
CCL4 Functions as a Biomarker of Type 2 Airway Inflammation

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Preparation
2.2. Quantitative RT-PCR
2.3. Cell Lysis and Western Blotting
2.4. Immunoassay for Chemokines and TSLP
2.5. Immunofluorescence Staining
2.6. Statistical Analysis
3. Results
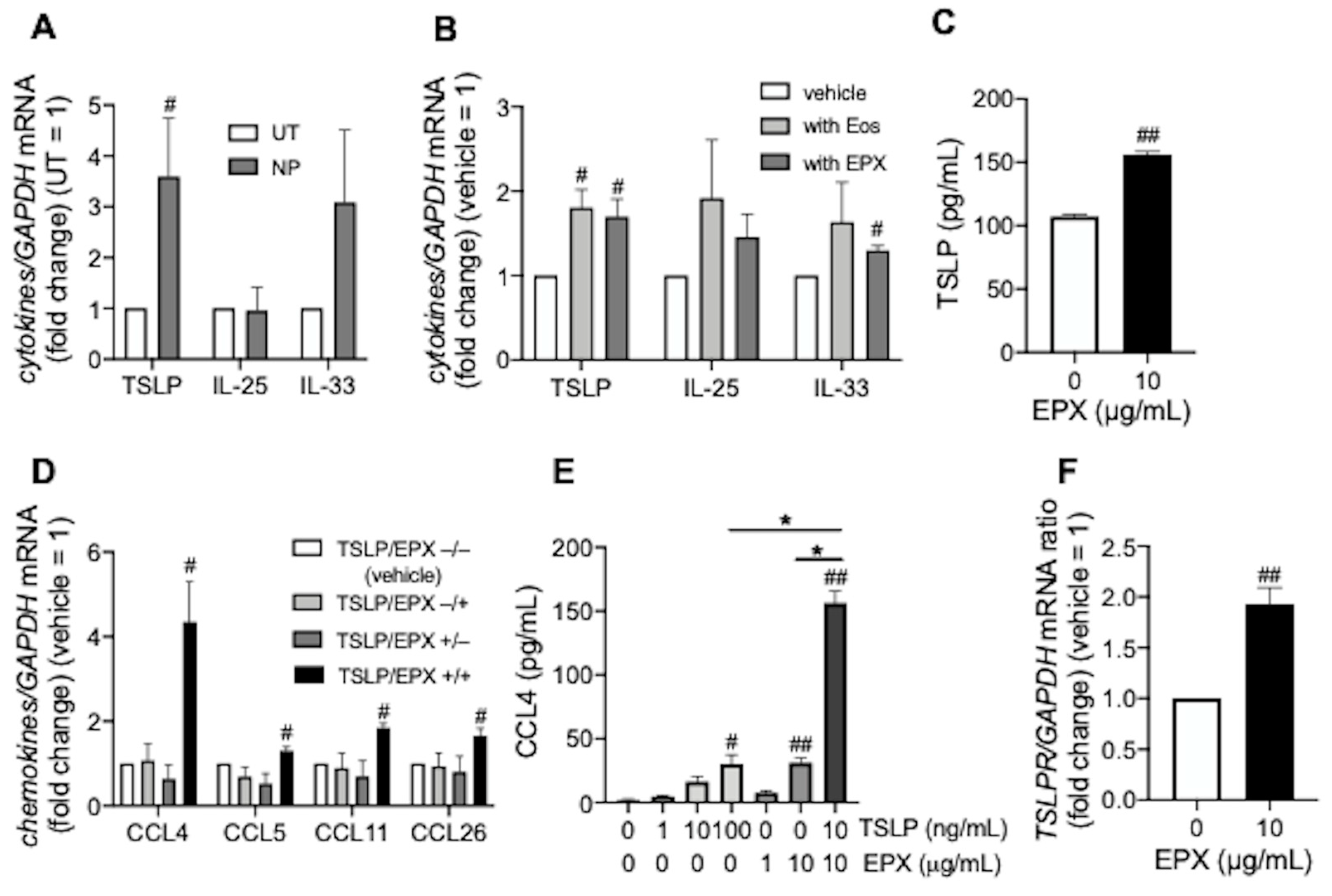
3.1. TSLP induces CCL4 Release under Eosinophilic Airway Inflammation
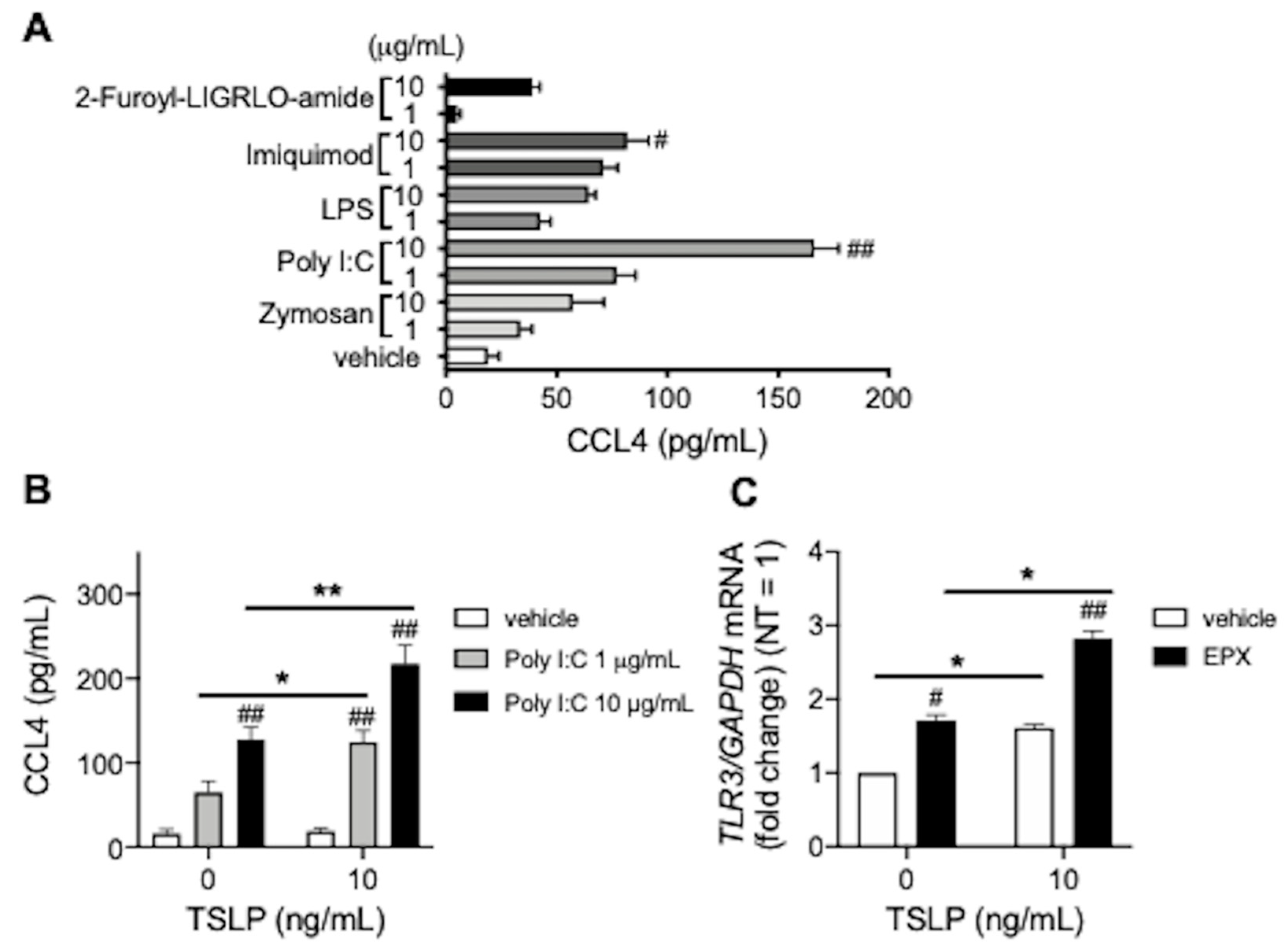
3.2. TSLP Enhances TLR3 Ligand-Mediated CCL4 Release
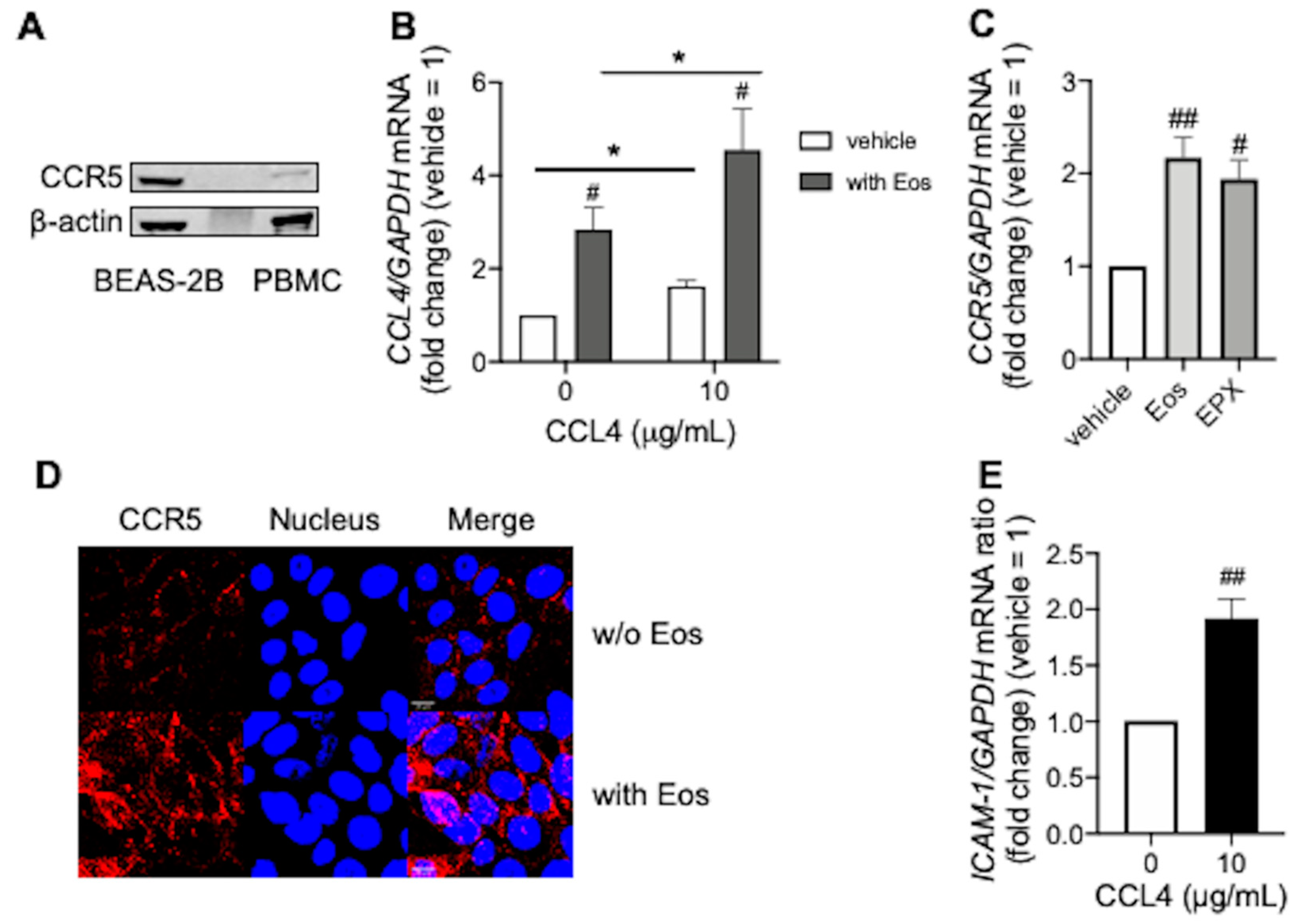
3.3. CCL4 Exerts Autocrine Effects during Eosinophilic Airway Inflammation
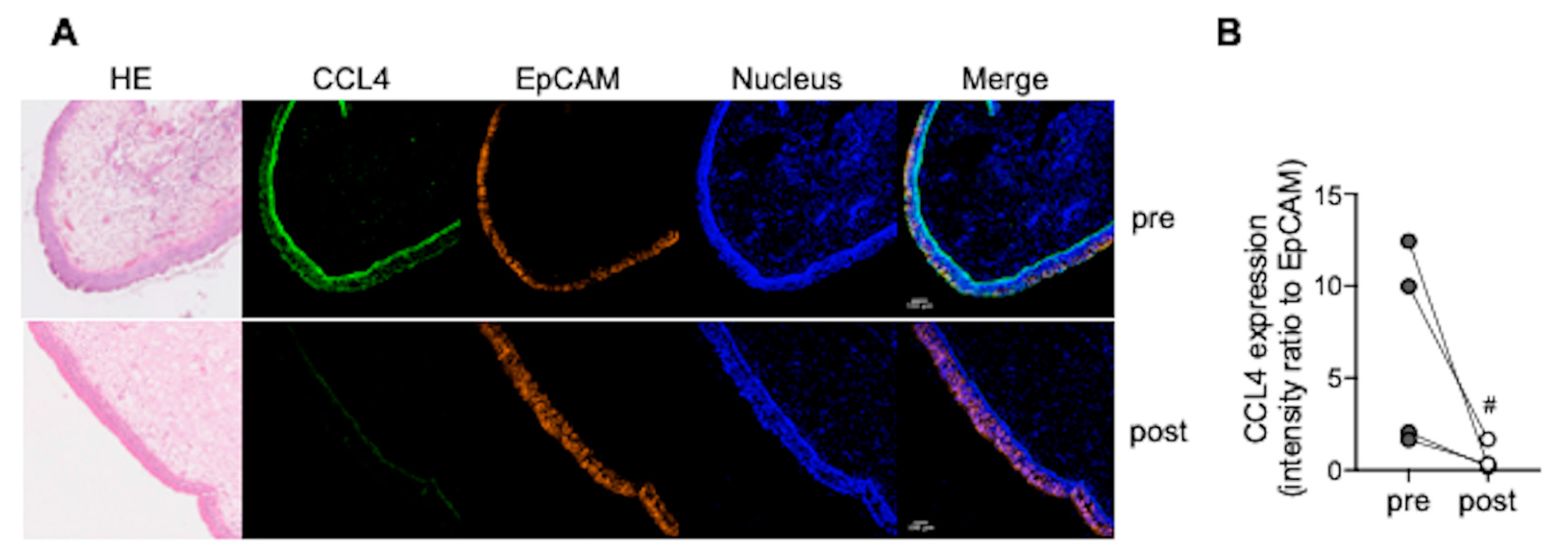
3.4. CCL4 Expression in Nasal Polyps Is Reduced after Dupilumab Treatment
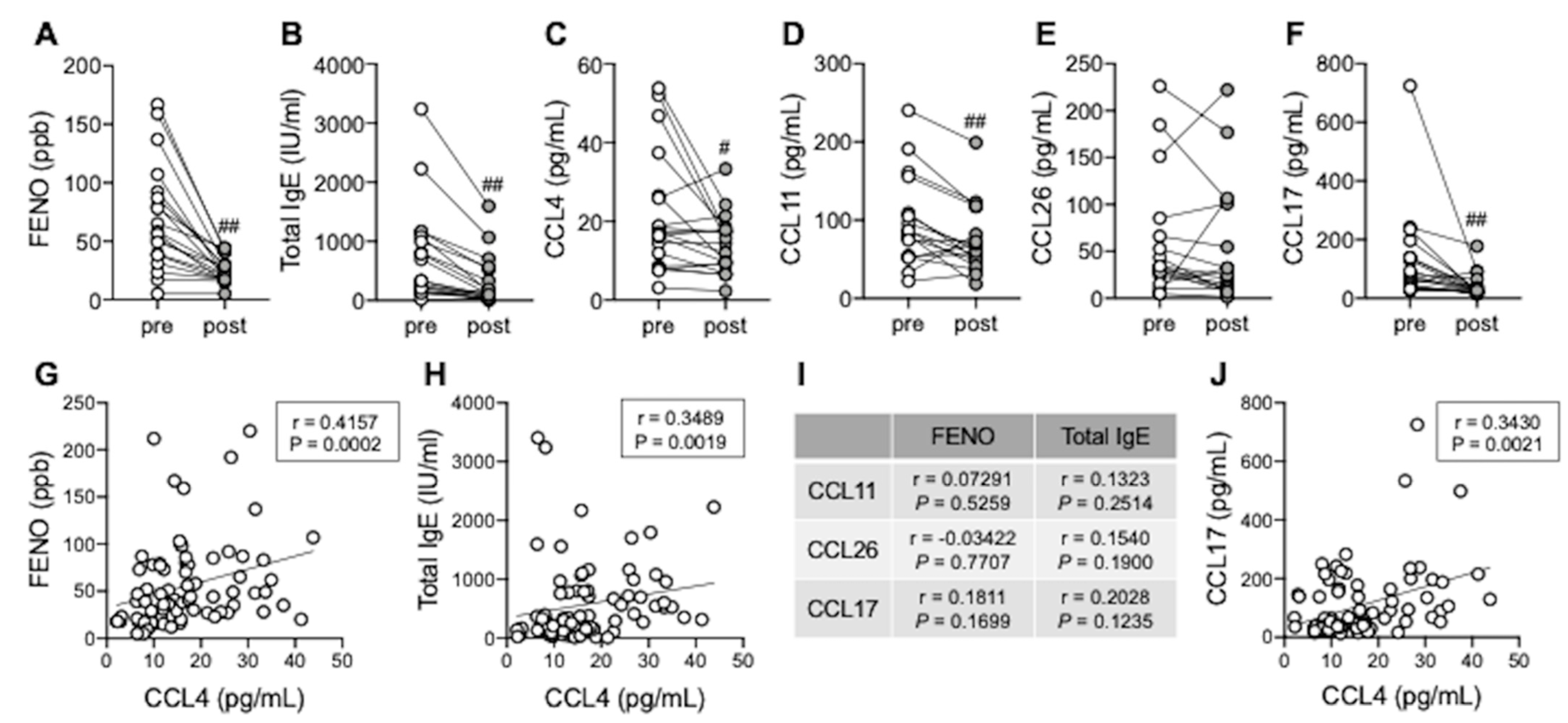
3.5. Serum CCL4 Correlates with Type 2 Inflammation Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Couillard, S.; Jackson, D.J.; Wechsler, M.E.; Pavord, I.D. Workup of Severe Asthma. Chest 2021, 160, 2019–2029. [Google Scholar] [CrossRef]
- Carr, T.F.; Zeki, A.A.; Kraft, M. Eosinophilic and Noneosinophilic Asthma. Am. J. Respir. Crit. Care Med. 2018, 197, 22–37. [Google Scholar] [CrossRef]
- Nagase, H. Severe asthma in Japan. Allergol. Int. 2019, 68, 167–171. [Google Scholar] [CrossRef]
