
New Insights into Cerebral Vessel Disease Landscapes at Single-Cell Resolution: Pathogenetic and Therapeutic Perspectives

 ,
, 
Abstract
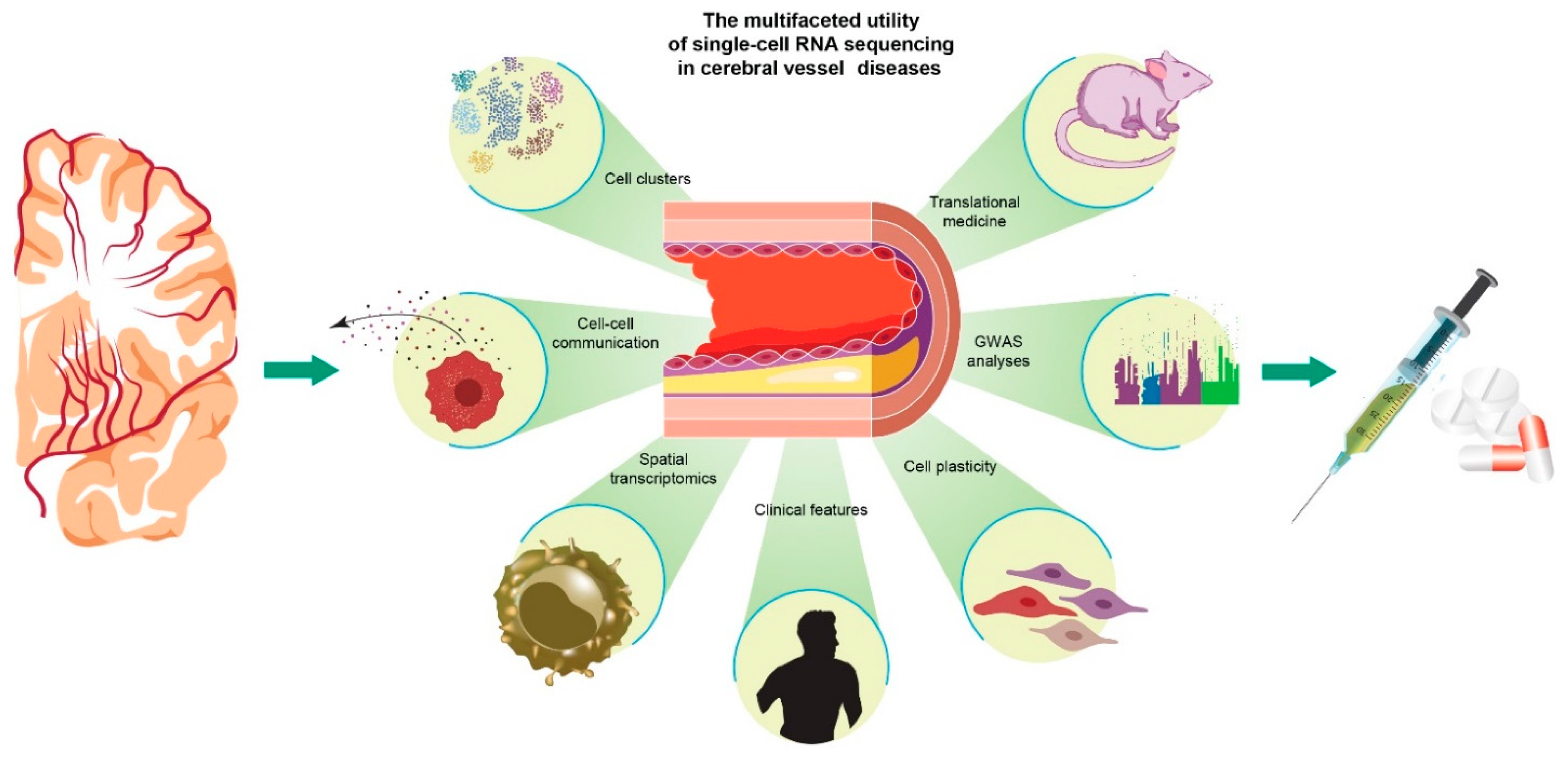
:1. Introduction
2. Single-Cell Omics Technologies
3. Single-Cell Sequencing in Preclinical Animal Models
Single-Cell Sequencing in Humans
4. Perspectives on the Application of Single-Cell Technology for Precision Medicine in Cerebrovascular Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Vos, T.; Nichols, E.; Owolabi, M.O.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: Translating evidence into policy. Lancet Neurol. 2019, 19, 255–265. [Google Scholar] [CrossRef]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Rabinstein, A.A. Update on Treatment of Acute Ischemic Stroke. Contin. Lifelong Learn. Neurol. 2020, 26, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cámara, R.; González-Santos, J.; González-Berna, J.; Trejo-Gabriel-Galán, J.M. Factors associated with a rapid call for assistance for patients with ischemic stroke. Emergencias 2020, 32, 33–39. [Google Scholar] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Cahan, P.; Cacchiarelli, D.; Dunn, S.-J.; Hemberg, M.; Lopes, S.M.C.D.S.; Morris, S.A.; Rackham, O.J.; del Sol, A.; Wells, C.A. Computational Stem Cell Biology: Open Questions and Guiding Principles. Cell Stem Cell 2021, 28, 20–32. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Choi, Y.H.; Kim, J.K. Dissecting Cellular Heterogeneity Using Single-Cell RNA Sequencing. Mol. Cells 2019, 42, 189–199. [Google Scholar] [CrossRef]
- See, P.; Lum, J.; Chen, J.; Ginhoux, F. A Single-Cell Sequencing Guide for Immunologists. Front. Immunol. 2018, 9, 2425. [Google Scholar] [CrossRef]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef]
- Slenders, L.; Tessels, D.E.; van der Laan, S.W.; Pasterkamp, G.; Mokry, M. The Applications of Single-Cell RNA Sequencing in Atherosclerotic Disease. Front. Cardiovasc. Med. 2022, 9, 826103. [Google Scholar] [CrossRef] [PubMed]
- McQueen, L.W.; Ladak, S.S.; Abbasciano, R.; George, S.J.; Suleiman, M.-S.; Angelini, G.D.; Murphy, G.J.; Zakkar, M. Next-Generation and Single-Cell Sequencing Approaches to Study Atherosclerosis and Vascular Inflammation Pathophysiology: A Systematic Review. Front. Cardiovasc. Med. 2022, 9, 849675. [Google Scholar] [CrossRef] [PubMed]
- Zappia, L.; Phipson, B.; Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. PLoS Comput. Biol. 2018, 14, e1006245. [Google Scholar] [CrossRef] [PubMed]
- Andrews, T.S.; Kiselev, V.Y.; McCarthy, D.; Hemberg, M. Tutorial: Guidelines for the computational analysis of single-cell RNA sequencing data. Nat. Protoc. 2020, 16, 1–9. [Google Scholar] [CrossRef]
- Ding, J.; Alavi, A.; Ebrahimkhani, M.R.; Bar-Joseph, Z. Computational tools for analyzing single-cell data in pluripotent cell differentiation studies. Cell Rep. Methods 2021, 1, 100087. [Google Scholar] [CrossRef]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2017, 18, 35–45. [Google Scholar] [CrossRef]
