–92. [Google Scholar] [CrossRef] [Green Version]
- Mack, K.; Doeker, M.; Grabowski, L.; Jupke, A.; Rother, D. Extractive in situ product removal for the application of naturally produced L-alanine as an amine donor in enzymatic metaraminol production. Green Chem. 2021, 23, 4892–4901. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.J.; Birmingham, W.R.; Zhuo, Y.; Thorpe, T.W.; Zucoloto da Costa, B.; Crawshaw, R.; Rowles, I.; Finnigan, J.D.; Young, C.; Holgate, G.M.; et al. An engineered cytidine deaminase for biocatalytic production of a key intermediate of the Covid-19 antiviral molnupiravir. J. Am. Chem. Soc. 2022, 144, 3761–3765. [Google Scholar] [CrossRef]
- McIntosh, J.A.; Benkovics, T.; Silverman, S.M.; Huffman, M.A.; Kong, J.; Maligres, P.E.; Itoh, T.; Yang, H.; Verma, D.; Pan, W.; et al. Engineered ribosyl-1-kinase enables concise synthesis of molnupiravir, an antiviral for COVID-19. ACS Cent. Sci. 2021, 7, 1980–1985. [Google Scholar] [CrossRef] [PubMed]
- Painter, G.R.; Bluemling, G.R.; Natchus, M.G.; Guthrie, D. N4-Hydroxycytidine and Derivatives and Anti-Viral Uses Related Thereto. International Patent WO 2016/106050 A1, 30 June 2016. [Google Scholar]