- Fujieda, S.; Imoto, Y.; Kato, Y.; Ninomiya, T.; Tokunaga, T.; Tsutsumiuchi, T.; Yoshida, K.; Kidoguchi, M.; Takabayashi, T. Eosinophilic chronic rhinosinusitis. Allergol. Int. 2019, 68, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Amelink, M.; de Groot, J.C.; de Nijs, S.B.; Lutter, R.; Zwinderman, A.H.; Sterk, P.J.; ten Brinke, A.; Bel, E.H. Severe adult-onset asthma: A distinct phenotype. J. Allergy Clin. Immunol. 2013, 132, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, T.; Sakashita, M.; Haruna, T.; Asaka, D.; Takeno, S.; Ikeda, H.; Nakayama, T.; Seki, N.; Ito, S.; Murata, J.; et al. Novel scoring system and algorithm for classifying chronic rhinosinusitis: The JESREC Study. Allergy 2015, 70, 995–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Suzukawa, M.; Inoue, N.; Imai, S.; Akazawa, M.; Matsui, H. Real-world benefits of biologics for asthma: Exacerbation events and systemic corticosteroid use. World Allergy Organ. J. 2021, 14, 100600. [Google Scholar] [CrossRef]
- Fujieda, S.; Matsune, S.; Takeno, S.; Ohta, N.; Asako, M.; Bachert, C.; Inoue, T.; Takahashi, Y.; Fujita, H.; Deniz, Y.; et al. Dupilumab efficacy in chronic rhinosinusitis with nasal polyps from SINUS-52 is unaffected by eosinophilic status. Allergy 2022, 77, 186–196. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kanda, A.; Yun, Y.; Dan Van, B.; Suzuki, K.; Sawada, S.; Asako, M.; Iwai, H. Reduced Local Response to Corticosteroids in Eosinophilic Chronic Rhinosinusitis with Asthma. Biomolecules 2020, 10, 326. [Google Scholar] [CrossRef] [Green Version]
