Abstract
Lysine acetylation is a highly conserved mechanism that affects several biological processes such as cell growth, metabolism, enzymatic activity, subcellular localization of proteins, gene transcription or chromatin structure. This post-translational modification, mainly regulated by lysine acetyltransferase (KAT) and lysine deacetylase (KDAC) enzymes, can occur on histone or non-histone proteins. Several studies have demonstrated that dysregulated acetylation is involved in cardiac dysfunction, associated with metabolic disorder or heart failure. Since the prevalence of obesity, type 2 diabetes or heart failure rises and represents a major cause of cardiovascular morbidity and mortality worldwide, cardiac acetylation may constitute a crucial pathway that could contribute to disease development. In this review, we summarize the mechanisms involved in the regulation of cardiac acetylation and its roles in physiological conditions. In addition, we highlight the effects of cardiac acetylation in physiopathology, with a focus on obesity, type 2 diabetes and heart failure. This review sheds light on the major role of acetylation in cardiovascular diseases and emphasizes KATs and KDACs as potential therapeutic targets for heart failure.
1. Introduction
Obesity is a worldwide epidemic associated with several public health challenges including type 2 diabetes, hypertension, obstructive sleep apnea, dyslipidemia and cardiovascular disease, and notably heart failure. The prevalence of these disorders (e.g., obesity, type 2 diabetes and heart failure) is still increasing and heart failure remains the most common cause of cardiovascular morbidity and mortality in the world [1]. Interestingly, although heart failure could provoke systolic or diastolic cardiac dysfunction, obesity and type 2 diabetes are more often characterized by cardiac hypertrophy and diastolic dysfunction [2]. Moreover, diastolic heart failure remains one of the more challenging diseases to treat and, thus, a better understanding of the physio(patho)logical mechanisms involved in metabolic heart disease is necessary to propose new pharmacological therapies. In this context, epigenetic or protein post-translational modifications, including phosphorylation, methylation, O-GlcNAcylation, ubiquitylation, and acetylation, could represent interesting targets since they modulate gene or protein expression during metabolic diseases [3].
Protein acetylation is a highly conserved mechanism that was first described 60 years ago, in which an acetyl group is covalently attached to the ε-amino group of lysine residues [4]. This modification induces important changes to the protein structure at its lysine residue, by altering its charge status and adding an extra structural moiety [5]. These post-translational modifications can regulate protein–protein interactions, stability and function. Acetylation was first described to regulate chromatin structure and histone activity, associated to epigenetic-mediated gene regulation [4]. Since then, several proteomic analyses enabled to identify more than a thousand acetylation sites in both histone and non-histone proteins [6,7,8], and, interestingly, revealed that the subcellular distribution of acetylation is tissue dependent [9]. Focusing on cardiovascular and metabolic diseases, Table 1 indicates some of the most common proteins modified by cardiac acetylation.

Table 1.
Most common proteins modified by cardiac acetylation in cardiovascular and metabolic diseases.
2. Cardiac Acetylation
2.1. Regulation of Lysine Acetylation
Lysine acetylation is a reversible mechanism that is regulated by the dynamic actions of lysine acetyltransferase (KATs) and lysine deacetylase (KDACs) enzymes [43]. There are three categories of KATs: the MYST family, the Gcn-5-related N-acetyltransferases (GNATs) and the E1A-associated protein of 300 kDa/CREB binding protein (p300/CBP) family [44]. KDACs are broadly categorized into four classes based on function and DNA sequence similarity. Class I (reduced potassium dependency 3 family), II (histone deacetylase 1 family) and IV (HDAC11) are considered as classical KDACs with zinc-dependent active sites, whereas class III enzymes are a family of silent information regulator 2-like nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases/mono-ADP-ribosyl transferases known as sirtuins 1 to 7 [44]. Interestingly, the level of expression of KATs and KDACs follows a specific tissue distribution, suggesting specific functions in these different organs [45]. For example, in physiological conditions, KATs are weakly expressed in vascular tissue, except for the lysine acetyltransferase KAT2B, which is highly expressed [45]. Conversely, a database mining approach analyzing the expression profile of 164 enzymes in human and murine tissues highlighted heart as tissue in which KATs and KDACs are in the highest varieties [45]. In addition to tissue-specific expression, both KATs and KDACs have precise subcellular localizations related to their specific targets (Figure 1) and can regulate each other. For example, cardiomyocyte overexpression of SIRT6 decreases P300 levels, where SIRT6 promotes ubiquitination and degradation of P300 [46]. Moreover, cardiomyocyte overexpression of SIRT3 increases SIRT1 expression [27].
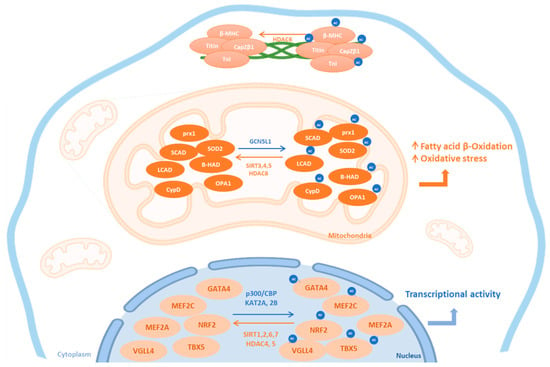
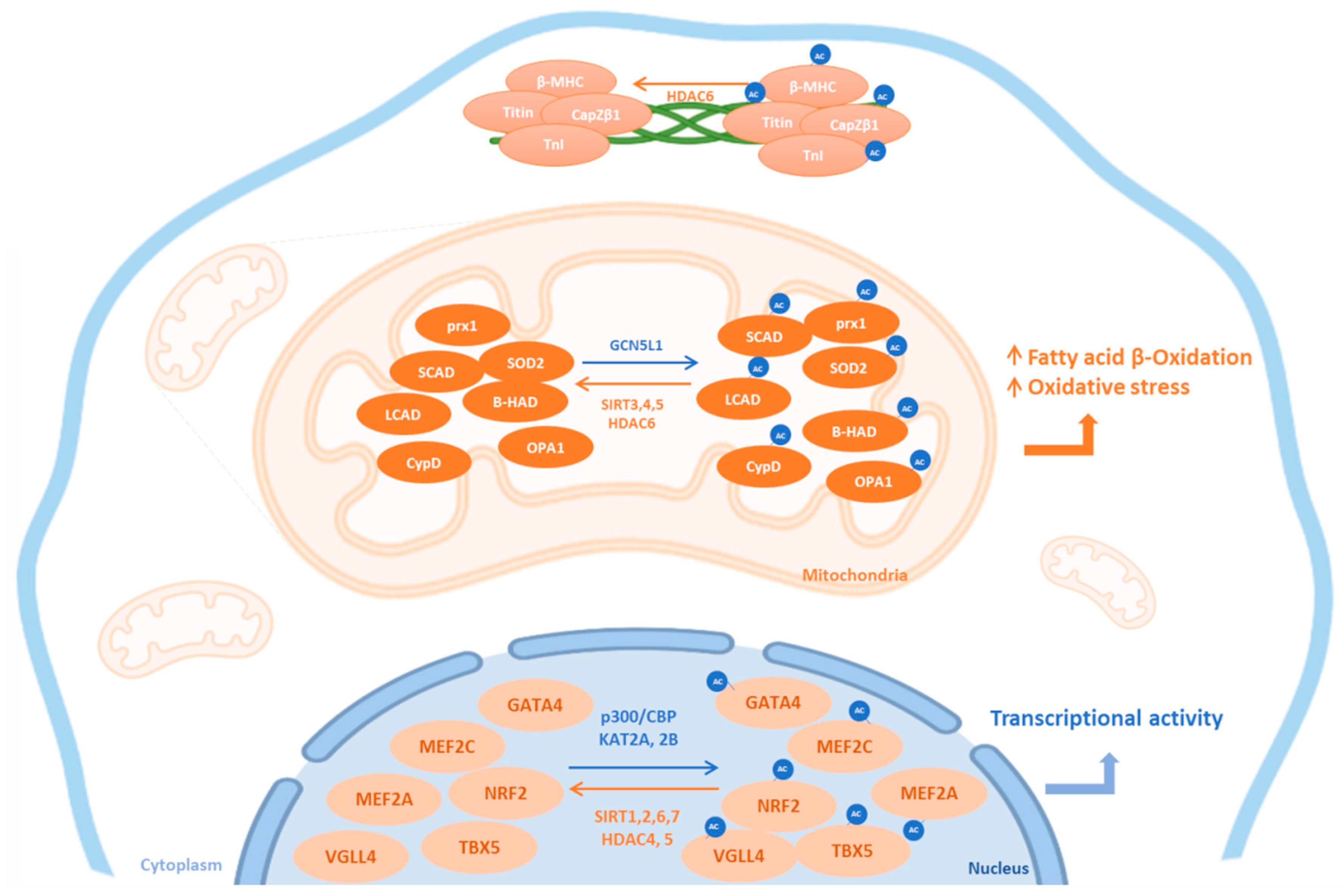
Figure 1.