- Upadhaya, S.; Sawai, C.M.; Papalexi, E.; Rashidfarrokhi, A.; Jang, G.; Chattopadhyay, P.; Satija, R.; Reizis, B. Kinetics of adult hematopoietic stem cell differentiation in vivo. J. Exp. Med. 2018, 215, 2815–2832. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 2020, 22, 71–88. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.-A.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, M.A.; Prange, K.H.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; De Jager, S.C.; Asselbergs, F.W.; De Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef]
- Tutino, V.M.; Fricano, S.; Frauens, K.; Patel, T.R.; Monteiro, A.; Rai, H.H.; Waqas, M.; Chaves, L.; Poppenberg, K.E.; Siddiqui, A.H. Isolation of RNA from Acute Ischemic Stroke Clots Retrieved by Mechanical Thrombectomy. Genes 2021, 12, 1617. [Google Scholar] [CrossRef]
- Williams, J.W.; Winkels, H.; Durant, C.P.; Zaitsev, K.; Ghosheh, Y.; Ley, K. Single Cell RNA Sequencing in Atherosclerosis Research. Circ. Res. 2020, 126, 1112–1126. [Google Scholar] [CrossRef]
- Hill, C.A.; Fernandez, D.M.; Giannarelli, C. Single cell analyses to understand the immune continuum in atherosclerosis. Atherosclerosis 2021, 330, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Miragaia, R.J.; Natarajan, K.N.; Teichmann, S.A. A rapid and robust method for single cell chromatin accessibility profiling. Nat. Commun. 2018, 9, 5345. [Google Scholar] [CrossRef] [PubMed]
- Placek, K.; Schultze, J.L.; Aschenbrenner, A.C. Epigenetic reprogramming of immune cells in injury, repair, and resolution. J. Clin. Investig. 2019, 129, 2994–3005. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, F.J.; Disteche, C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, D.M.; Giannarelli, C. Immune cell profiling in atherosclerosis: Role in research and precision medicine. Nat. Rev. Cardiol. 2021, 19, 43–58. [Google Scholar] [CrossRef]
- Kimball, A.K.; Oko, L.M.; Bullock, B.L.; Nemenoff, R.A.; Van Dyk, L.F.; Clambey, E.T. A Beginner’s Guide to Analyzing and Visualizing Mass Cytometry Data. J. Immunol. 2018, 200, 3–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.; Vallmitjana, A.; Gu, J.; La, K.; Xu, Q.; Flores, J.; Zimak, J.; Shiu, J.; Hosohama, L.; Wu, J.; et al. Spatial transcriptomics using combinatorial fluorescence spectral and lifetime encoding, imaging and analysis. Nat. Commun. 2022, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, D.; Song, D.; Liu, X.; Zhang, Y.; Xu, X.; Wang, X. Clinical and translational values of spatial transcriptomics. Signal. Transduct. Target. Ther. 2022, 7, 111. [Google Scholar] [CrossRef]
- Kumar, S.; Andueza, A.; Kim, J.; Kang, D.-W.; Jo, H. Isolation of Endothelial Cells from the Lumen of Mouse Carotid Arteries for Single-cell Multi-omics Experiments. J. Vis. Exp. 2021, 4, 176. [Google Scholar] [CrossRef]
- Andueza, A.; Kumar, S.; Kim, J.; Kang, D.-W.; Mumme, H.L.; Perez, J.I.; Villa-Roel, N.; Jo, H. Endothelial Reprogramming by Disturbed Flow Revealed by Single-Cell RNA and Chromatin Accessibility Study. Cell Rep. 2020, 33, 108491. [Google Scholar] [CrossRef]
- Williams, D.; Mahmoud, M.; Liu, R.; Andueza, A.; Kumar, S.; Kang, D.-W.; Zhang, J.; Tamargo, I.; Villa-Roel, N.; Baek, K.-I.; et al. Stable flow-induced expression of KLK10 inhibits endothelial inflammation and atherosclerosis. eLife 2022, 11, 72579. [Google Scholar] [CrossRef]