- Bisset, L.R.; Schmid-Grendelmeier, P. Chemokines and their receptors in the pathogenesis of allergic asthma: Progress and perspective. Curr. Opin. Pulm. Med. 2005, 11, 35–42. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Konno, Y.; Kanda, A.; Yamada, Y.; Yasuba, H.; Sakata, Y.; Fukuchi, M.; Tomoda, K.; Iwai, H.; Ueki, S. Critical role of CCL4 in eosinophil recruitment into the airway. Clin. Exp. Allergy 2019, 49, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kanda, A.; Bui, D.V.; Yun, Y.; Nguyen, L.M.; Chu, H.H.; Mitani, A.; Suzuki, K.; Asako, M.; Iwai, H. Omalizumab Restores Response to Corticosteroids in Patients with Eosinophilic Chronic Rhinosinusitis and Severe Asthma. Biomedicines 2021, 9, 787. [Google Scholar] [CrossRef] [PubMed]
- Hastie, A.T.; Steele, C.; Dunaway, C.W.; Moore, W.C.; Rector, B.M.; Ampleford, E.; Li, H.; Denlinger, L.C.; Jarjour, N.; Meyers, D.A.; et al. Complex association patterns for inflammatory mediators in induced sputum from subjects with asthma. Clin. Exp. Allergy 2018, 48, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Liao, B.; Cao, P.P.; Zeng, M.; Zhen, Z.; Wang, H.; Zhang, Y.N.; Hu, C.Y.; Ma, J.; Li, Z.Y.; Song, J.; et