
Skeletal Muscle Cells Derived from Induced Pluripotent Stem Cells: A Platform for Limb Girdle Muscular Dystrophies

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture and Differentiation
2.3. Flow Cytometry
2.4. Multiplex Fluorescence In Situ Hybridization (m-FISH) Karyotype Analysis
2.5. Immunofluorescence Analysis
2.6. Time-Lapse Imaging of skMt Differentiation
2.7. Quantitative PCR
2.8. Western Immunoblotting
2.9. Statistical Analysis
3. Results
3.1. Generation and Characterization of Skeletal Muscle Cells from Human Induced Pluripotent Stem Cells
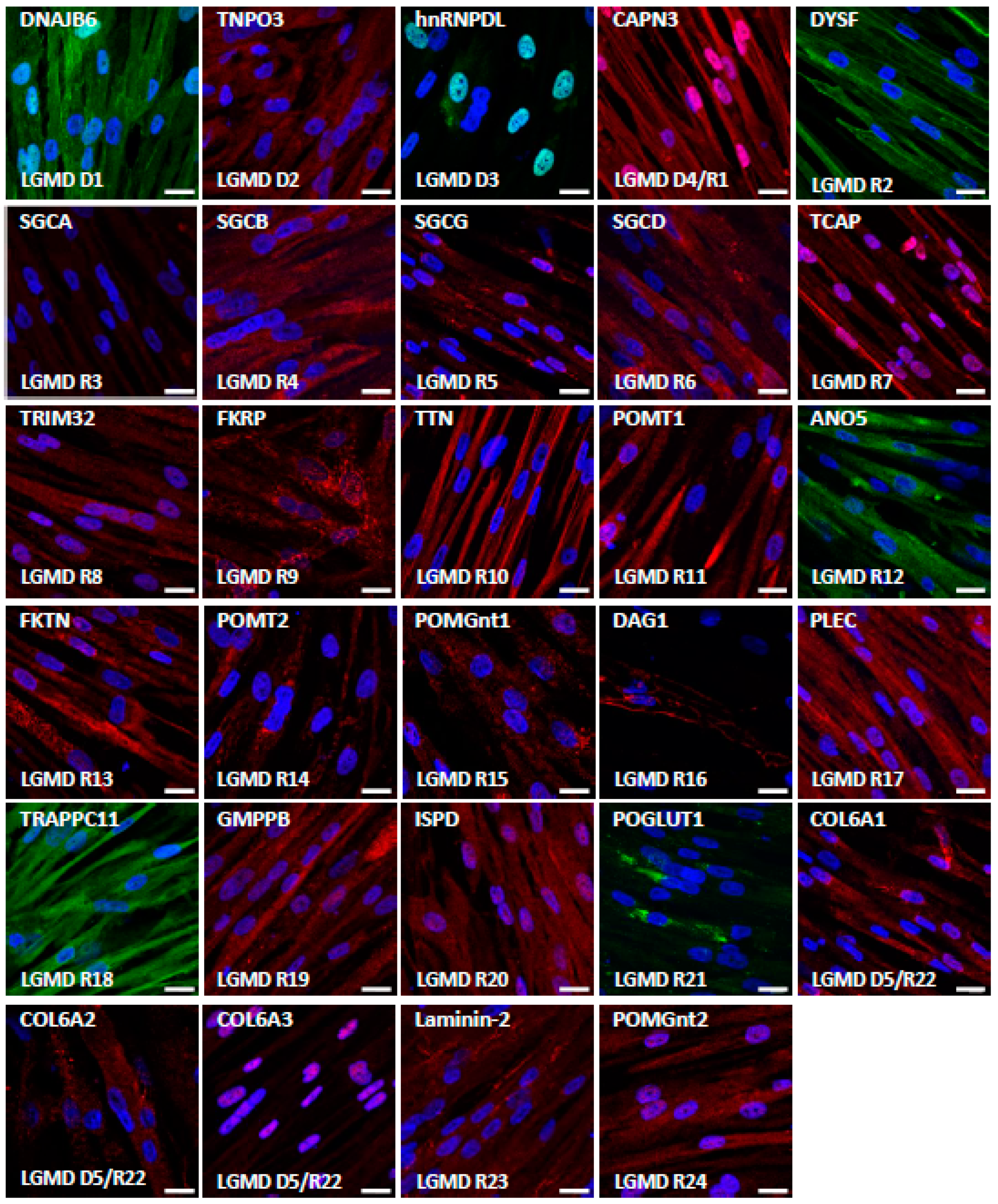
3.2. Characterization of LGMD Genes and Proteins in Myotubes Derived from Human Induced Pluripotent Stem Cells
3.3. Derivation and Characterization of Myotubes from FKRP-Deficient Human Induced Pluripotent Stem Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guglieri, M.; Straub, V.; Bushby, K.; Lochmüller, H. Limb–Girdle Muscular Dystrophies. Curr. Opin. Neurol. 2008, 21, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Savarese, M. Genetic Basis of Limb-Girdle Muscular Dystrophies: The 2014 Update. Acta Myol. 2014, 33, 1–12. [Google Scholar] [PubMed]
- Walton, J.N.; Nattrass, F.J. On the Classification, Natural History and Treatment of the Myopathies. Brain 1954, 77, 169–231. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K.; Korngut, L.; Fiest, K.M.; Dykeman, J.; Day, L.J.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-Analysis on the Epidemiology of the Muscular Dystrophies. Can. J. Neurol. Sci. 2016, 43, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Liewluck, T.; Milone, M. Untangling the Complexity of Limb-girdle Muscular Dystrophies. Muscle Nerve 2018, 58, 167–177. [Google Scholar] [CrossRef]
- Wicklund, M.P.; Kissel, J.T. The Limb-Girdle Muscular Dystrophies. Neurol. Clin. 2014, 32, 729–749. [Google Scholar] [CrossRef]
- Richard, I.; Broux, O.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; Hillaire, D.; et al. Mutations in the Proteolytic Enzyme Calpain 3 Cause Limb-Girdle Muscular Dystrophy Type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef]
- Straub, V.; Murphy, A.; Udd, B.; Corrado, A.; Aymé, S.; Bönneman, C.; de Visser, M.; Hamosh, A.; Jacobs, L.; Khizanishvili, N.; et al. 229th ENMC International Workshop: Limb Girdle Muscular Dystrophies—Nomenclature and Reformed Classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Holt, K.H.; Lim, L.E.; Straub, V.; Venzke, D.P.; Duclos, F.; Anderson, R.D.; Davidson, B.L.; Campbell, K.P. Functional Rescue of the Sarcoglycan Complex in the BIO 14.6 Hamster Using δ-Sarcoglycan Gene Transfer. Mol. Cell 1998, 1, 841–848. [Google Scholar] [CrossRef]
- Li, J.; Dressman, D.; Tsao, Y.P.; Sakamoto, A.; Hoffman, E.P.; Xiao, X. RAAV Vector-Mediated Sarcogylcan Gene Transfer in a Hamster Model for Limb Girdle Muscular Dystrophy. Gene 1999, 6, 74–82. [Google Scholar] [CrossRef]