- Valliere, M.A.; Korman, T.P.; Woodall, N.B.; Khitrov, G.A.; Taylor, R.E.; Baker, D.; Bowie, J.U. A cell-free platform for the prenylation of natural products and application to cannabinoid production. Nat. Commun. 2019, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Valliere, M.A.; Korman, T.P.; Arbing, M.A.; Bowie, J.U. A bio-inspired cell-free system for cannabinoid production from inexpensive inputs. Nat. Chem. Biol. 2020, 16, 1427–1433. [Google Scholar] [CrossRef]
- Becker, M.; Nikel, P.; Andexer, J.N.; Lütz, S.; Rosenthal, K. A multi-enzyme cascade reaction for the production of 2′3′-cGAMP. Biomolecules 2021, 11, 590. [Google Scholar] [CrossRef]
- McIntosh, J.A.; Liu, Z.; Andresen, B.M.; Marzijarani, N.S.; Moore, J.C.; Marshall, N.M.; Borra-Garske, M.; Obligacion, J.V.; Fier, P.S.; Peng, F.; et al. A kinase-cGAS cascade to synthesize a therapeutic STING activator. Nature 2022, 603, 439–444. [Google Scholar] [CrossRef]
- Merck & Co. Inc. Merck Press Release. Merck Announces Clinical Holds on Studies Evaluating Islatravir for the Treatment and Prevention of HIV-1 Infection; Merck & Co., Inc.: Whitehouse Station, NJ, USA, 2021. [Google Scholar]
- Schrot, R.J.; Hubbard, J.R. Cannabinoids: Medical implications. Ann. Med. 2016, 48, 128–141. [Google Scholar] [CrossRef]