Subcellular localization of cardiac KATs and KDACs. This figure summarizes the subcellular localization of the most common lysine acetyltransferases (KATs, blue) and lysine deacetylases (KDAC, orange) and their cardiac targets. Ac: acetylated form; HDAC: histone deacetylase; SIRT: sirtuins; β-MHC: beta-myosin heavy chain; TnI: Troponin I; prx1: peroxiredoxin 1; LCAD: long chain acyl CoA dehydrogenase; SCAD: short chain acyl CoA dehydrogenase; β-HAD: L-3-hydroxy acyl-CoA dehydrogenase; SOD2: superoxide dismutase 2; CypD: Cyclophilin D; OPA1: optic atrophic 1; TBX5: T-Box transcription factor 5; VGLL4: vestigial-like 4; GATA4: GATA-binding factor 4; MEF: myocyte enhancer factor; Nrf2: nuclear factor erythroid-2-related factor 2; GCN5L1: general control of amino acid synthesis 5 like-1; CBP: CREB binding protein.
In addition, cofactors play an essential role to modulate acetylation. Several short-chain acyl-CoA products, such as acetyl-coenzyme A (acetyl-CoA), are important metabolite intermediates generated during catabolism of various energy fuels that modulate cardiac acetylation. Indeed, acetyl-CoA acts as a substrate for KATs. Moreover, auto-acetylation of mitochondrial proteins can occur efficiently at the pH of the matrix (pH ≥ 7.5) in actively respiring mitochondria [9]. On the other hand, levels of NAD+, an important cofactor for sirtuins, may impact protein acetylation [5]. Indeed, NAD+ levels, synthesized from metabolic pathways, connect their enzymatic activity to metabolism and provide a metabolic link between the sirtuins activity, energy homeostasis and stress responses.
2.2. Physiological Roles of Lysine Acetylation
Lysine acetylation can modulate cell growth, metabolism, protein–protein interactions, protein stability, subcellular localization, gene transcription, chromatin structure or enzymatic activity [6,7]. This post-translational modification plays an essential role in cardiac physiology (Figure 2).
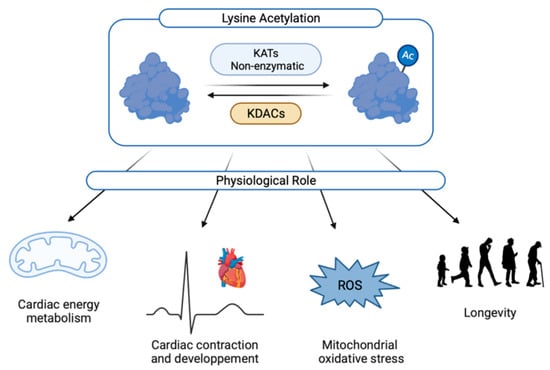
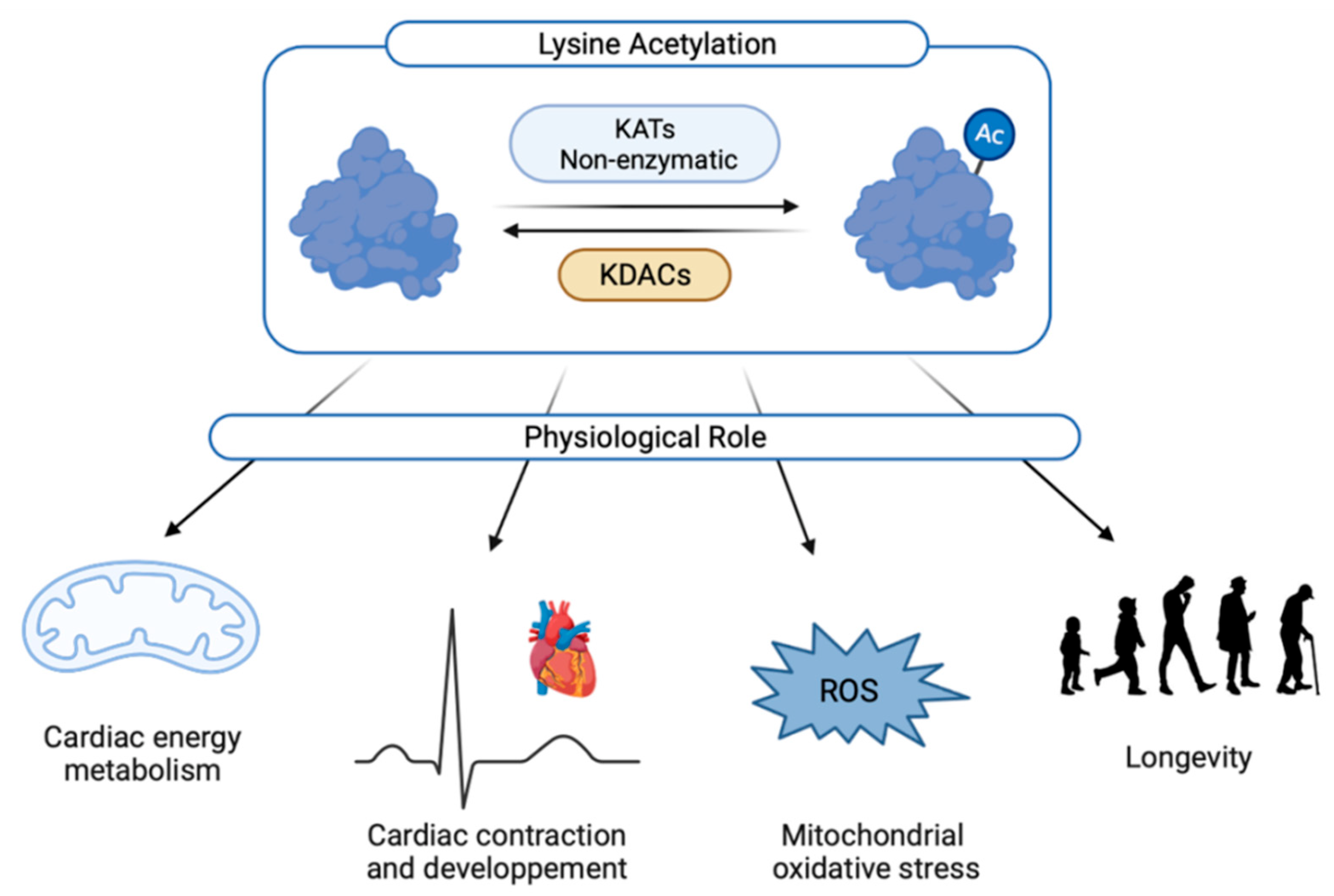
Figure 2.