- Xiao, X.; Li, J.; Tsao, Y.-P.; Dressman, D.; Hoffman, E.P.; Watchko, J.F. Full Functional Rescue of a Complete Muscle (TA) in Dystrophic Hamsters by Adeno-Associated Virus Vector-Directed Gene Therapy. J. Virol. 2000, 74, 1436–1442. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allamand, V.; Donahue, K.M.; Straub, V.; Davisson, R.L.; Davidson, B.L.; Campbell, K.P. Early Adenovirus-Mediated Gene Transfer Effectively Prevents Muscular Dystrophy in Alpha-Sarcoglycan-Deficient Mice. Gene 2000, 7, 1385–1391. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bartoli, M.; Roudaut, C.; Martin, S.; Fougerousse, F.; Suel, L.; Poupiot, J.; Gicquel, E.; Noulet, F.; Danos, O.; Richard, I. Safety and Efficacy of AAV-Mediated Calpain 3 Gene Transfer in a Mouse Model of Limb-Girdle Muscular Dystrophy Type 2A. Mol. Ther. 2006, 13, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, P.C.; Griffin, D.A.; Pozsgai, E.R.; Johnson, R.W.; Grose, W.E.; Heller, K.N.; Shontz, K.M.; Montgomery, C.L.; Liu, J.; Clark, K.R.; et al. AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann. Clin. Transl. Neurol. 2015, 2, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Allamand, V. Animal Models for Muscular Dystrophy: Valuable Tools for the Development of Therapies. Hum. Mol. Genet. 2000, 9, 2459–2467. [Google Scholar] [CrossRef]
- Ng, R.; Banks, G.B.; Hall, J.K.; Muir, L.A.; Ramos, J.N.; Wicki, J.; Odom, G.L.; Konieczny, P.; Seto, J.; Chamberlain, J.R.; et al. Animal Models of Muscular Dystrophy. Prog. Mol. Biol. Transl. Sci. 2012, 105, 83–111. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal Models of Duchenne Muscular Dystrophy: From Basic Mechanisms to Gene Therapy. Dis. Models Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef]
- Ackroyd, M.R.; Skordis, L.; Kaluarachchi, M.; Godwin, J.; Prior, S.; Fidanboylu, M.; Piercy, R.J.; Muntoni, F.; Brown, S.C. Reduced Expression of Fukutin Related Protein in Mice Results in a Model for Fukutin Related Protein Associated Muscular Dystrophies. Brain 2008, 132, 439–451. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Reis, A.; Keers, S.; Harrison, R.; Anderson, L.V.B.; Raffelsberger, T.; Ivanova, S.; Hoger, H.; Bittner, R.E.; Bushby, K.; et al. Cloning of the Mouse Dysferlin Gene and Genomic Characterization of the SJL-Dysf Mutation. Neuroreport 2001, 12, 625–629. [Google Scholar] [CrossRef]
- Aas, V.; Bakke, S.S.; Feng, Y.Z.; Kase, E.T.; Jensen, J.; Bajpeyi, S.; Thoresen, G.H.; Rustan, A.C. Are Cultured Human Myotubes Far from Home? Cell Tissue Res. 2013, 354, 671–682. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Gerli, M.F.M.; Di Stefano, B.; Almada, A.E.; Galvin, A.; Coffey, A.; Huebner, A.J.; Feige, P.; Verheul, C.; Cheung, P.; et al. Direct Reprogramming of Mouse Fibroblasts into Functional Skeletal Muscle Progenitors. Stem Cell Rep. 2018, 10, 1505–1521. [Google Scholar] [CrossRef] [PubMed]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized Pathological Human Myoblasts: Towards a Universal Tool for the Study of Neuromuscular Disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Thorley, M.; Duguez, S.; Mazza, E.M.C.; Valsoni, S.; Bigot, A.; Mamchaoui, K.; Harmon, B.; Voit, T.; Mouly, V.; Duddy, W. Skeletal Muscle Characteristics Are Preserved in HTERT/Cdk4 Human Myogenic Cell Lines. Skelet. Muscle 2016, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Lasa-Elgarresta, J.; Mosqueira-Martín, L.; González-Imaz, K.; Marco-Moreno, P.; Gerenu, G.; Mamchaoui, K.; Mouly, V.; López de Munain, A.; Vallejo-Illarramendi, A. Targeting the Ubiquitin-Proteasome System in Limb-Girdle Muscular Dystrophy With CAPN3 Mutations. Front. Cell Dev. Biol. 2022, 10, 822563. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Bevilacqua, J.A.; Arriagada, C.; Cárdenas, A.M.; Bigot, A.; Mouly, V.; Sáez, J.C.; Caviedes, P. The Absence of Dysferlin Induces the Expression of Functional Connexin-Based Hemichannels in Human Myotubes. BMC Cell Biol. 2016, 17, 15. [Google Scholar] [CrossRef]
- Carmeille, R.; Bouvet, F.; Tan, S.; Croissant, C.; Gounou, C.; Mamchaoui, K.; Mouly, V.; Brisson, A.R.; Bouter, A. Membrane Repair of Human Skeletal Muscle Cells Requires Annexin-A5. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2267–2279. [Google Scholar] [CrossRef]
- Philippi, S.; Bigot, A.; Marg, A.; Mouly, V.; Spuler, S.; Zacharias, U. Dysferlin-Deficient Immortalized Human Myoblasts and Myotubes as a Useful Tool to Study Dysferlinopathy. PLoS Curr. 2012, 4, RRN1298. [Google Scholar] [CrossRef]
- Chandra, G.; Defour, A.; Mamchoui, K.; Pandey, K.; Mishra, S.; Mouly, V.; Sreetama, S.; Mahad Ahmad, M.; Mahjneh, I.; Morizono, H.; et al. Dysregulated Calcium Homeostasis Prevents Plasma Membrane Repair in Anoctamin 5/TMEM16E-Deficient Patient Muscle Cells. Cell Death Discov. 2019, 5, 118. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in Culture of Pluripotential Cells from Mouse Embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a Pluripotent Cell Line from Early Mouse Embryos Cultured in Medium Conditioned by Teratocarcinoma Stem Cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef]
- Shamblott, M.J.; Axelman, J.; Wang, S.; Bugg, E.M.; Littlefield, J.W.; Donovan, P.J.; Blumenthal, P.D.; Huggins, G.R.; Gearhart, J.D. Derivation of Pluripotent Stem Cells from Cultured Human Primordial Germ Cells. Dev. Biol. 1998, 95, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Sci. New Ser. 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Wobus, A.M.; Boheler, K.R. Embryonic Stem Cells: Prospects for Developmental Biology and Cell Therapy. Physiol. Rev. 2005, 85, 635–678. [Google Scholar] [CrossRef]
- Laustriat, D.; Gide, J.; Barrault, L.; Chautard, E.; Benoit, C.; Auboeuf, D.; Boland, A.; Battail, C.; Artiguenave, F.; Deleuze, J.-F.; et al. In Vitro and In Vivo Modulation of Alternative Splicing by the Biguanide Metformin. Mol. Ther. Nucleic Acids 2015, 4, e262. [Google Scholar] [CrossRef]