al. Interaction of thymic stromal lymphopoietin, IL-33, and their receptors in epithelial cells in eosinophilic chronic rhinosinusitis with nasal polyps. Allergy 2015, 70, 1169–1180. [Google Scholar] [CrossRef]
- Ramu, S.; Calvén, J.; Michaeloudes, C.; Menzel, M.; Akbarshahi, H.; Chung, K.F.; Uller, L. TLR3/TAK1 signalling regulates rhinovirus-induced interleukin-33 in bronchial smooth muscle cells. ERJ Open Res. 2020, 6, 00147–2020. [Google Scholar] [CrossRef]
- Hwang, J.W.; Lee, K.J.; Choi, I.H.; Han, H.M.; Kim, T.H.; Lee, S.H. Decreased expression of type I (IFN-β) and type III (IFN-λ) interferons and interferon-stimulated genes in patients with chronic rhinosinusitis with and without nasal polyps. J. Allergy Clin. Immunol. 2019, 144, 1551–1565. [Google Scholar] [CrossRef] [Green Version]
- Golebski, K.; van Tongeren, J.; van Egmond, D.; de Groot, E.J.; Fokkens, W.J.; van Drunen, C.M. Specific Induction of TSLP by the Viral RNA Analogue Poly(I:C) in Primary Epithelial Cells Derived from Nasal Polyps. PLoS ONE 2016, 11, e0152808. [Google Scholar] [CrossRef]
- Porsbjerg, C.; Nieto-Fontarigo, J.J.; Cerps, S.; Ramu, S.; Menzel, M.; Hvidtfeldt, M.; Silberbrandt, A.; Froessing, L.; Klein, D.; Sverrild, A.; et al. Phenotype and severity of asthma determines bronchial epithelial immune responses to a viral mimic. Eur. Respir. J. 2021, in press. [Google Scholar] [CrossRef]