- Li, F.; Yan, K.; Wu, L.; Zheng, Z.; Du, Y.; Liu, Z.; Zhao, L.; Li, W.; Sheng, Y.; Ren, L.; et al. Single-cell RNA-seq reveals cellular heterogeneity of mouse carotid artery under disturbed flow. Cell Death Discov. 2021, 7, 180. [Google Scholar] [CrossRef]
- Gao, X.-F.; Chen, A.-Q.; Wang, Z.-M.; Wang, F.; Luo, S.; Chen, S.-Y.; Gu, Y.; Kong, X.-Q.; Zuo, G.-F.; Chen, Y.; et al. Single-Cell RNA Sequencing of the Rat Carotid Arteries Uncovers Potential Cellular Targets of Neointimal Hyperplasia. Front. Cardiovasc. Med. 2021, 8, 751525. [Google Scholar] [CrossRef]
- Eberhardt, N.; Giannarelli, C. How Single-Cell Technologies Have Provided New Insights Into Atherosclerosis. Arter. Thromb. Vasc. Biol. 2022, 42, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Slenders, L.; Landsmeer, L.P.L.; Cui, K.; Depuydt, M.A.C.; Verwer, M.; Mekke, J.; Timmerman, N.; Dungen, N.A.M.V.D.; Kuiper, J.; de Winther, M.P.J.; et al. Intersecting single-cell transcriptomics and genome-wide association studies identifies crucial cell populations and candidate genes for atherosclerosis. Eur. Heart J. Open 2021, 2, oeab043. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Tabas, I. The Cellular Biology of Macrophages in Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, L.S.; Lareau, C.A.; Ulirsch, J.C.; Christian, E.; Muus, C.; Li, L.H.; Pelka, K.; Ge, W.; Oren, Y.; Brack, A.; et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 2019, 176, 1325–1339.e22. [Google Scholar] [CrossRef] [Green Version]
- Misra, A.; Rehan, R.; Lin, A.; Patel, S.; Fisher, E.A. Emerging Concepts of Vascular Cell Clonal Expansion in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2022, 42, e74–e84. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis as an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Li, F.; Du, Y.; Hong, L.; Liu, Z.; Yan, K.; Liu, C.; Zhu, Z.; Lu, Q.; Tang, C.; Zhu, L. Single-cell transcriptional profiling of human carotid plaques reveals a subpopulation of endothelial cells associated with stroke incidences. J. Cell. Mol. Med. 2022, 26, 3446–3459. [Google Scholar] [CrossRef]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsaigh, T.; Evans, D.; Frankel, D.; Torkamani, A. Decoding the transcriptome of atherosclerotic plaque at single-cell resolution. bioRXiv 2020. [Google Scholar] [CrossRef]
- Bersano, A.; Markus, H.S.; Quaglini, S.; Arbustini, E.; Lanfranconi, S.; Micieli, G.; Boncoraglio, G.B.; Taroni, F.; Gellera, C.; Baratta, S.; et al. Clinical Pregenetic Screening for Stroke Monogenic Diseases. Stroke 2016, 47, 1702–1709. [Google Scholar] [CrossRef] [Green Version]
- Chou, E.L.; Cardenas, C.L.L.; Chaffin, M.; Arduini, A.D.; Juric, D.; Stone, J.R.; LaMuraglia, G.M.; Eagleton, M.J.; Conrad, M.F.; Isselbacher, E.M.; et al. Vascular smooth muscle cell phenotype switching in carotid atherosclerosis. JVS Vasc. Sci. 2021, 3, 41–47. [Google Scholar] [CrossRef]
- Winkler, E.A.; Kim, C.N.; Ross, J.M.; Garcia, J.H.; Gil, E.; Oh, I.; Chen, L.Q.; Wu, D.; Catapano, J.S.; Raygor, K.; et al. A single-cell atlas of the normal and malformed human brain vasculature. Science 2022, 375, eabi7377. [Google Scholar] [CrossRef]