- Nissan, X.; Blondel, S.; Navarro, C.; Maury, Y.; Denis, C.; Girard, M.; Martinat, C.; De Sandre-Giovannoli, A.; Levy, N.; Peschanski, M. Unique Preservation of Neural Cells in Hutchinson- Gilford Progeria Syndrome Is Due to the Expression of the Neural-Specific MiR-9 MicroRNA. Cell Rep. 2012, 2, 1–9. [Google Scholar] [CrossRef]
- Allouche, J.; Bellon, N.; Saidani, M.; Stanchina-Chatrousse, L.; Masson, Y.; Patwardhan, A.; Gilles-Marsens, F.; Delevoye, C.; Domingues, S.; Nissan, X.; et al. In Vitro Modeling of Hyperpigmentation Associated to Neurofibromatosis Type 1 Using Melanocytes Derived from Human Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 9034–9039. [Google Scholar] [CrossRef]
- Estronca, L.; Francisco, V.; Pitrez, P.; Honório, I.; Carvalho, L.; Vazão, H.; Blersch, J.; Rai, A.; Nissan, X.; Simon, U.; et al. Induced Pluripotent Stem Cell-Derived Vascular Networks to Screen Nano–Bio Interactions. Nanoscale Horiz. 2021, 6, 245–259. [Google Scholar] [CrossRef]
- Lo Cicero, A.; Saidani, M.; Allouche, J.; Egesipe, A.L.; Hoch, L.; Bruge, C.; Sigaudy, S.; De Sandre-Giovannoli, A.; Levy, N.; Baldeschi, C.; et al. Pathological Modelling of Pigmentation Disorders Associated with Hutchinson-Gilford Progeria Syndrome (HGPS) Revealed an Impaired Melanogenesis Pathway in IPS-Derived Melanocytes. Sci. Rep. 2018, 8, 9112. [Google Scholar] [CrossRef]
- Moreau, A.; Reisqs, J.; Delanoe-Ayari, H.; Pierre, M.; Janin, A.; Deliniere, A.; Bessière, F.; Meli, A.C.; Charrabi, A.; Lafont, E.; et al. Deciphering DSC2 Arrhythmogenic Cardiomyopathy Electrical Instability: From Ion Channels to ECG and Tailored Drug Therapy. Clin. Transl. Med. 2021, 11, e319. [Google Scholar] [CrossRef] [PubMed]
- Egesipe, A.-L.; Blondel, S.; Lo Cicero, A.; Jaskowiak, A.-L.; Navarro, C.; Sandre-Giovannoli, A.D.; Levy, N.; Peschanski, M.; Nissan, X. Metformin Decreases Progerin Expression and Alleviates Pathological Defects of Hutchinson–Gilford Progeria Syndrome Cells. npj Aging Mech. Dis. 2016, 2, 16026. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Lévy, N. MG 132-induced Progerin Clearance Is Mediated by Autophagy Activation and Splicing Regulation. EMBO Mol. Med. 2017, 9, 1294–1313. [Google Scholar] [CrossRef] [PubMed]
- Lo Cicero, A.; Jaskowiak, A.-L.; Egesipe, A.-L.; Tournois, J.; Brinon, B.; Pitrez, P.R.; Ferreira, L.; de Sandre-Giovannoli, A.; Levy, N.; Nissan, X. A High Throughput Phenotypic Screening Reveals Compounds That Counteract Premature Osteogenic Differentiation of HGPS IPS-Derived Mesenchymal Stem Cells. Sci. Rep. 2016, 6, 34798. [Google Scholar] [CrossRef] [PubMed]