Physiological roles of cardiac acetylation. Ac: acetylated form; KATs: lysine acetyltransferases; KDACs: lysine deacetylase; ROS: reactive oxygen species.
2.2.1. Heart Development and Cardiac Ageing
Acetylation can regulate embryonic heart development and cardiac progenitor fate [47,48]. TBX5, a T-box family transcription factor, is involved in heart and forelimb development and is acetylated by KAT2A and KAT2B at lysine 339, leading to stimulate its transcriptional activity [18]. Knockdown of KAT2A and KAT2B in zebrafish severely impairs heart development and induces pericardial oedema by inhibiting TBX5 [18]. Besides KATs, the lysine deacetylase enzymes HDAC4 and HDAC5 are also able to interact with TBX5 to exert a repressive role on cardiac genes transcription through deacetylation [19]. Furthermore, TBX1, another T-box family transcription factor expressed in cardiac progenitors, represses myocyte enhancer factor 2c (Mef2c) gene expression by reducing histone 3 acetylation on lysine 27 [48]. Moreover, acetylation of vestigial-like 4 (VGLL4) at K225 by P300 negatively regulates its binding to the transcription factor TEA Domain Transcription Factor 1 (TEAD1), leading to decreased neonatal cardiomyocytes proliferation and cardiomyocytes necrosis [20]. Inhibition of HDAC1 was also described to decrease the proliferation cardiomyocyte in zebrafish [49]. Indeed, mutation in HDAC1 gene that induces protein instability is associated with decreased cardiomyocyte proliferation, suggesting an important role of HDAC1 during heart growth [49].
In addition, sirtuins are well-described regulators of aging and have been associated with longevity [50,51,52]. Indeed, mice deficient for SIRT1 [53,54], SIRT3 [55] or SIRT6 [56] display a shorter life span with severe cardiac damage, such as hypertrophy and fibrosis. SIRT1 is well known to be cardioprotective by decreasing oxidative stress and inflammation, which promotes cardiomyocyte survival [54]. Moreover, SIRT3 is required for cardiomyocyte survival under several stress conditions such as serum starvation, genotoxic and oxidative stress [57]. Altogether, these studies demonstrate that KDACs and KATs exert crucial functions during heart development and cardiac ageing.
2.2.2. Cardiac Contraction
Recently, reversible acetylation of sarcomeric proteins has been described as a mechanism regulating cardiac function. The comparison of lysine acetylation patterns from rats as well as from human skeletal muscle biopsies revealed that 80% of the proteins involved in muscle contraction were acetylated [9]. Moreover, proteomic analyses suggested that HDAC6 is localized in Z-disks and acts as a sarcomeric protein desacetylase [58]. Among them, acetylation can impact the β-myosin heavy chain (lysine 34, lysine 58, lysine 213, lysine 429, lysine 951 and lysine 1195) [31], titin [33], CapZβ1 (lysine 199) [34] or cardiac troponin I [35], directly affecting cardiac contraction.
2.2.3. Role of Lysine Acetylation in Cardiac Energy Metabolism and Mitochondrial Activity
Proteomic analyses suggested that the subcellular distribution of lysine-acetylated proteins is tissue dependent [9]. For example, the heart and muscles, both high energy-demanding organs, are tissues with the largest fraction of mitochondrial protein acetylation [9]. Calorie restriction or changes in nutrition specifically affect the mitochondrial acetylome, but not the cytosol or nucleus [59], suggesting different roles for lysine acetylation in the mitochondria, nucleus and cytoplasm. Indeed, mitochondrial lysine acetylation was described to modulate cell metabolism by regulating fatty acid β-oxidation, the tricarboxylic acid cycle, the urea cycle, and oxidative phosphorylation [60].
In the early newborn period, an important change in myocardial energy substrate metabolism occurs with an increase in fatty acid β-oxidation [47], associated with an increase in long chain acyl CoA dehydrogenase (LCAD) and L-3-hydroxy acyl-CoA dehydrogenase (β-HAD) acetylation and activation [12]. At the cardiac level, mitochondrial general control of amino acid synthesis 5 like-1 (GCN5L1) triggers LCAD and β-HAD acetylation [10,11] whereas SIRT3 deacetylates both LCAD and β-HAD [13,14]. The implication of GCN5L1 in cardiac metabolism was confirmed by genetic deletion approaches. Indeed, knockdown of GCN5L1 in H9c2 cardiomyoblasts decreases maximal mitochondrial respiration and activity of proteins involved in fatty acid oxidation [12].
However, there is no consensus about the effects of lysine acetylation on fatty acid β-oxidation. Indeed, some studies suggested an inhibitory effect [43] whereas others suggested a stimulatory effect [13,14]. As an example, it was reported that an impaired fatty acid β-oxidation was linked to a reduced acetylation and activity of LCAD in the liver [13] or heart [14] of SIRT3 knockout mice. On the other hand, cardiac mitochondrial protein hyperacetylation induced by SIRT3 deletion is associated with an increased fatty acid β-oxidation rates [43]. This discrepancy could be due to organ, physio(patho)logical status (e.g., diabetes, obesity or newborn period) or acetylated proteins themselves, and are well described in the following review [43].
The implication of sirtuins in metabolic regulation is well detailed in a recent review [61]. Indeed, SIRT6 was described as a gatekeeper of glucose metabolism in cardiomyocytes. SIRT6 partial depletion decreased mitochondrial respiration whereas SIRT6 overexpression enhanced basal oxygen consumption [62]. Conversely, fatty acid uptake increases in SIRT6-deficient cardiomyocytes and is decreased in SIRT6-overexpressed cardiomyocytes [63]. Of note, proteins involved in mitochondrial biogenesis can also be acetylated, such as optic atrophy 1 (OPA1) through SIRT3 which is able to activate OPA1 by deacetylation of the residues lysine 926 and lysine 931 [15]. Finally, a cross-talk between SIRT1, peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) and AMP-activated protein kinase (AMPK) has been described to regulate cardiac metabolism [54]. Indeed, PGC-1α is a transcription factor playing a central role in the regulation of cellular energy metabolism, mitochondrial biogenesis and oxidative phosphorylation. SIRT1 could interact with PGC-1α and increase its expression levels [16]. Moreover, AMPK increases cellular NAD+ levels and subsequently increases SIRT1 activity, resulting in PGC-1α deacetylation and activation [64].
2.2.4. Mitochondrial Oxidative Stress
It is well described that several mitochondrial proteins involved in oxidative stress are acetylated in the heart [60]. Oxidative stress is defined by a production of reactive oxygen species (ROS) higher than anti-oxidant capacities. Indeed, although different molecular processes may contribute to global oxidative stress, the majority of ROS originates from the mitochondrial compartment in the heart. Excessive ROS production occurs during mitochondrial dysfunction and induces irreversible damage to mitochondria, defining them as significant contributors to the development of cardiovascular disease [65,66]. In this context, we recently described that SIRT3 deacetylates and activates the mitochondrial superoxide dismutase 2 (SOD2), but there is a decreased interaction between SIRT3 and SOD2 induced SOD2 acetylation on lysine 68 and its subsequent inactivation, leading to mitochondrial oxidative stress and dysfunction in hypertrophied cardiomyocytes [27]. Moreover, SIRT3 inhibition increased oxidative stress in neonatal rat cardiomyocytes [27] or human aortic endothelial cells [26].
Other anti-oxidant enzymes were described to be acetylated in the heart, such as peroxiredoxin 1 [28] in the mitochondria or nuclear factor erythroid 2-related factor 2 [29] in the nucleus.
3. Implication of Cardiac Acetylation in Metabolic Heart Disease
Post-translational acetylation triggers modification of the activity of several proteins that occurs in obesity, diabetes and early stage of heart failure as detailed below [67,68]. Table 2 summarizes the cardiac modulation of KATs and KDACs expression levels reported in metabolic heart disease.