- Daemen, M.J.; Gijsen, F.J.H.; Van der Heiden, K.; Hoogendoorn, A. Animal models for plaque rupture: A biomechanical assessment. Thromb. Haemost. 2016, 115, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.F.; Hodonsky, C.J.; Turner, A.W.; Wong, D.; Song, Y.; Mosquera, J.V.; Ligay, A.V.; Slenders, L.; Gancayco, C.; Pan, H.; et al. Enhanced single-cell RNA-seq workflow reveals coronary artery disease cellular cross-talk and candidate drug targets. Atherosclerosis 2021, 340, 12–22. [Google Scholar] [CrossRef]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.-X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: How to proceed? Nat. Rev. Cardiol. 2022, 31, 1–21. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Data ID | Population | Cells | The Focus of the Paper | Findings |
|---|---|---|---|---|
| Fernandez_2019 Carotid Artery Plaque Endarterectomy | 6 patients | 7.169 CD45+ cells | Immune cells in the plaques | T cell exhaustion and different IL-1 signaling patterns in symptomatic patients |
| Depuit_2020 Carotid Artery Plaque Endarterectomy | 18 patients | 3.282 | All the cells in the plaques | IL12-IFNγ axis, an important feature of T-cell activation in the plaque |
| Pan_2020 Carotid Artery Plaque Endarterectomy | 3 patients | 8.867 | Vascular smooth muscle cell | Retinoic acid signaling modulates SMC and atherosclerosis progression |
| Alencar_2020 Carotid Artery Plaque Endarterectomy | 18 patients | 1.287 | Vascular smooth muscle cells | SMC phenotypic is regulated by Klf4 and Oct4 |
| Alsaigh_2020 Carotid Artery Plaque Endarterectomy + Adjacent | 3 patients | 51.981 | All the cells in the plaques | TNFa pathways are active in both endothelial and SMC |
| Slenders_2021 Carotid Artery Plaque Endarterectomy | 38 patients | 5.633 | Genetic risk factors | New gene target in cerebrovascular disease: ESAM LMNA, SLC44A2. |
| Chou_2021 Carotid Artery Plaque Endarterectomy | 7 patients | 6.049 | Vascular smooth muscle cells | HDAC9 modifies VSMC phenotype and immune cell recruitment in carotid disease |
| Winkler_2022 Large and small arteries and veins Epileptic patients that underwent lobectomy | 5 patients | 74.535 | Normal human brain vasculature. | Cellular and molecular profiles of the adult human cerebrovasculature |
| Winkler_2022 arteriovenous malformation (AVM) | 5 patients | 106.853 | Malformed human brain vasculature | AVM rupture is leaded by GPNMB+ monocytes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meneri, M.; Bonato, S.; Gagliardi, D.; Comi, G.P.; Corti, S. New Insights into Cerebral Vessel Disease Landscapes at Single-Cell Resolution: Pathogenetic and Therapeutic Perspectives. Biomedicines 2022, 10, 1693. https://doi.org/10.3390/biomedicines10071693
Meneri M, Bonato S, Gagliardi D, Comi GP, Corti S. New Insights into Cerebral Vessel Disease Landscapes at Single-Cell Resolution: Pathogenetic and Therapeutic Perspectives. Biomedicines. 2022; 10(7):1693. https://doi.org/10.3390/biomedicines10071693
Chicago/Turabian StyleMeneri, Megi, Sara Bonato, Delia Gagliardi, Giacomo P. Comi, and Stefania Corti. 2022. "New Insights into Cerebral Vessel Disease Landscapes at Single-Cell Resolution: Pathogenetic and Therapeutic Perspectives" Biomedicines 10, no. 7: 1693. https://doi.org/10.3390/biomedicines10071693