- Luo, X.; Reiter, M.A.; D’Espaux, L.; Wong, J.; Denby, C.M.; Lechner, A.; Zhang, Y.; Grzybowski, A.T.; Harth, S.; Lin, W.; et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 2019, 567, 123–126. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogoi, H.; Mansouri, S.; Jin, L. The age of cyclic dinucleotide vaccine adjuvants. Vaccines 2020, 8, 453. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, T.; Becker, M.; Rolf, J.; Rosenthal, K.; Lütz, S. Biotechnological production of cyclic dinucleotides—Challenges and opportunities. Biotechnol. Bioeng. 2022, 119, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, K.; Becker, M.; Rolf, J.; Siedentop, R.; Hillen, M.; Nett, M.; Lütz, S. Catalytic promiscuity of cGAS: A facile enzymatic synthesis of 2′-3′-linked cyclic dinucleotides. ChemBioChem 2020, 21, 3225–3228. [Google Scholar] [CrossRef] [PubMed]
- Novotná, B.; Vaneková, L.; Zavřel, M.; Buděšínský, M.; Dejmek, M.; Smola, M.; Gutten, O.; Tehrani, Z.A.; Pimková Polidarová, M.; Brázdová, A.; et al. Enzymatic preparation of 2′-5′,3′-5′-cyclic dinucleotides, their binding properties to stimulator of interferon genes adaptor protein, and structure/activity correlations. J. Med. Chem. 2019, 62, 10676–10690. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Losada-Garcia, N.; Cabrera, Z.; Urrutia, P.; Garcia-Sanz, C.; Andreu, A.; Palomo, J.M. Recent advances in enzymatic and chemoenzymatic cascade processes. Catalysts 2020, 10, 1258. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Palomo, J.M.; Mateo, C. Tandem reactions combining biocatalysts and chemical catalysts for asymmetric synthesis. Catalysts 2016, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Glassman, A.H. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002, 288, 701. [Google Scholar] [CrossRef]
- Doogan, D.P.; Caillard, V. Sertraline in the prevention of depression. Br. J. Psychiatry 1992, 160, 217–222. [Google Scholar] [CrossRef]
- Basanta, B.; Bick, M.J.; Bera, A.K.; Norn, C.; Chow, C.M.; Carter, L.P.; Goreshnik, I.; Dimaio, F.; Baker, D. An enumerative algorithm for de novo design of proteins with diverse pocket structures. Proc. Natl. Acad. Sci. USA 2020, 117, 22135–22145. [Google Scholar] [CrossRef]
- Risso, V.A.; Romero-Rivera, A.; Gutierrez-Rus, L.I.; Ortega-Muñoz, M.; Santoyo-Gonzalez, F.; Gavira, J.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C.L. Enhancing a de novo enzyme activity by computationally-focused ultra-low-throughput screening. Chem. Sci. 2020, 11, 6134–6148. [Google Scholar] [CrossRef] [PubMed]