Table 2.
Modulation of KATs and KDACs expression in heart failure and metabolic diseases.
3.1. Cardiac Hypertrophy
Cardiac hypertrophy is a consequence of genetic, mechanic or neurohormonal changes contributing to heart failure progression. Interestingly, total KAT activity was found to be increased in the hearts of mice subjected to phenylephrine (PE), an hypertrophic stimulus [23]. In this context, the increased expression of P300 and KAT2B acetylases may explain hypertrophied cardiomyocytes under PE [23,25,46,69]. In addition, PE-induced hypertrophy also decreases SIRT6 expression, leading to an increase in histone H3 acetylation on lysine 9 (H3K9) [25,46,69]. Interestingly, PE-induced hypertrophy is decreased by treatment with polyunsaturated fatty acid [69] or anacardic acid [23] in association with a decreased H3K9 acetylation. In parallel, overexpression of P300 induces an increased size of cardiomyocytes and a modification of their myofibrillar organization [69], whereas inhibition of P300 by siRNA attenuates hypertrophic response in neonatal rat cardiomyocytes [46]. On the other hand, overexpression of SIRT6 [46] or SIRT3 [27] decreased cardiomyocytes hypertrophy. As an example, NAD inhibits oxidative stress and hypertrophy induced by PE in cardiomyocytes or by angiotensin II in mice [36]. Indeed, NAD requires SIRT3 to deacetylate and activate liver kinase B1 (LKB1) and its target AMPK [36]. Furthermore, a decreased SIRT2 protein expression was reported in hypertrophic hearts from mice whereas cardiac-specific SIRT2 overexpression protected the hearts against angiotensin II–induced hypertrophy [37]. As shown for SIRT3, SIRT2 deacetylates LKB1 at lysine 48, these promote the phosphorylation of LKB1 and the subsequent activation of AMPK signaling [37]. Moreover, SIRT7 levels increase in myocardial tissues after pressure overload induced by transverse aortic constriction in mice [21]. Cardiomyocyte-specific deletion of SIRT7 exacerbates hypertrophy and fibrosis induced by transverse aortic constriction, suggesting an antihypertrophic role of SIRT7 [21]. Indeed, SIRT7 deacetylates the hypertrophy response transcription factors such as GATA-binding factor 4 (GATA4) in cardiomyocytes [21]. Conversely, P300 acetylates GATA4 and activates its DNA binding activity, inducing cardiac hypertrophy and heart failure [22].
Finally, HDAC3 expression and activity increase during hypertrophy [80] whereas a decreased binding of HDAC3 to myofibrils is observed in PE-induced hypertrophy in neonatal rat cardiomyocytes that increases the acetylation of CapZβ1 at lysine 199 and alters myofibril growth during cardiac hypertrophy [34].
3.2. Cardiac Fibrosis
Cardiac fibrosis, defined as an excessive deposition of extracellular matrix in the cardiac muscle, is an important contributor to several heart diseases. Interestingly, in isoproterenol-injected mice, KAT2B activity was found to be increased only in cardiac fibroblasts, whereas no effects were observed in cardiomyocytes isolated from these mice [39]. In this model, KAT2B induces acetylation of SMAD2 and, subsequently, activates SMAD2 and the transforming growth factor β signaling pathway [39]. SMAD3 could also be acetylated during cardiac fibrosis [41,42]. Activation of SIRT1 by resveratrol [41], nicotinamide mononucleotide [42] or geniposide [70] could decrease SMAD3 acetylation and fibrosis. Moreover, inhibition of P300 appears to exert anti-fibrotic functions [82].
Decreased α-tubulin acetylation and increased HDAC6 were also described in cardiac fibroblasts or the heart from isoproterenol-induced fibrosis. Interestingly, treatment of cardiac fibroblasts with tubastatin A, an HDAC6 inhibitor, restored α-tubulin acetylation and decreased fibrosis [83].
3.3. Heart Failure
Cardiac hypertrophy and fibrosis are both involved in heart failure development by activation of several intracellular signaling pathways, leading to left ventricular remodeling with systolic dysfunction. Despite the best modern therapeutic management, left ventricular remodeling remains independently associated with heart failure and cardiovascular death at long-term follow-up after myocardial infarction [84]. Moreover, alterations in cardiac energy metabolism, both in terms of changes in energy substrate preference and decreased mitochondrial oxidative metabolism and ATP production, are key contributors to heart failure development [5].
An increase in P300 expression is observed during ischemia/reperfusion injury [85] and myocardial infarction [69]. Interestingly, it was described that activation of P300 acetylase, leading to an increase in acetylated H3K9 induced by myocardial infarction, is reversed by treatment with polyunsaturated fatty acid [69]. Indeed, eicosapentaenoic acid and docosahexaenoic acid directly blocks the histone acetyltransferase activity of P300 and subsequently reduces both hypertrophy and fibrosis induced by myocardial infarction in rats [69]. Other P300 inhibitors were also described to be cardioprotective, such as Ecklonia stolonifera Okamura extract [86], curcumin [87] and metformin [88]. Moreover, KAT2B levels are also increased by ischemia/reperfusion injury [73].
The expression and activity of sirtuins are also highly impacted during heart failure. As an example, a decrease in SIRT3 and SIRT6 expressions were observed in human failing hearts that induces a global increase in protein acetylation [63,77]. Proteomic analysis also identified an increased acetylation of mitochondrial proteins induced by transverse aortic constriction [89].
Inhibition of SIRT3 was also described to induce mitochondrial oxidative stress and hypertrophy, notably by increasing the inactive form of SOD2 (acetylated on lysine 68) in hypertrophy [27] or following ischemia/reperfusion [90]. Moreover, deletion of SIRT2 exacerbates cardiac hypertrophy and fibrosis and decreases cardiac ejection fraction and fractional shortening in angiotensin II-infused mice by inhibition of AMPK activation, whereas cardiac-specific SIRT2 overexpression reversed this phenotype [37]. SIRT1 expression and activity are decreased in the heart following ischemia/reperfusion [30]. SIRT1 inhibition was also reported to increase endoplasmic reticulum stress-induced cardiac injury by decreasing eukaryotic initiation factor 2 alpha (eIF2α) deacetylation on lysine 143 in cardiomyocytes and in adult-inducible SIRT1 knockout mice [91].
Acetylation of contractile proteins is also modified during heart failure. For example, acetylation of β-MHC on lysine 951 was decreased in both ischemic and non-ischemic failing hearts [31].
3.4. Obesity
Obesity is a major cause of disability and is often associated with cardiac hypertrophy, fibrosis, type 2 diabetes, obstructive sleep apnea and alteration of cardiac metabolism. It has notably described an increase in genes involved in fatty acid oxidation [11]. Moreover, several studies have suggested an increase in acetylation raised from the non-enzymatic reaction of high levels of acetyl-CoA generated during a high-fat diet (HFD) and obesity [5]. Furthermore, hyperacetylation of mitochondrial proteins and metabolic inflexibility was reported in response to HFD or obesity. SIRT3 expression is decreased in left ventricles of obese patients and is associated with an increased level of brain natriuretic peptide (BNP), a marker of cardiac dysfunction and of protein acetylation [17]. It was also reported that exposure of cardiomyoblasts H9c2 to palmitate led to an increase in both SIRT3 and GCN5L1 RNA levels [11]. At the metabolic level, HFD induced an increase in SCAD and LCAD acetylation and activity [11]. Moreover, the acetylation level of α-tubulin on lysine 40 is increased in the hearts of HFD mice and a pharmacological activation of α-tubulin acetylation decreases glucose transport [92].