- Pitrez, P.R.; Estronca, L.; Monteiro, L.M.; Colell, G.; Vazão, H.; Santinha, D.; Harhouri, K.; Thornton, D.; Navarro, C.; Egesipe, A.-L.; et al. Vulnerability of Progeroid Smooth Muscle Cells to Biomechanical Forces Is Mediated by MMP13. Nat. Commun. 2020, 11, 4110. [Google Scholar] [CrossRef]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C.R. Human ES- and IPS-Derived Myogenic Progenitors Restore DYSTROPHIN and Improve Contractility upon Transplantation in Dystrophic Mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Gerli, M.F.M.; Ragazzi, M.; Dastidar, S.; Benedetti, S.; Loperfido, M.; VandenDriessche, T.; Chuah, M.K.; Tedesco, F.S. Efficient Derivation and Inducible Differentiation of Expandable Skeletal Myogenic Cells from Human ES and Patient-Specific IPS Cells. Nat. Protoc. 2015, 10, 941–958. [Google Scholar] [CrossRef]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and Reproducible Myogenic Differentiation from Human IPS Cells: Prospects for Modeling Miyoshi Myopathy In Vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Borchin, B.; Chen, J.; Barberi, T. Derivation and FACS-Mediated Purification of PAX3+/PAX7+ Skeletal Muscle Precursors from Human Pluripotent Stem Cells. Stem Cell Rep. 2013, 1, 620–631. [Google Scholar] [CrossRef]
- Caron, L.; Kher, D.; Lee, K.L.; McKernan, R.; Dumevska, B.; Hidalgo, A.; Li, J.; Yang, H.; Main, H.; Ferri, G.; et al. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem Cells Transl. Med. 2016, 5, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of Pluripotent Stem Cells to Muscle Fiber to Model Duchenne Muscular Dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquié, O. Generation of Human Muscle Fibers and Satellite-like Cells from Human Pluripotent Stem Cells in Vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef]
- Shelton, M.; Metz, J.; Liu, J.; Carpenedo, R.L.; Demers, S.-P.; Stanford, W.L.; Skerjanc, I.S. Derivation and Expansion of PAX7-Positive Muscle Progenitors from Human and Mouse Embryonic Stem Cells. Stem Cell Rep. 2014, 3, 516–529. [Google Scholar] [CrossRef]
- Shelton, M.; Kocharyan, A.; Liu, J.; Skerjanc, I.S.; Stanford, W.L. Robust Generation and Expansion of Skeletal Muscle Progenitors and Myocytes from Human Pluripotent Stem Cells. Methods 2016, 101, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Swartz, E.W.; Baek, J.; Pribadi, M.; Wojta, K.J.; Almeida, S.; Karydas, A.; Gao, F.-B.; Miller, B.L.; Coppola, G. A Novel Protocol for Directed Differentiation of C9orf72-Associated Human Induced Pluripotent Stem Cells Into Contractile Skeletal Myotubes. Stem Cells Transl. Med. 2016, 5, 1461–1472. [Google Scholar] [CrossRef]
- Xu, C.; Tabebordbar, M.; Iovino, S.; Ciarlo, C.; Liu, J.; Castiglioni, A.; Price, E.; Liu, M.; Barton, E.R.; Kahn, C.R.; et al. A Zebrafish Embryo Culture System Defines Factors That Promote Vertebrate Myogenesis across Species. Cell 2013, 155, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons. Sci. New Ser. 2008, 321, 1218–1221. [Google Scholar] [CrossRef]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced Pluripotent Stem Cells from a Spinal Muscular Atrophy Patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.L.