- Jendrusch, M.; Korbel, J.O.; Sadiq, S.K. AlphaDesign: A de novo protein design framework based on AlphaFold. bioRxiv 2021, 2021.10.11.463937. [Google Scholar] [CrossRef]
- Burton, A.J.; Thomson, A.R.; Dawson, W.M.; Brady, R.L.; Woolfson, D.N. Installing hydrolytic activity into a completely de novo protein framework. Nat. Chem. 2016, 8, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, E. Building enzymes from scratch. Nat. Chem. 2022, 14, 246–248. [Google Scholar] [CrossRef]
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine learning in enzyme engineering. ACS Catal. 2020, 10, 1210–1223. [Google Scholar] [CrossRef]
- Marshall, J.R.; Mangas-Sanchez, J.; Turner, N.J. Expanding the synthetic scope of biocatalysis by enzyme discovery and protein engineering. Tetrahedron 2021, 82, 131926. [Google Scholar] [CrossRef]
- Wittmann, B.J.; Johnston, K.E.; Wu, Z.; Arnold, F.H. Advances in machine learning for directed evolution. Curr. Opin. Struct. Biol. 2021, 69, 11–18. [Google Scholar] [CrossRef]










| Enzyme | Source Organism | Modification | Biocatalyst | Substrate | Product | Process Performance | Reference |
|---|---|---|---|---|---|---|---|
