Abstract
Lysosomal storage diseases (LSDs) are a heterogeneous group of rare multisystem metabolic disorders occurring mostly in infancy and childhood, characterized by a gradual accumulation of non-degraded substrates inside the cells. Although biochemical enzymatic assays are considered the gold standard for diagnosis of symptomatic patients, genotyping is a requirement for inclusion in enzyme replacement programs and is a prerequisite for carrier tests in relatives and DNA-based prenatal diagnosis. The emerging next-generation sequencing (NGS) technologies are now offering a powerful diagnostic tool for genotyping LSDs patients by providing faster, cheaper, and higher-resolution testing options, and are allowing to unravel, in a single integrated workflow SNVs, small insertions and deletions (indels), as well as major structural variations (SVs) responsible for the pathology. Here, we summarize the current knowledge about the most recurrent and private SVs involving LSDs-related genes, review advantages and drawbacks related to the use of the NGS in the SVs detection, and discuss the challenges to bring this type of analysis in clinical diagnostics.
1. Introduction
Lysosomal storage diseases (LSDs) are inborn metabolic disorders caused by the absence or deficiency of one specific component in the lysosomal pathway, resulting in the accumulation of undegraded substrates in multiple organs [1]. These disorders encompass a group of about 70 different entities that can be subclassified according to the enzyme deficit and the specific involved substrate: lipid storage diseases, mucopolysaccharidoses, glycoproteinoses, lipofuscinosis, lysosomal integral membrane proteins diseases, post-translational modifications dysfunctions and lysosomal related organelles (LRO) disorders [2]. Although each disorder per se is rare, with estimated incidences ranging from 1 in 50,000 to 1 in 250,000 live births, they are relatively common disorders when considered collectively (1:5000 live births) [2].
The storage process leads to a broad spectrum of clinical manifestations depending on the specific substrate and site of accumulation [3]. Signs and symptoms may occur from prenatal period to adulthood, and develop progressively over time from mild to extremely severe forms, and could include organomegaly, pulmonary and cardiac problems, facial dysmorphisms, musculoskeletal abnormalities, cognitive decline, deafness, blindness and movement problems, all having a severe impact on patients’ prognosis and quality of life [1,3,4].
From a genetic point of view, LSDs are Mendelian monogenic disorders, inherited mainly in an autosomal recessive or in an X-linked manner (i.e., Fabry, Hunter, and Danon diseases), and are caused by mutations in genes encoding for acid hydrolases, integral membrane proteins, activators and transporter proteins, or other accessory proteins related to lysosomal function [2,5]. Although biochemical enzyme assays are considered the gold standard for diagnosis of symptomatic patients, genotyping is a requirement for inclusion in enzyme replacement programs and is a prerequisite for carrier tests in blood relatives and DNA-based prenatal diagnosis. Routine diagnostic DNA analysis usually focuses on genetic coding regions using PCR reactions and subsequent Sanger sequencing and is successful in identifying single nucleotide variants (SNVs) disrupting protein function [2]. Despite this, a number of patients with suspected LSDs have other types of genomic variations and do not achieve a definitive clinical diagnosis for many years. The current advancement in next generation sequencing (NGS) technologies is now providing a powerful diagnostic tool for patients affected by LSDs who escape the diagnosis by traditional genetic investigation, and is offering a range of new opportunities for genomic medicine [6,7]. Thanks to these high-throughput sequencing techniques, geneticists and clinicians can go beyond the search for single nucleotide variants (SNVs) hotspot mutations in the coding sequence of responsible genes, and more easily identify small insertions and deletions (indels) and major structural variations (SVs) responsible for the pathology [8,9,10].
SVs, in particular, include genetic variants greater than 50 base pairs (bp) in length and comprise chromosomal inversions, insertions, translocations, further complex rearrangements and genomic imbalances (duplication or deletions) commonly referred to as copy number variants (CNVs) [11]. Improvement in SV detection is enhancing our understanding of the intricate interplay between genetic makeup and associated phenotype, unraveling their contribution in disease etiology, gene expression regulation and phenotypic diversity in rare diseases, including LSDs. During the last years, great progress has been made in order to standardize the detection of SVs with sufficient accuracy and precision for use in clinical diagnostics, but the challenge still remains open [11].
Here we summarize the current knowledge about the most recurrent and private genomic SVs involving LSDs-related genes, review advantages and drawbacks related to the use of NGS in the SVs detection, and discuss the challenges to bring this type of analysis in clinical diagnostics.
2. CNVs Are the Major SVs
Copy number variants (CNVs), encompassing losses or gains of genomic DNA segments larger than 50 bp, are the most common type of SVs in the human genome and represent a major source of human genetic variation accounting for disease and population diversity [11,12]. Recent studies revealed that de novo locus-specific mutation rates appear much higher for CNVs than for SNVs [13,14,15], and that the frequency of a CNV shows strong anticorrelation with its size and gene density [16].
Although several genomic copy number imbalances are presumably benign, their role in the pathogenesis of various diseases has been recently gaining increasing attention. Benign CNVs are frequently small, intergenic, or comprise genes that can tolerate copy number changes, while pathogenic CNVs are significantly enriched in dosage sensitive-genes or in regions with constrained evolutionary patterns of gene duplication and loss [17]. In the early era of CNV detection, large CNVs (>500 kb) appeared to be associated with genomic disorders only. However, it is now clear that these types of alterations may cause or influence Mendelian diseases as well complex traits [12,18].