- Kopecka, J.; Rozkova, D.; Sediva, A. Plasmacytoid DCs, exposed to TSLP in synergy with TLR ligands, acquire significant potential towards Th2 polarization. Med. Sci. Monit. Basic Res. 2013, 19, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Rabe, K.F.; Nair, P.; Brusselle, G.; Maspero, J.F.; Castro, M.; Sher, L.; Zhu, H.; Hamilton, J.D.; Swanson, B.N.; Khan, A.; et al. Efficacy and Safety of Dupilumab in Glucocorticoid-Dependent Severe Asthma. N. Engl. J. Med. 2018, 378, 2475–2485. [Google Scholar] [CrossRef] [PubMed]
- Kam, J.C.; Szefler, S.J.; Surs, W.; Sher, E.R.; Leung, D.Y. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J. Immunol. 1993, 151, 3460–3466. [Google Scholar] [PubMed]
- Okada, N.; Fukagawa, K.; Takano, Y.; Dogru, M.; Tsubota, K.; Fujishima, H.; Matsumoto, K.; Nakajima, T.; Saito, H. The implications of the upregulation of ICAM-1/VCAM-1 expression of corneal fibroblasts on the pathogenesis of allergic keratopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4512–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, C.D.; Gundel, R.H.; Reilly, P.; Haynes, N.; Letts, L.G.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of asthma. Science 1990, 247, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, K.; Nagata, M. Involvement and Possible Role of Eosinophils in Asthma Exacerbation. Front. Immunol. 2018, 9, 2220. [Google Scholar] [CrossRef]