| 4-Chlorobenzoate ATP-dependent CoA ligase (CBL) and serotonin hydroxycinnamoyltransferase (66CaAT) | Alcaligenes sp.; Capsicum annuum | - | Whole-cell | 6-Chloronicotinic acid; neopentylamine | 6-Chloro-N-neopentylnicotinamide (losmapimod key intermediate) | 83% conversion, 74% isolated yield | [16] |
| ATP-dependent amide bond synthetase McbA | Marinactinospora thermotolerans | - | Isolated enzyme | 4-Chlorobenzoic acid; 4-(2-aminoethyl) morpholine | Moclobemide | 70% conversion, 64% isolated yield | [17] |
| Carboxylic acidreductase CARmm-A | Mycobacterium marinum | Truncated enzyme variant | Isolated enzyme | 3,4,5-trimethoxycinnamic acid; piperazine acetic acid pyrrolidine | Cinepazide | 18% isolated yield | [18] |
| SpRedAm-R3-V6 | Streptomyces purpureus | Engineered variant (four amino acid exchanges) | Isolated enzyme | Isopropyl 3-oxocyclobutane-1-carboxylate; monomethylamine | Isopropyl 3-(methylamino) cyclobutane-1-carboxylate (abrocritinib key intermediate) | 60 g L−1 d−1, purity >99.5%, selectivity >99:1 cis:trans | [19] |
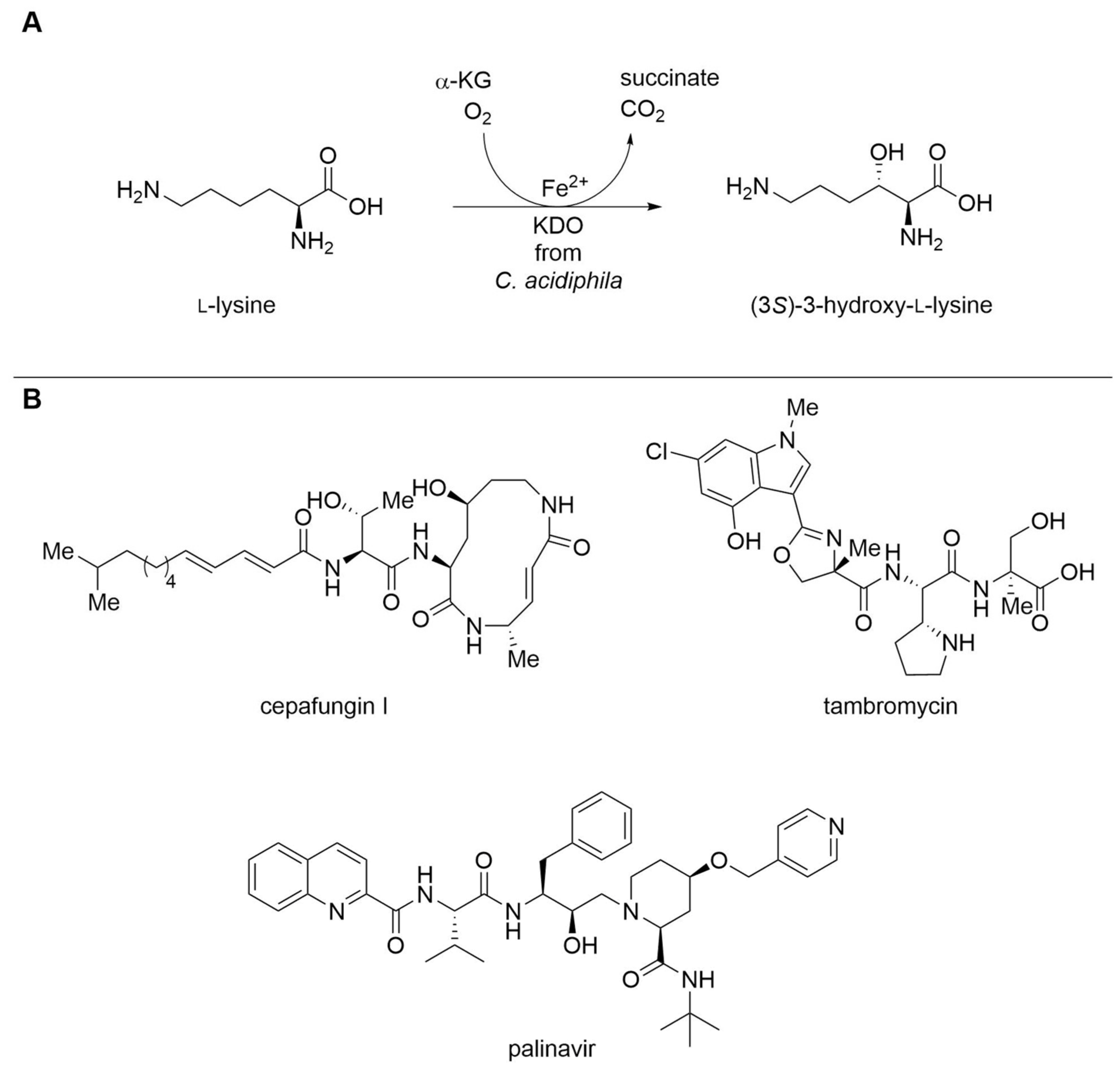
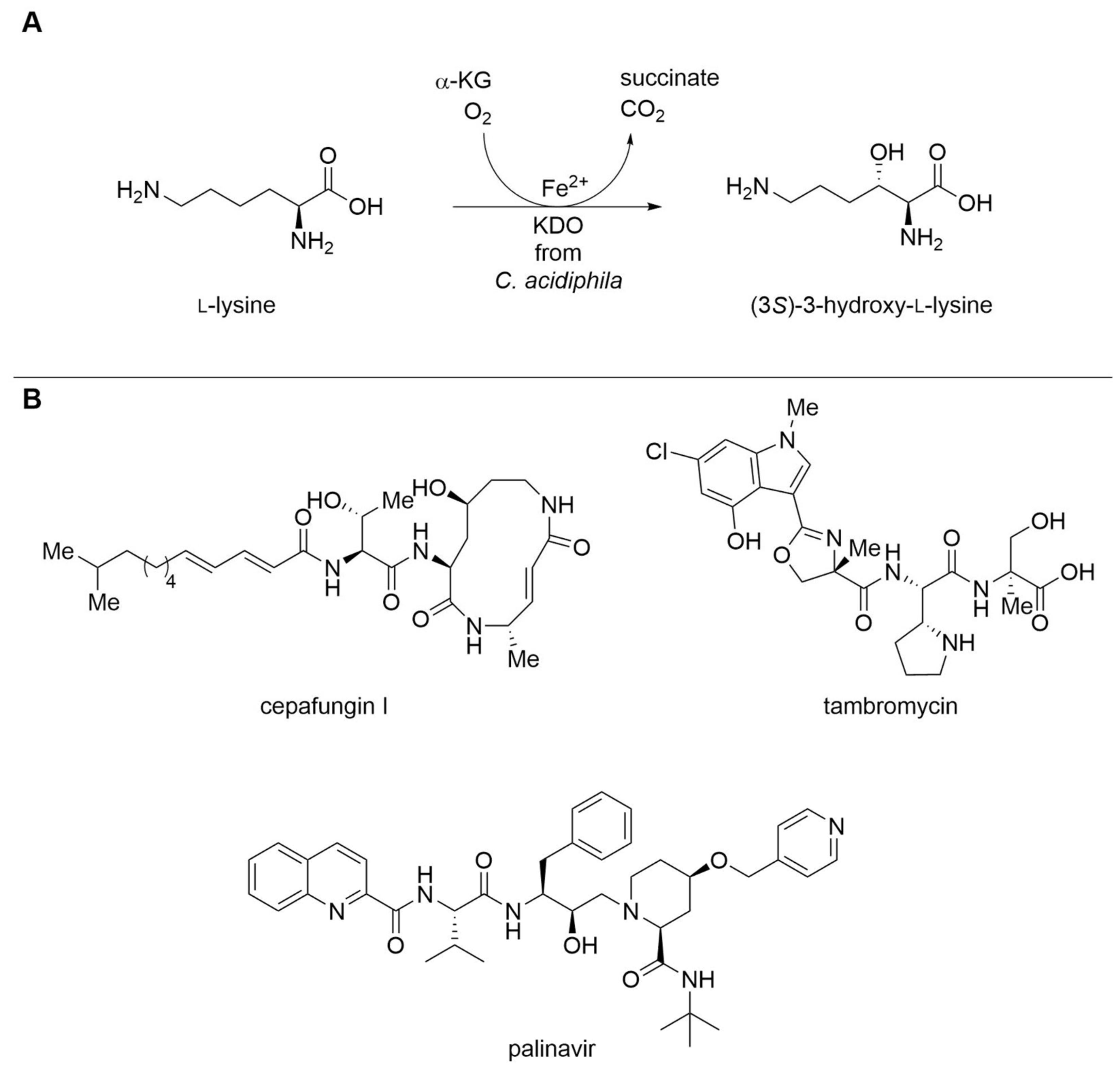
| Lysine dioxygenase (KDO) | Catenulispora acidiphila | Immobilization with HaloTag® | Isolated enzyme | l-Lysine | (3S)-3-Hydroxy- l-lysine | 32.4 g L−1, 100 g L−1 h−1 per gimmobilized enzyme | [20] |
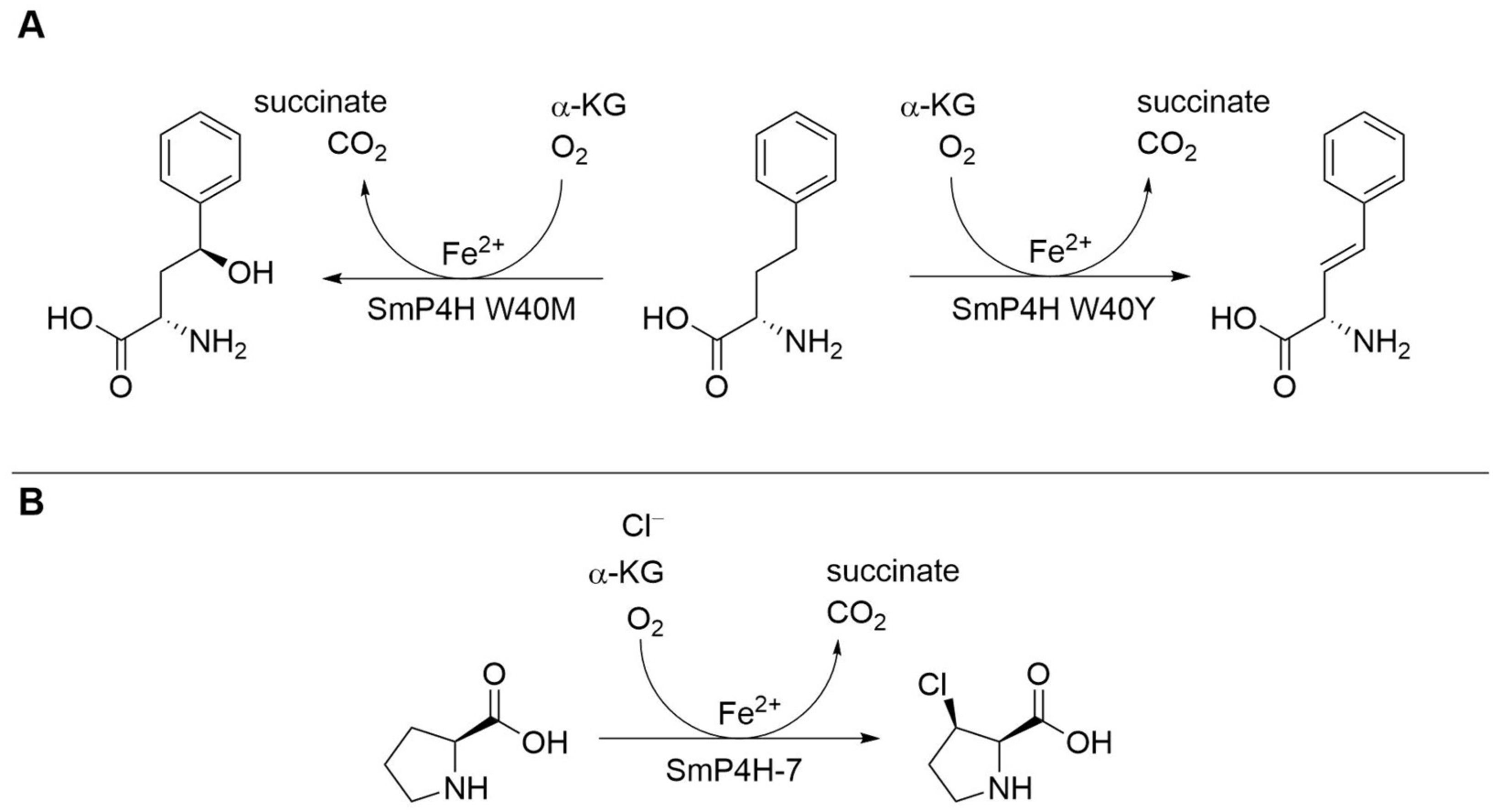