In the other hand, cardiac specific inhibition of SIRT6 in mice exposed to HFD-induced cardiac hypertrophy and lipid accumulation [93]. In this context, SIRT6 activated the expression of endonuclease G and SOD2, that could decrease oxidative stress and hypertrophy [93].
3.5. Type 2 Diabetes
As obesity, type 2 diabetes is also associated with several cardiac dysfunctions such as cardiac hypertrophy and fibrosis, and acetylation may be involved in these mechanisms. Indeed, SIRT1 expression and activity are decreased in the heart of diabetic rats, induced by HFD and streptozotocin injection and, conversely, an up-regulation of SIRT1 by adenovirus attenuates cardiac dysfunction and oxidative stress [30]. Moreover, in a model of sucrose-fed rats, cardiac dysfunction is associated with a decreased SIRT3 protein expression (despite an increase at RNA level) and an increased GCN5L1 (protein and RNA) and mitochondrial protein acetylation [11,17]. Decreased SIRT3 levels are also associated with an increased acetylation of SOD2, and an increased oxidative stress and apoptosis in heart of diabetic mice [79]. Interestingly, exposure of cardiomyoblasts H9c2 to high glucose concentration decreased SIRT3 [78] and increased GCN5L1, oxidative stress and autophagy mediated by cytoplasmic Forkhead box O1 (FOXO1) acetylation [11,76]. Conversely, expression of GCN5L1 is decreased in hearts of diabetic ZSF1 rats, a model characterized by obese animals with hyperglycemia, hyperinsulinemia and cardiac dysfunction [10]. This decreased GCN5L1 expression and activity, with no modulation of SIRT3, induced a decrease of short- and long-chain acyl CoA dehydrogenases and a reduced respiratory capacity [10]. One explanation could be the transition from prediabetes, in which GCN5L1 becomes elevated to promote mitochondrial fatty acids oxidation, to an overt diabetic state, in which GCN5L1 expression is downregulated leading to an overall decrease in mitochondrial fuel oxidation [10].
On the other hand, OVE26 mice, a mouse model of type 1 diabetes, develop cardiac dysfunction with increased left ventricle diameters and fibrosis and decreased ejection fraction associated with an increase in HDAC3 activity, oxidative stress and inflammation [81]. Interestingly, the selective HDAC3 inhibitor RGFP966 reversed the cardiac phenotype in these mice [81]. In another model of type 1 diabetes, injection of streptozotocin in rats induced cardiac dysfunction, cardiac hypertrophy, fibrosis and inflammation [72] as well as oxidative stress [28]. This oxidative stress is due to an increased HDAC6 activity leading to a decrease in the acetylated form of peroxiredoxin 1, suggesting that acetylation of peroxiredoxin 1 is involved in this activation [28]. In this context, tubastatin A, a highly selective inhibitor of HADC6, may represent an interesting pharmacological target since treatment of diabetic rats with tubastatin A increased acetylation of peroxiredoxin 1 and decreased oxidative stress and cardiac dysfunction [28]. Conversely, SIRT1 expression and activity appear to be decreased in the left ventricles of streptozotocin-induced diabetic rats [72] and mice [38]. In these models, reduced SIRT1 expression is associated with acetylation of endothelial nitric oxide synthase [72] or Akt [38]. In addition, SIRT6 expression is also decreased in the hearts of diabetic mice [63], suggesting that sirtuin family members may play a crucial role in type 1 diabetes-associated cardiac dysfunctions.
3.6. Pharmacological Modulation of Cardiac Acetylation
Due to the major implication of KATs and KDACs in cardiac dysfunction induced by obesity, type 2 diabetes or heart failure, targeting these enzymes could be beneficial for patients.
First, inhibition of P300 appears to be a promising approach to inhibit cardiac hypertrophy and fibrosis induced by several stimuli. As an example, oral administration of eicosapentaenoic acid (1 g/kg) or docosahexaenoic acid (1 g/kg) one week after myocardial infarction preserved fractional shortening and decreased cardiac hypertrophy and perivascular fibrosis [68]. Ecklonia stolonifera Okamura extract, an algae traditionally used in Japanese foods, inhibits PE-induced hypertrophy in neonatal rat cardiomyocytes and restores fractional shortening and reduced cardiac hypertrophy and fibrosis by oral administration one week after myocardial infarction in rats [73]. Oral administration of curcumin (50 mg/kg/day) acts also as a P300 inhibitor and decreases posterior wall thickness, cardiac hypertrophy and perivascular fibrosis induced by hypertension [74]. Finally, metformin, that directly inhibits P300-mediated acetylation of H3K9, blocks PE-induced cardiomyocytes hypertrophy [75].
Based on the cardioprotective effects of SIRT3, it seems essential to develop new therapeutic strategy to increase its expression or activity. Recent reviews have well described these molecules [94,95]. For example, exogenous NAD is able to inhibit hypertrophy induced by PE in cardiomyocytes or by angiotensin II in mice [36]. Resveratrol, an activator of both SIRT1 and SIRT3, is also described to be cardioprotective [41].
4. Conclusions
Despite available therapies, cardiovascular diseases, and particularly ischemic diseases, still remain the first cause of mortality and morbidity in the world. Obesity and type 2 diabetes are some of the major risk factors of cardiovascular diseases. Acetylation is an essential mechanism involved in several processes contributing to cardiac diseases such as cell metabolism, gene transcription or enzymatic activity. In this context, targeting enzymes responsible of acetylation/deacetylation could be beneficial and is a very promising research area, as demonstrated by the development of inhibitor targeting P300 [82], modulators of SIRT6 [96] or agonists of SIRT3 [94,95] to treat cardiac dysfunctions.
Author Contributions
Writing—original draft preparation, E.D.-D., Y.E.M., A.T., F.P. and J.-S.A., funding acquisition, P.A., F.P. and J.-S.A. All authors have read and agreed to the published version of the manuscript.