- Weston, C.A.; Rana, B.M.J.; Cousins, D.J. Differential expression of functional chemokine receptors on human blood and lung group 2 innate lymphoid cells. J. Allergy Clin. Immunol. 2019, 143, 410–413. [Google Scholar] [CrossRef] [Green Version]
- Di Sciascio, M.B.; Vianale, G.; Verna, N.; Petrarca, C.; Perrone, A.; Toniato, E.; Muraro, R.; Conti, P.; Di Gioacchino, M. Eosinophil recruiting chemokines are down-regulated in peripheral blood mononuclear cells of allergic patients treated with deflazacort or desloratadine. Int. J. Immunopathol. Pharmacol. 2007, 20, 745–751. [Google Scholar] [CrossRef]
- Suzukawa, M.; Ohshima, N.; Tashimo, H.; Asari, I.; Kobayashi, N.; Shoji, S.; Tohma, S.; Ohta, K. A Low Serum CCL4/MIP-1β Level May Predict a Severe Asthmatic Responsiveness to Mepolizumab. Intern. Med. 2020, 59, 2849–2855. [Google Scholar] [CrossRef]
- Goleva, E.; Hauk, P.J.; Hall, C.F.; Liu, A.H.; Riches, D.W.; Martin, R.J.; Leung, D.Y. Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. J. Allergy Clin. Immunol. 2008, 122, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Brusselle, G.G.; Koppelman, G.H. Biologic Therapies for Severe Asthma. N. Engl. J. Med. 2022, 386, 157–171. [Google Scholar] [CrossRef]