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and Study of the Mechanism of Dilated Cardiomyopathy Using Induced Pluripotent Stem Cells Derived from Individuals with Duchenne Muscular Dystrophy. Dis. Models Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early Pathogenesis of Duchenne Muscular Dystrophy Modelled in Patient-Derived Human Induced Pluripotent Stem Cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Li, X.; Yücel, G.; Lang, S.; Sattler, K.; Schünemann, J.-D.; Zimmermann, W.-H.; Cyganek, L.; et al. Ion Channel Dysfunctions in Dilated Cardiomyopathy in Limb-Girdle Muscular Dystrophy. Circ. Genom. Precis. Med. 2018, 11, e001893. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Aierdi, A.J.; Dehesa-Etxebeste, M.; Goicoechea, M.; Aiastui, A.; Richaud-Patin, Y.; Jiménez-Delgado, S.; Raya, A.; Naldaiz-Gastesi, N.; López de Munain, A. Patient-Specific IPSC-Derived Cellular Models of LGMDR1. Stem Cell Res. 2021, 53, 102333. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Gerli, M.F.M.; Perani, L.; Benedetti, S.; Ungaro, F.; Cassano, M.; Antonini, S.; Tagliafico, E.; Artusi, V.; Longa, E.; et al. Transplantation of Genetically Corrected Human IPSC-Derived Progenitors in Mice with Limb-Girdle Muscular Dystrophy. Sci. Transl. Med. 2012, 4, 140ra89. [Google Scholar] [CrossRef]
- Wu, J.; Hunt, S.D.; Matthias, N.; Servián-Morilla, E.; Lo, J.; Jafar-Nejad, H.; Paradas, C.; Darabi, R. Generation of an Induced Pluripotent Stem Cell Line (CSCRMi001-A) from a Patient with a New Type of Limb-Girdle Muscular Dystrophy (LGMD) Due to a Missense Mutation in POGLUT1 (Rumi). Stem Cell Res. 2017, 24, 102–105. [Google Scholar] [CrossRef]
- Boissart, C.; Poulet, A.; Georges, P.; Darville, H.; Julita, E.; Delorme, R.; Bourgeron, T.; Peschanski, M.; Benchoua, A. Differentiation from Human Pluripotent Stem Cells of Cortical Neurons of the Superficial Layers Amenable to Psychiatric Disease Modeling and High-Throughput Drug Screening. Transl. Psychiatry 2013, 3, e294. [Google Scholar] [CrossRef]
- Darville, H.; Poulet, A.; Rodet-Amsellem, F.; Chatrousse, L.; Pernelle, J.; Boissart, C.; Héron, D.; Nava, C.; Perrier, A.; Jarrige, M.; et al. Human Pluripotent Stem Cell-Derived Cortical Neurons for High Throughput Medication Screening in Autism: A Proof of Concept Study in SHANK3 Haploinsufficiency Syndrome. EBioMedicine 2016, 9, 293–305. [Google Scholar] [CrossRef]
- Rao, L.; Qian, Y.; Khodabukus, A.; Ribar, T.; Bursac, N. Engineering Human Pluripotent Stem Cells into a Functional Skeletal Muscle Tissue. Nat. Commun. 2018, 9, 126. [Google Scholar] [CrossRef]
- Mercuri, E.; Brockington, M.; Straub, V.; Quijano-Roy, S.; Yuva, Y.; Herrmann, R.; Brown, S.C.; Torelli, S.; Dubowitz, V.; Blake, D.J.; et al. Phenotypic Spectrum Associated with Mutations in the Fukutin-Related Protein Gene. Ann. Neurol. 2003, 53, 537–542. [Google Scholar] [CrossRef]