Funding
E.D.D. was supported by grants from region Hauts de France, CPER “Longévité” of Institut Pasteur de Lille and “Fondation Lefoulon Delalande”. F.P. and E.D.D. received a grant by the French Government, managed by the National Research Agency (ANR) under the program “Investissements d’avenir” with the reference ANR-16-RHUS-0003. Y.E.M., E.D.D. and F.P. are members of FHU-CARNAVAL. This work was supported by Programme Hubert Curien AMADEUS (France-Autriche), ANR grant BETAPLASTICITY (ANR-17-CE14-0034 to J-S.A.), INSERM, CNRS, Institut Pasteur de Lille (CPER CTRL Melodie), Université de Lille, Fondation pour la Recherche Médicale (EQU202103012732), Conseil Régional Hauts de France and Métropole Européenne de Lille and Société Francophone du Diabète.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update. Circulation 2021, 143, E254–E743. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, R.W.; Vogel, H.; Schürmann, A. Genetic and epigenetic control of metabolic health. Mol. Metab. 2013, 2, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketema, E.B.; Lopaschuk, G.D. Post-translational Acetylation Control of Cardiac Energy Metabolism. Front. Cardiovasc. Med. 2021, 8, 723996. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic Analysis of Lysine Acetylation Sites in Rat Tissues Reveals Organ Specificity and Subcellular Patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Hosp, F.; Lassowskat, I.; Santoro, V.; De Vleesschauwer, D.; Fliegner, D.; Redestig, H.; Mann, M.; Christian, S.; Hannah, M.A.; Finkemeier, I. Lysine acetylation in mitochondria: From inventory to function. Mitochondrion 2017, 33, 58–71. [Google Scholar] [CrossRef]
- Thapa, D.; Zhang, M.; Manning, J.R.; Guimarães, D.A.; Stoner, M.W.; Lai, Y.; Shiva, S.; Scott, I. Loss of GCN5L1 in cardiac cells limits mitochondrial respiratory capacity under hyperglycemic conditions. Physiol. Rep. 2019, 7, e14054. [Google Scholar] [CrossRef] [Green Version]
- Thapa, D.; Zhang, M.; Manning, J.R.; Guimarães, D.A.; Stoner, M.W.; O’Doherty, R.M.; Shiva, S.; Scott, I. Acetylation of mitochondrial proteins by GCN5L1 promotes enhanced fatty acid oxidation in the heart. Am. J. Physiol. Circ. Physiol. 2017, 313, H265–H274. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, A.; Alrob, O.A.; Zhang, L.; Wagg, C.S.; Altamimi, T.; Rawat, S.; Rebeyka, I.M.; Kantor, P.F.; Lopaschuk, G.D. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am. J. Physiol. Circ. Physiol. 2016, 311, H347–H363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Alrob, O.A.; Sankaralingam, S.; Ma, C.; Wagg, C.S.; Fillmore, N.; Jaswal, J.S.; Sack, M.N.; Lehner, R.; Gupta, M.P.; Michelakis, E.D.; et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc. Res. 2014, 103, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor γ co-activator 1 α (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Rivas, G.; Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Treviño-Saldaña, N.; Guerrero-Beltrán, C.E.; Bernal-Ramírez, J.; Torres-Quintanilla, A.; García, N.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell. Physiol. Biochem. 2019, 53, 465–479. [Google Scholar]
- Ghosh, T.K.; Aparicio-Sánchez, J.J.; Buxton, S.; Ketley, A.; Mohamed, T.; Rutland, C.S.; Loughna, S.; Brook, J.D. Acetylation of TBX5 by KAT2B and KAT2A regulates heart and limb development. J. Mol. Cell. Cardiol. 2018, 114, 185–198. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Aparicio-Sánchez, J.J.; Buxton, S.; Brook, J.D. HDAC4 and 5 repression of TBX5 is relieved by protein kinase D1. Sci. Rep. 2019, 9, 17992. [Google Scholar] [CrossRef]
- Lin, Z.; Guo, H.; Cao, Y.; Zohrabian, S.; Zhou, P.; Ma, Q.; VanDusen, N.; Guo, Y.; Zhang, J.; Stevens, S.M.; et al. Acetylation of VGLL4 Regulates Hippo-YAP Signaling and Postnatal Cardiac Growth. Dev. Cell 2016, 39, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, S.; Izumiya, Y.; Araki, S.; Nakamura, T.; Kimura, Y.; Hanatani, S.; Yamada, T.; Ishida, T.; Yamamoto, M.; Onoue, Y.; et al. Cardiomyocyte Sirt (Sirtuin) 7 Ameliorates Stress-Induced Cardiac Hypertrophy by Interacting with and Deacetylating GATA4. Hypertension 2020, 75, 98–108. [Google Scholar] [CrossRef]
- Shimizu, S.; Sunagawa, Y.; Hajika, N.; Yorimitsu, N.; Katanasaka, Y.; Funamoto, M.; Miyazaki, Y.; Sari, N.; Shimizu, K.; Hasegawa, K.; et al. Multimerization of the GATA4 transcription factor regulates transcriptional activity and cardiomyocyte hypertrophic response. Int. J. Biol. Sci. 2022, 18, 1079–1095. [Google Scholar] [CrossRef]
- Peng, C.; Luo, X.; Li, S.; Sun, H. Phenylephrine-induced cardiac hypertrophy is attenuated by a histone acetylase inhibitor anacardic acid in mice. Mol. Biosyst. 2017, 13, 714–724. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, Z.; Alibin, C.P.; Acosta, C.; Anderson, H.D. Liganded Peroxisome Proliferator-Activated Receptors (PPARs) Preserve Nuclear Histone Deacetylase 5 Levels in Endothelin-Treated Sprague-Dawley Rat Cardiac Myocytes. PLoS ONE 2014, 9, e115258. [Google Scholar] [CrossRef] [Green Version]
- Mao, Q.; Wu, S.; Peng, C.; Peng, B.; Luo, X.; Huang, L.; Zhang, H. Interactions between the ERK1/2 signaling pathway and PCAF play a key role in PE-induced cardiomyocyte hypertrophy. Mol. Med. Rep. 2021, 24, 636. [Google Scholar] [CrossRef]
- Winnik, S.; Gaul, D.S.; Siciliani, G.; Lohmann, C.; Pasterk, L.; Calatayud, N.; Weber, J.; Eriksson, U.; Auwerx, J.; van Tits, L.J.; et al. Mild endothelial dysfunction in Sirt3 knockout mice fed a high-cholesterol diet: Protective role of a novel C/EBP-β-dependent feedback regulation of SOD2. Basic Res. Cardiol. 2016, 111, 33. [Google Scholar] [CrossRef] [Green Version]
- Peugnet, V.; Chwastyniak, M.; Mulder, P.; Lancel, S.; Bultot, L.; Fourny, N.; Renguet, E.; Bugger, H.; Beseme, O.; Loyens, A.; et al. Mitochondrial-Targeted Therapies Require Mitophagy to Prevent Oxidative Stress Induced by SOD2 Inactivation in Hypertrophied Cardiomyocytes. Antioxidants 2022, 11, 723. [Google Scholar] [CrossRef]
- Leng, Y.; Wu, Y.; Lei, S.; Zhou, B.; Qiu, Z.; Wang, K.; Xia, Z. Inhibition of HDAC6 Activity Alleviates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats: Potential Role of Peroxiredoxin 1 Acetylation and Redox Regulation. Oxid. Med. Cell. Longev. 2018, 2018, 9494052. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.-J.; Cui, J.; Lin, Q.; Chen, X.-Y.; Zhang, J.; Gao, E.-H.; Wei, B.; Zhao, W. Protection of the enhanced Nrf2 deacetylation and its downstream transcriptional activity by SIRT1 in myocardial ischemia/reperfusion injury. Int. J. Cardiol. 2021, 342, 82–93. [Google Scholar] [CrossRef]
- Ding, M.; Lei, J.; Han, H.; Li, W.; Qu, Y.; Fu, E.; Fu, F.; Wang, X. SIRT1 protects against myocardial ischemia–reperfusion injury via activating eNOS in diabetic rats. Cardiovasc. Diabetol. 2015, 14, 143. [Google Scholar] [CrossRef] [Green Version]
- Landim-Vieira, M.; Childers, M.C.; Wacker, A.L.; Garcia, M.R.; He, H.; Singh, R.; Brundage, E.A.; Johnston, J.R.; Whitson, B.A.; Chase, P.B.; et al. Post-translational modification patterns on β-myosin heavy chain are altered in ischemic and nonischemic human hearts. eLife 2022, 11, e74919. [Google Scholar] [CrossRef]