- Kavanagh, J.E.; Hearn, A.P.; Jackson, D.J. A pragmatic guide to choosing biologic therapies in severe asthma. Breathe 2021, 17, 210144. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, Y.; Chu, H.H.; Kanda, A.; Yun, Y.; Shimono, M.; Nguyen, L.M.; Mitani, A.; Suzuki, K.; Asako, M.; Iwai, H. CCL4 Functions as a Biomarker of Type 2 Airway Inflammation. Biomedicines 2022, 10, 1779. https://doi.org/10.3390/biomedicines10081779
Kobayashi Y, Chu HH, Kanda A, Yun Y, Shimono M, Nguyen LM, Mitani A, Suzuki K, Asako M, Iwai H. CCL4 Functions as a Biomarker of Type 2 Airway Inflammation. Biomedicines. 2022; 10(8):1779. https://doi.org/10.3390/biomedicines10081779
Chicago/Turabian StyleKobayashi, Yoshiki, Hanh Hong Chu, Akira Kanda, Yasutaka Yun, Masami Shimono, Linh Manh Nguyen, Akitoshi Mitani, Kensuke Suzuki, Mikiya Asako, and Hiroshi Iwai. 2022. "CCL4 Functions as a Biomarker of Type 2 Airway Inflammation" Biomedicines 10, no. 8: 1779. https://doi.org/10.3390/biomedicines10081779
APA StyleKobayashi, Y., Chu, H. H., Kanda, A., Yun, Y., Shimono, M., Nguyen, L. M., Mitani, A., Suzuki, K., Asako, M., & Iwai, H. (2022). CCL4 Functions as a Biomarker of Type 2 Airway Inflammation. Biomedicines, 10(8), 1779. https://doi.org/10.3390/biomedicines10081779