- Poppe, M.; Bourke, J.; Eagle, M.; Frosk, P.; Wrogemann, K.; Greenberg, C.; Muntoni, F.; Voit, T.; Straub, V.; Hilton-Jones, D.; et al. Cardiac and Respiratory Failure in Limb-Girdle Muscular Dystrophy 2I. Ann. Neurol. 2004, 56, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Valero de Bernabe, D. Mutations in the FKRP Gene Can Cause Muscle-Eye-Brain Disease and Walker-Warburg Syndrome. J. Med. Genet. 2004, 41, e61. [Google Scholar] [CrossRef] [PubMed]
- Brockington, M.; Blake, D.J.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Ponting, C.P.; Estournet, B.; Romero, N.B.; Mercuri, E.; et al. Mutations in the Fukutin-Related Protein Gene (FKRP) Cause a Form of Congenital Muscular Dystrophy with Secondary Laminin A2 Deficiency and Abnormal Glycosylation of a-Dystroglycan. Am. J. Hum. Genet. 2001, 69, 1198–1209. [Google Scholar] [CrossRef]
- Cataldi, M.P.; Lu, P.; Blaeser, A.; Lu, Q.L. Ribitol Restores Functionally Glycosylated α-Dystroglycan and Improves Muscle Function in Dystrophic FKRP-Mutant Mice. Nat. Commun. 2018, 9, 3448. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.F.; Gicquel, E.; Marsolier, J.; Richard, I. Functional and Cellular Localization Diversity Associated with Fukutin-related Protein Patient Genetic Variants. Hum. Mutat. 2019, 40, 1874–1885. [Google Scholar] [CrossRef]
- Alhamidi, M.; Brox, V.; Stensland, E.; Liset, M.; Lindal, S.; Nilssen, Ø. Limb Girdle Muscular Dystrophy Type 2I: No Correlation between Clinical Severity, Histopathology and Glycosylated α-Dystroglycan Levels in Patients Homozygous for Common FKRP Mutation. Neuromuscul. Disord. 2017, 27, 619–626. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruge, C.; Geoffroy, M.; Benabides, M.; Pellier, E.; Gicquel, E.; Dhiab, J.; Hoch, L.; Richard, I.; Nissan, X. Skeletal Muscle Cells Derived from Induced Pluripotent Stem Cells: A Platform for Limb Girdle Muscular Dystrophies. Biomedicines 2022, 10, 1428. https://doi.org/10.3390/biomedicines10061428
Bruge C, Geoffroy M, Benabides M, Pellier E, Gicquel E, Dhiab J, Hoch L, Richard I, Nissan X. Skeletal Muscle Cells Derived from Induced Pluripotent Stem Cells: A Platform for Limb Girdle Muscular Dystrophies. Biomedicines. 2022; 10(6):1428. https://doi.org/10.3390/biomedicines10061428
Chicago/Turabian StyleBruge, Celine, Marine Geoffroy, Manon Benabides, Emilie Pellier, Evelyne Gicquel, Jamila Dhiab, Lucile Hoch, Isabelle Richard, and Xavier Nissan. 2022. "Skeletal Muscle Cells Derived from Induced Pluripotent Stem Cells: A Platform for Limb Girdle Muscular Dystrophies" Biomedicines 10, no. 6: 1428. https://doi.org/10.3390/biomedicines10061428
APA StyleBruge, C., Geoffroy, M., Benabides, M., Pellier, E., Gicquel, E., Dhiab, J., Hoch, L., Richard, I., & Nissan, X. (2022). Skeletal Muscle Cells Derived from Induced Pluripotent Stem Cells: A Platform for Limb Girdle Muscular Dystrophies. Biomedicines, 10(6), 1428. https://doi.org/10.3390/biomedicines10061428