| Proline hydroxylase SmP4H | Sinorhizobium meliloti | Engineered variant (two amino acid exchanges) | Isolated enzyme | l-Homophenylalanine | γ-Hydroxylated l-homophenylalanine | kcat 1.680 ± 0.068 min−1, 35.3% yield | [21] |
| Proline hydroxylase SmP4H | Sinorhizobium meliloti | One amino acid exchange (W40Y) | Isolated enzyme | l-Homophenylalanine | 3,4-Desaturated l-homophenylalanine | kcat 0.83 ± 0.02 min−1, 50.7% yield | [21] |
| Proline hydroxylase SmP4H | Sinorhizobium meliloti | Engineered variant (six amino acid exchanges) | Cell lysate | l-Proline | cis-3-Chloro- l-proline | 4.86 ± 0.16% yield, >98.5%-ee | [22] |
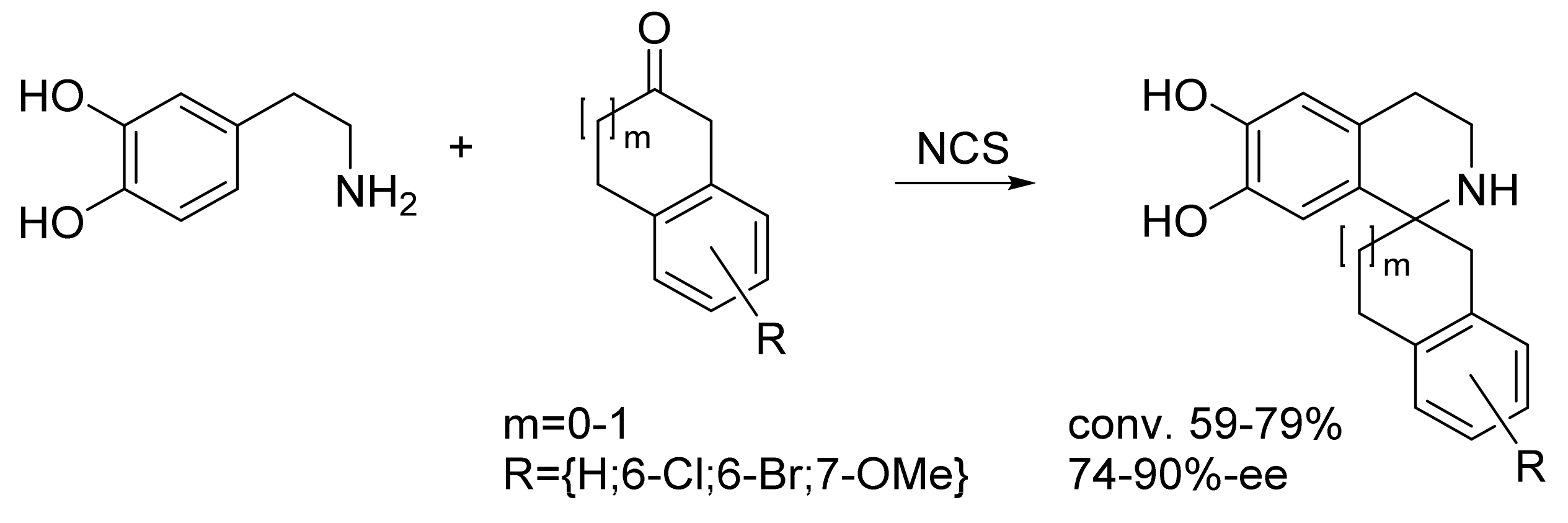
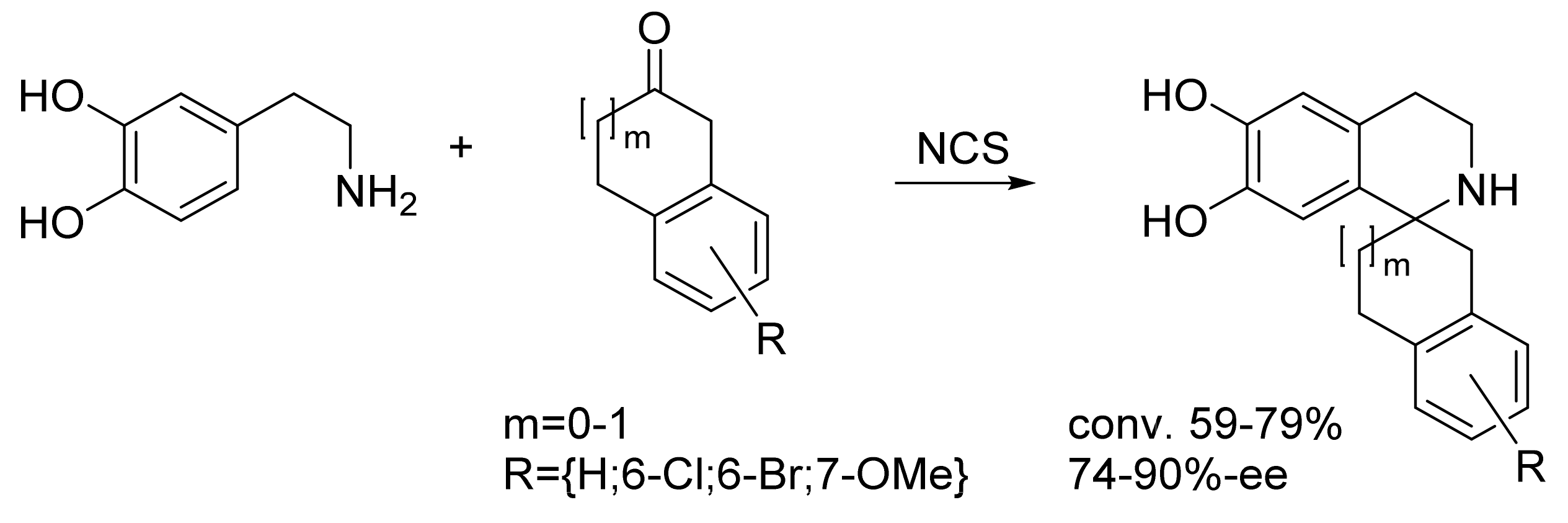
| Norcoclaurine synthase (tfNCS) | Thalictrum flavum | Directed evolution | Cell lysate | Aromatic β-amine; ketone | Tetrahydroisoquinolines | 59-79% conversion 74-90%-ee | [23] |
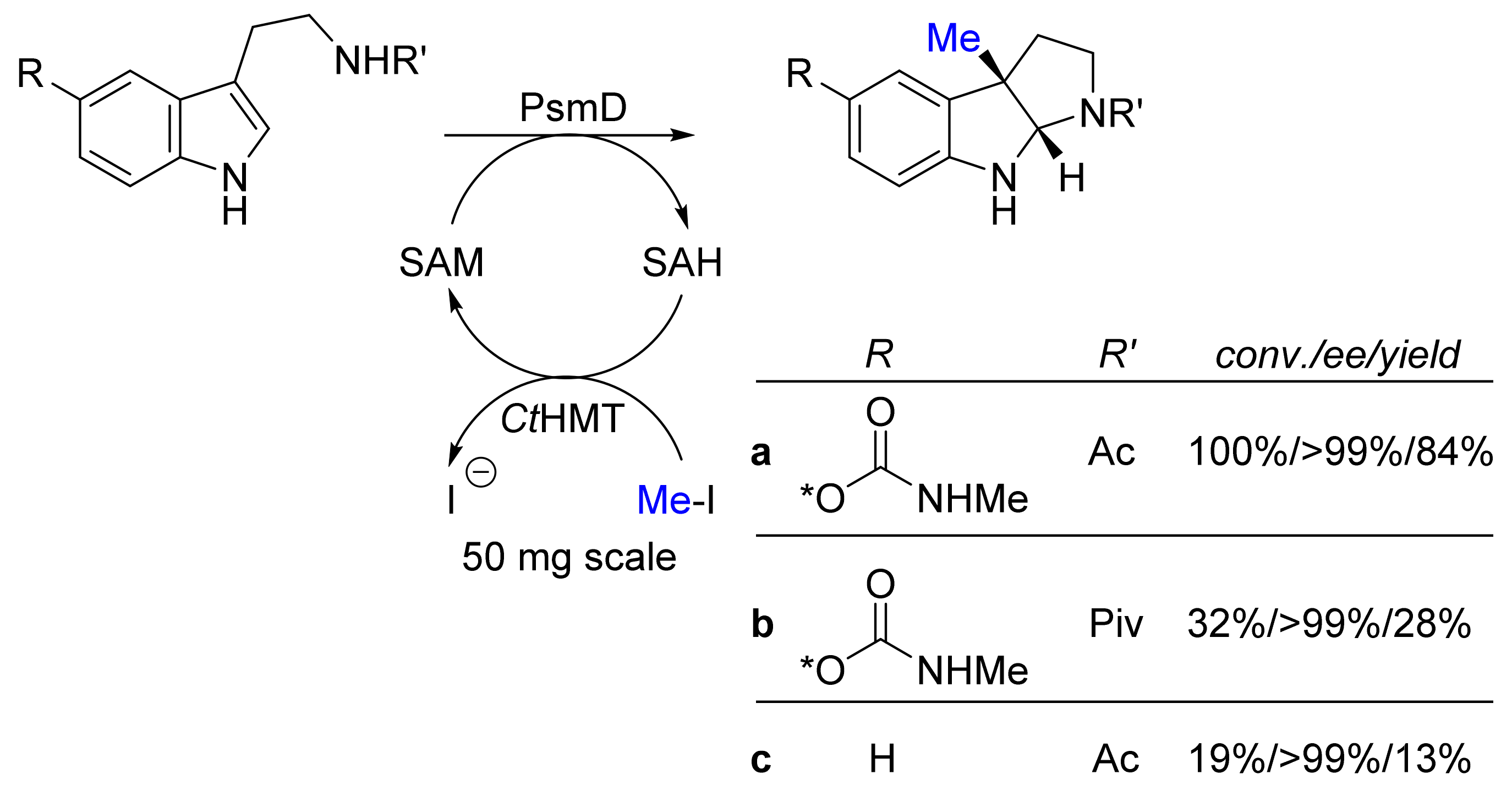
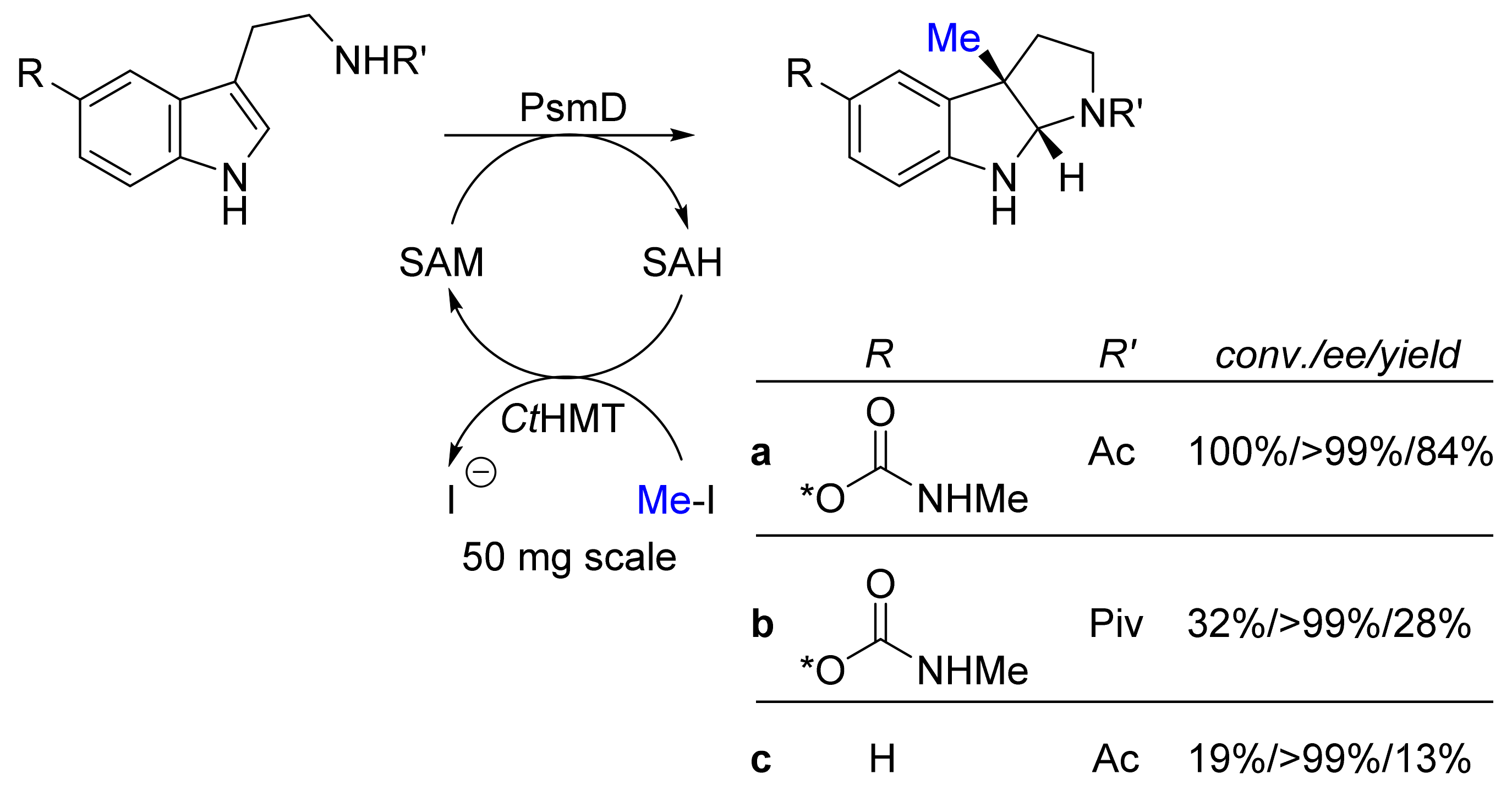
| Methyl transferase PsmD; halide methyl transferase (ctHMT) | Streptomyces griseofuscus; Chloracidobacterium thermophilum | - | Isolated enzymes including SAM- cofactor recycling | Tryptamines | Physostigmines | 13-84% isolated yield >99%-ee | [24] |
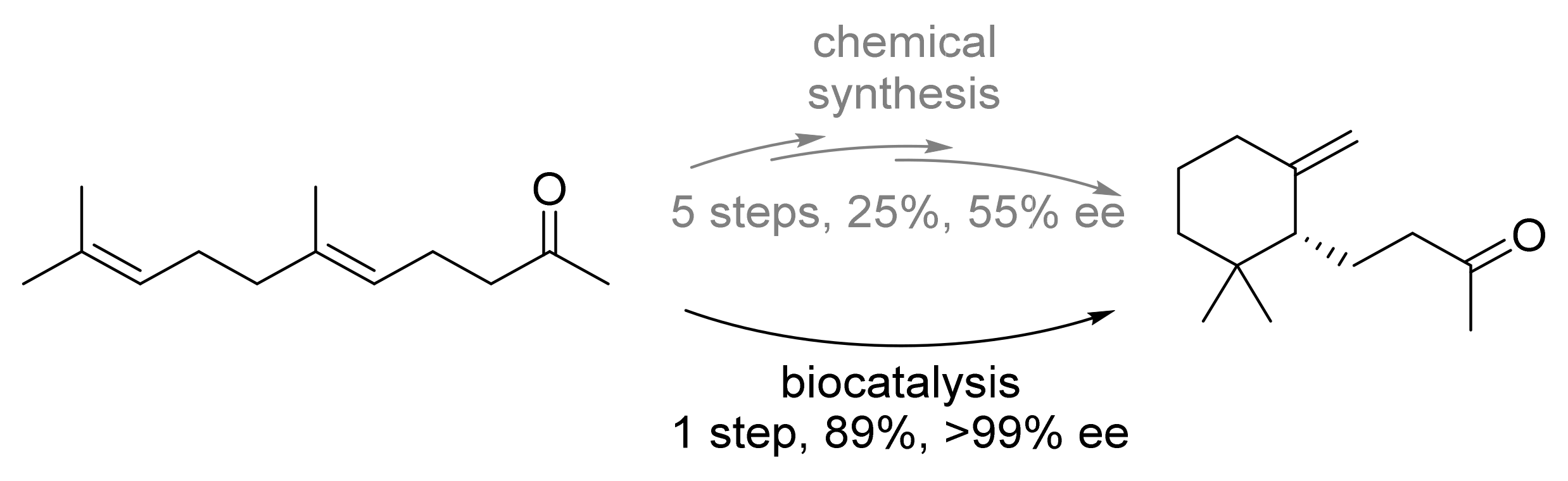