- Samant, S.A.; Pillai, V.B.; Sundaresan, N.R.; Shroff, S.G.; Gupta, M.P. Histone Deacetylase 3 (HDAC3)-dependent Reversible Lysine Acetylation of Cardiac Myosin Heavy Chain Isoforms Modulates Their Enzymatic and Motor Activity. J. Biol. Chem. 2015, 290, 15559–15569. [Google Scholar] [CrossRef] [Green Version]
- Loescher, C.M.; Hobbach, A.J.; Linke, W.A. Titin (TTN): From molecule to modifications, mechanics, and medical significance. Cardiovasc. Res. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Warren, C.M.; Li, J.; McKinsey, T.A.; Russell, B. Myofibril growth during cardiac hypertrophy is regulated through dual phosphorylation and acetylation of the actin capping protein CapZ. Cell. Signal. 2016, 28, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Schmidt, W.; Fritz, K.S.; Jeong, M.Y.; Cammarato, A.; Foster, D.B.; Biesiadecki, B.J.; McKinsey, T.A.; Woulfe, K.C. Site-specific acetyl-mimetic modification of cardiac troponin I modulates myofilament relaxation and calcium sensitivity. J. Mol. Cell. Cardiol. 2020, 139, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD Blocks Cardiac Hypertrophic Response via Activation of the SIRT3-LKB1-AMP-activated Kinase Pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Chen, X.-F.; Wang, N.-Y.; Wang, X.-M.; Liang, S.-T.; Zheng, W.; Lu, Y.-B.; Zhao, X.; Hao, D.-L.; Zhang, Z.-Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Hu, L.; Yang, H.; Gao, C.; Zeng, K.; Yu, M.; Feng, J.; Qiu, J.; Liu, C.; Fu, F.; et al. Reduction of SIRT1 blunts the protective effects of ischemic post-conditioning in diabetic mice by impairing the Akt signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Jeong, A.; Kwon, D.-H.; Lee, Y.-U.; Kim, Y.-K.; Ahn, Y.; Kook, T.; Park, W.-J.; Kook, H. P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. Int. J. Mol. Sci. 2021, 22, 9944. [Google Scholar] [CrossRef] [PubMed]
- Bugyei-Twum, A.; Ford, C.; Civitarese, R.; Seegobin, J.; Advani, S.L.; Desjardins, J.-F.; Kabir, G.; Zhang, Y.; Mitchell, M.; Switzer, J.; et al. Sirtuin 1 activation attenuates cardiac fibrosis in a rodent pressure overload model by modifying Smad2/3 transactivation. Cardiovasc. Res. 2018, 114, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zeng, Y.; Yang, X.; Wu, Y.; Zhang, S.; Huang, S.; Zhong, Y.; Chen, M. Resveratrol ameliorates myocardial fibrosis by regulating Sirt1/Smad3 deacetylation pathway in rat model with dilated cardiomyopathy. BMC Cardiovasc. Disord. 2022, 22, 17. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, B.; Lin, Q.; Xu, W.; Zuo, W.; Li, J.; Liu, N.; Tu, T.; Zhang, B.; Xiao, Y.; et al. Nicotinamide mononucleotide attenuates isoproterenol-induced cardiac fibrosis by regulating oxidative stress and Smad3 acetylation. Life Sci. 2021, 274, 119299. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Mitochondrial Acetylation and Diseases of Aging. J. Aging Res. 2011, 2011, 234875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Chernaya, V.; Johnson, C.; Yang, W.Y.; Cueto, R.; Sha, X.; Zhang, Y.; Qin, X.; Sun, J.; Choi, E.T.; et al. Metabolic Diseases Downregulate the Majority of Histone Modification Enzymes, Making a Few Upregulated Enzymes Novel Therapeutic Targets—“Sand Out and Gold Stays”. J. Cardiovasc. Transl. Res. 2016, 9, 49–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, P.; Feng, X.; Zhang, X.; Huang, X.; Liu, S.; Lu, X.; Li, J.; You, J.; Lu, J.; Li, Z.; et al. SIRT6 suppresses phenylephrine-induced cardiomyocyte hypertrophy though inhibiting p300. J. Pharmacol. Sci. 2016, 132, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, Y.; Ren, J. Acetylation in cardiovascular diseases: Molecular mechanisms and clinical implications. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165836. [Google Scholar] [CrossRef]
- Pane, L.S.; Fulcoli, F.G.; Cirino, A.; Altomonte, A.; Ferrentino, R.; Bilio, M.; Baldini, A. Tbx1 represses Mef2c gene expression and is correlated with histone 3 deacetylation of the anterior heart field enhancer. Dis. Model. Mech. 2018, 11, dmm029967. [Google Scholar] [CrossRef] [Green Version]
- Bühler, A.; Gahr, B.M.; Park, D.-D.; Bertozzi, A.; Boos, A.; Dalvoy, M.; Pott, A.; Oswald, F.; Kovall, R.A.; Kühn, B.; et al. Histone deacetylase 1 controls cardiomyocyte proliferation during embryonic heart development and cardiac regeneration in zebrafish. PLOS Genet. 2021, 17, e1009890. [Google Scholar] [CrossRef]
- Cencioni, C.; Spallotta, F.; Mai, A.; Martelli, F.; Farsetti, A.; Zeiher, A.M.; Gaetano, C. Sirtuin function in aging heart and vessels. J. Mol. Cell. Cardiol. 2015, 83, 55–61. [Google Scholar] [CrossRef]
- Watroba, M.; Szukiewicz, D. Sirtuins at the Service of Healthy Longevity. Front. Physiol. 2021, 12, 724506. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Schwer, B.; Alt, F.W.; Mostoslavsky, R. SIRT6 in DNA repair, metabolism and ageing. J. Intern. Med. 2008, 263, 128–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M. Longevity genes, cardiac ageing, and the pathogenesis of cardiomyopathy: Implications for understanding the effects of current and future treatments for heart failure. Eur. Heart J. 2020, 41, 3856–3861. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Conti, S.; Perico, L.; Corna, D.; Cerullo, D.; Zentilin, L.; Zoja, C.; Perna, A.; Lionetti, V.; et al. Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxid. Redox Signal. 2019, 31, 1255–1271. [Google Scholar] [CrossRef] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic Instability and Aging-like Phenotype in the Absence of Mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 Is a Stress-Responsive Deacetylase in Cardiomyocytes That Protects Cells from Stress-Mediated Cell Death by Deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-H.; Major, J.L.; Liebner, T.; Hourani, Z.; Travers, J.G.; Wennersten, S.A.; Haefner, K.R.; Cavasin, M.A.; Wilson, C.E.; Jeong, M.Y.; et al. HDAC6 modulates myofibril stiffness and diastolic function of the heart. J. Clin. Investig. 2022, 132, e148333. [Google Scholar] [CrossRef]
- Pougovkina, O.; te Brinke, H.; Ofman, R.; van Cruchten, A.G.; Kulik, W.; Wanders, R.J.A.; Houten, S.M.; de Boer, V.C.J. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 2014, 23, 3513–3522. [Google Scholar] [CrossRef] [Green Version]
- Tomczyk, M.M.; Cheung, K.G.; Xiang, B.; Tamanna, N.; Fonseca Teixeira, A.L.; Agarwal, P.; Kereliuk, S.M.; Spicer, V.; Lin, L.; Treberg, J.; et al. Mitochondrial Sirtuin-3 (SIRT3) Prevents Doxorubicin-Induced Dilated Cardiomyopathy by Modulating Protein Acetylation and Oxidative Stress. Circ. Heart Fail. 2022, 15, e008547. [Google Scholar] [CrossRef]
- Maissan, P.; Mooij, E.J.; Barberis, M. Sirtuins-mediated system-level regulation of mammalian tissues at the interface between metabolism and cell cycle: A systematic review. Biology 2021, 10, 194. [Google Scholar] [CrossRef]
- Khan, D.; Sarikhani, M.; Dasgupta, S.; Maniyadath, B.; Pandit, A.S.; Mishra, S.; Ahamed, F.; Dubey, A.; Fathma, N.; Atreya, H.S.; et al. SIRT6 deacetylase transcriptionally regulates glucose metabolism in heart. J. Cell. Physiol. 2018, 233, 5478–5489. [Google Scholar] [CrossRef]