| Terpene cyclase (AacSHC) | Alicyclobacillus acidocaldarius | Semi-rational design | Isolated enzyme | Geranyl acetone | γ-Dihydroionone | 89% isolated yield, >99%-ee | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinner, A.; Nerke, P.; Siedentop, R.; Steinmetz, T.; Classen, T.; Rosenthal, K.; Nett, M.; Pietruszka, J.; Lütz, S. Recent Advances in Biocatalysis for Drug Synthesis. Biomedicines 2022, 10, 964. https://doi.org/10.3390/biomedicines10050964
Kinner A, Nerke P, Siedentop R, Steinmetz T, Classen T, Rosenthal K, Nett M, Pietruszka J, Lütz S. Recent Advances in Biocatalysis for Drug Synthesis. Biomedicines. 2022; 10(5):964. https://doi.org/10.3390/biomedicines10050964
Chicago/Turabian StyleKinner, Alina, Philipp Nerke, Regine Siedentop, Till Steinmetz, Thomas Classen, Katrin Rosenthal, Markus Nett, Jörg Pietruszka, and Stephan Lütz. 2022. "Recent Advances in Biocatalysis for Drug Synthesis" Biomedicines 10, no. 5: 964. https://doi.org/10.3390/biomedicines10050964
APA StyleKinner, A., Nerke, P., Siedentop, R., Steinmetz, T., Classen, T., Rosenthal, K., Nett, M., Pietruszka, J., & Lütz, S. (2022). Recent Advances in Biocatalysis for Drug Synthesis. Biomedicines, 10(5), 964. https://doi.org/10.3390/biomedicines10050964