- Khan, D.; Ara, T.; Ravi, V.; Rajagopal, R.; Tandon, H.; Parvathy, J.; Gonzalez, E.A.; Asirvatham-Jeyaraj, N.; Krishna, S.; Mishra, S.; et al. SIRT6 transcriptionally regulates fatty acid transport by suppressing PPARγ. Cell Rep. 2021, 35, 109190. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1525–1534. [Google Scholar] [CrossRef]
- Hou, Y.-S.; Wang, J.-Z.; Shi, S.; Han, Y.; Zhang, Y.; Zhi, J.-X.; Xu, C.; Li, F.-F.; Wang, G.-Y.; Liu, S.-L. Identification of epigenetic factor KAT2B gene variants for possible roles in congenital heart diseases. Biosci. Rep. 2020, 40, BSR20191779. [Google Scholar] [CrossRef] [Green Version]
- Sunagawa, Y.; Katayama, A.; Funamoto, M.; Shimizu, K.; Shimizu, S.; Sari, N.; Katanasaka, Y.; Miyazaki, Y.; Hosomi, R.; Hasegawa, K.; et al. The polyunsaturated fatty acids, EPA and DHA, ameliorate myocardial infarction-induced heart failure by inhibiting p300-HAT activity in rats. J. Nutr. Biochem. 2022, 106, 109031. [Google Scholar] [CrossRef]
- Li, N.; Zhou, H.; Ma, Z.G.; Zhu, J.X.; Liu, C.; Song, P.; Kong, C.Y.; Wu, H.M.; Deng, W.; Tang, Q.Z. Geniposide alleviates isoproterenol-induced cardiac fibrosis partially via SIRT1 activation in vivo and in vitro. Front. Pharmacol. 2018, 9, 854. [Google Scholar] [CrossRef]
- Lu, T.M.; Tsai, J.Y.; Chen, Y.C.; Huang, C.Y.; Hsu, H.L.; Weng, C.F.; Shih, C.C.; Hsu, C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014, 21, 57. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, A.S.; El-Kott, A.F.; Eleawa, S.M.; El-Gerbed, M.S.A.; Khalifa, H.S.; El-Kenawy, A.E.; Albadrani, G.M.; Abdel-Daim, M.M. Kaempferol protects against streptozotocin-induced diabetic cardiomyopathy in rats by a hypoglycemic effect and upregulating sirt1. J. Physiol. Pharmacol. 2021, 72, 339–355. [Google Scholar]
- Qiu, L.; Xu, C.; Xia, H.; Chen, J.; Liu, H.; Jiang, H. Downregulation of P300/CBP-Associated Factor Attenuates Myocardial Ischemia-Reperfusion Injury Via Inhibiting Autophagy. Int. J. Med. Sci. 2020, 17, 1196–1206. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhan, L.; Zhou, Q.Y.; Zhang, L.L.; Chen, X.M.; Hu, X.M.; Yuan, X.C. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur. J. Pharmacol. 2015, 764, 554–561. [Google Scholar] [CrossRef]
- Turdi, S.; Li, Q.; Lopez, F.L.; Ren, J. Catalase alleviates cardiomyocyte dysfunction in diabetes: Role of Akt, Forkhead transcriptional factor and silent information regulator 2. Life Sci. 2007, 81, 895–905. [Google Scholar] [CrossRef]
- Lai, Y.C.; Tabima, D.M.; Dube, J.J.; Hughan, K.S.; Vanderpool, R.R.; Goncharov, D.A.; St. Croix, C.M.; Garcia-Ocanã, A.; Goncharova, E.A.; Tofovic, S.P.; et al. SIRT3-AMP-Activated Protein Kinase Activation by Nitrite and Metformin Improves Hyperglycemia and Normalizes Pulmonary Hypertension Associated with Heart Failure with Preserved Ejection Fraction. Circulation 2016, 133, 717–731. [Google Scholar]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Möbius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef]
- Li, L.; Zeng, H.; He, X.; Chen, J. Sirtuin 3 Alleviates Diabetic Cardiomyopathy by Regulating TIGAR and Cardiomyocyte Metabolism. J. Am. Heart Assoc. 2021, 10, e018913. [Google Scholar] [CrossRef]
- Dai, L.; Xie, Y.; Zhang, W.; Zhong, X.; Wang, M.; Jiang, H.; He, Z.; Liu, X.; Zeng, H.; Wang, H. Weighted Gene Co-Expression Network Analysis Identifies ANGPTL4 as a Key Regulator in Diabetic Cardiomyopathy via FAK/SIRT3/ROS Pathway in Cardiomyocyte. Front. Endocrinol. 2021, 12, 705154. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Gao, B.; Yang, Y.; Jia, S.-B.; Ma, X.-P.; Zhang, M.-H.; Wang, L.-J.; Ma, A.-Q.; Zhang, Q.-N. Histone deacetylase 3 suppresses the expression of SHP-1 via deacetylation of DNMT1 to promote heart failure. Life Sci. 2022, 292, 119552. [Google Scholar] [CrossRef]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.; Chen, S.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Yang, J.-J.; Shi, K.-H.; Li, J. Epigenetic factors MeCP2 and HDAC6 control α-tubulin acetylation in cardiac fibroblast proliferation and fibrosis. Inflamm. Res. 2016, 65, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Bauters, C.; Dubois, E.; Porouchani, S.; Saloux, E.; Fertin, M.; de Groote, P.; Lamblin, N.; Pinet, F. Long-term prognostic impact of left ventricular remodeling after a first myocardial infarction in modern clinical practice. PLoS ONE 2017, 12, e0188884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chen, X.; Bi, Z.; Liao, J.; Zhao, W.; Huang, W. Curcumin attenuates renal ischemia reperfusion injury via JNK pathway with the involvement of p300/CBP-mediated histone acetylation. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2021, 25, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Sunagawa, Y.; Maekawa, T.; Funamoto, M.; Shimizu, S.; Shimizu, K.; Katanasaka, Y.; Komiyama, M.; Hawke, P.; Hara, H.; et al. Ecklonia stolonifera Okamura Extract Suppresses Myocardial Infarction-Induced Left Ventricular Systolic Dysfunction by Inhibiting p300-HAT Activity. Nutrients 2022, 14, 580. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Funamoto, M.; Shimizu, K.; Shimizu, S.; Sari, N.; Katanasaka, Y.; Miyazaki, Y.; Kakeya, H.; Hasegawa, K.; Morimoto, T. Curcumin, an inhibitor of p300-hat activity, suppresses the development of hypertension-induced left ventricular hypertrophy with preserved ejection fraction in dahl rats. Nutrients 2021, 13, 2608. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Shimizu, K.; Katayama, A.; Funamoto, M.; Shimizu, K.; Nurmila, S.; Shimizu, S.; Miyazaki, Y.; Katanasaka, Y.; Hasegawa, K.; et al. Metformin suppresses phenylephrine-induced hypertrophic responses by inhibiting p300-HAT activity in cardiomyocytes. J. Pharmacol. Sci. 2021, 147, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C., Jr.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 1, e84897. [Google Scholar] [CrossRef]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602. [Google Scholar] [CrossRef] [Green Version]
- Prola, A.; Pires Da Silva, J.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2α deacetylation. Cell Death Differ. 2017, 24, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Renguet, E.; De Loof, M.; Fourny, N.; Ginion, A.; Bouzin, C.; Poüs, C.; Horman, S.; Beauloye, C.; Bultot, L.; Bertrand, L. α-Tubulin acetylation on lysine 40 controls cardiac glucose uptake. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H1032–H1043. [Google Scholar] [CrossRef]
- Gao, S.; Yang, Q.; Peng, Y.; Kong, W.; Liu, Z.; Li, Z.; Chen, J.; Bao, M.; Li, X.; Zhang, Y.; et al. SIRT6 regulates obesity-induced oxidative stress via ENDOG/SOD2 signaling in the heart. Cell Biol. Toxicol. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, Q.; Sun, X.; Qian, H.; Lyu, S.; Chen, R.; Xia, H.; Yuan, W. Sirtuin 3: Emerging therapeutic target for cardiovascular diseases. Free Radic. Biol. Med. 2022, 180, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, S.; Zhang, B.; Liu, J. SIRT3 as a potential therapeutic target for heart failure. Pharmacol. Res. 2021, 165, 105432. [Google Scholar] [CrossRef] [PubMed]
- Saiyang, X.; Deng, W.; Qizhu, T. Sirtuin 6: A potential therapeutic target for cardiovascular diseases. Pharmacol. Res. 2021, 163, 105214. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).