Disease-causing genomic rearrangements can be either recurrent or non-recurrent. The main fraction of recurrent CNVs arises from nonallelic homologous recombination (NAHR) mechanisms mediated by high-identity segmental duplications or Alu repeats [13,19]. These genomic disorders are highly penetrant, can occur in different haplotype backgrounds in multiple unrelated individuals in a relatively short period of time, and are under strong negative selection. CNVs can also arise from non-recurrent mechanisms, which include non-homologous end-joining (NHEJ), Fork Stalling and Template Switching (FoSTeS), and microhomology/microsatellite-mediated break-induced repair. These rearrangements do occur at a relatively lower frequency and assessing their clinical relevance is problematic, since it requires the screening of many thousands of patients to establish pathogenicity [13,19].
The molecular mechanisms by which a CNV causes abnormal phenotypes include gene-dosage sensitivity, gene interruption, gene fusion at the breakpoint junctions, deletion of a regulatory element, the unmasking of a recessive allele or a functional polymorphism [20], and alteration of non-coding regulatory elements (promoters or enhancers) [18]. The advances in whole-genome technologies are enabling the discovery and characterization of large and intermediate SVs, but the landscape of CNVs still remains largely unexplored, especially for those embedded within complex regions of the human genome.
3. SVs in LSDs
More than 65 genes responsible for the wide spectrum of LSDs have been described so far [1,2]. For most of these conditions, clinically relevant homozygous or compound heterozygous SVs (either recurrent or private) have been described in LSD patients in single case reports or in cohort studies and are collected in international reference databases (e.g., ClinVar, Decipher, DGV, GnomAD), where the reader is referred to for a complete and comprehensive consultation. Here, to the best of our knowledge, we will briefly report both past and recently identified SVs altering LSDs-related genes (Supplementary Table S1), although the creation of an extensive repository is far beyond the scope of the present work. For simplicity, pathologies will be discussed separately in subgroups.
3.1. Enzyme Deficiency Disorders
3.1.1. Lipid Storage Diseases
Lipid storage diseases include both the large group of sphingolipidoses and Wolman disease, which is caused by lysosomal acid lipase deficiency.
Sphingolipidoses are characterized by abnormal storage of various phospholipids with a sphingosine group, and include: Fabry (GLA), Niemann–Pick A/B (SMPD1), Krabbe (GALC), Gaucher (GBA), Tay–Sachs (HEXA), Farber (ASAH1), Sandhoff (HEXB) and Metachromatic Leukodystrophy (ARSA) [2]. A number of disease-causing gene CNVs have been described, including: (i) a gross deletion involving ASAH1 (g.728_18197del (c.126-3941_382 + 1358del) in a child with severe Farber disease [21]; (ii) a whole-gene deletion of ARSA in a patient with infantile Metachromatic Leukodystrophy [22]; (iii) two single-exon deletion involving GALC exon 12 and 14 and multiple contiguous exons loss (exons 11–17) in patients with Krabbe disease [23]; (iv) a recurrent 5′-end 16 kb deletion comprising HEXB promoter region, exons 1–5 and part of intron 5 in Sandhoff disease patients [24,25], and further partial deletions comprising intron 1-exon 2 [25] and exons 1–5 [26]; (v) multiple complex pathogenic duplications or deletions of SMPD1 in patients with acid sphingomyelinase deficiency [27].
Two genes of this subgroup (GBA and GLA) undergo frequent SVs, as better discussed here below.
The GBA gene is located in an unstable chromosomal region, structurally subject to misalignments, reciprocal/nonreciprocal homologous recombination events and DNA rearrangements (deletions, duplications, inversions and fusions), representing the main cause of pitfalls in Gaucher disease mutational analysis [9,28]. This gene-rich locus encompasses seven genes, Alu sequences and a highly homologous 5.7-kb pseudogene (GBAP1) located approximately 16 kb downstream GBA showing an exons/intron organization similar to the functional gene [9]. Total gene deletion and more than 20 recombinant alleles (including Alu-mediated large deletion) have been characterized to date, with RecNcil and Recdelta55 being the most common either alone or in combination with additional point mutations [9,28,29,30,31,32,33]. In particular, RecNcil derives from a cross-over junction area from intron 9 to exon 10, resulting in the incorporation of a segment of GBAP1 that includes several missense mutations into the functional GBA, whereas Recdelta55 encompasses a 55-bp deletion in exon 9 of the GBA corresponding to the deleted portion of the pseudogene. Given this highly unstable locus, the need to adopt adequate strategies to avoid misdetection of GBA recombinant mutations is clear. To this end, different strategies were described to provide proper GBA genotyping and genetic counseling, such as specific procedures for NGS-based targeted library preparation and data analysis [9] and clinical exome sequencing in combination with MLPA assays [10].
Regarding GLA, among the approximately 1000 gene mutations documented in the Human Gene Mutation Database, about 30 are SVs from 0.1 to several kb of size and predominantly include gross deletions, resulting from various recombination events (e.g., short regions of homology, short inverted repeat sequences, Alu-Alu recombination, insertion of retrotransposon L1, double strand breaks repaired by a non-homologous end-joining mechanism) [34]. Southern hybridization, restriction analyses, MLPA, and multiplex PCR assays have been useful in detecting these complex rearrangements [34,35,36,37,38], which are often missed by routinary DNA sequencing (especially in female Fabry disease patients). Recently, the use of NGS-based read amplicon technology proved successful in resolving a complex case [39].
Wolman’s disease (WD) is a rare and fatal infantile metabolic disorder caused by lysosomal acid lipase deficiency. Very recently, a 4-month-old infant presenting with hepatosplenomegaly, failure to thrive and other abnormalities carrying a homozygous deletion of exons 9–10 of the in LIPA gene has been described [40].
3.1.2. Mucopolysaccharidoses
Mucopolysaccharidoses (MPS) are a group of inherited LSDs characterized by accumulation of undegraded glycosaminoglycans (GAGs or mucopolysaccharides) in the lysosomes of cells, resulting in severe symptoms including coarse facial features, cognitive retardation, hepatosplenomegaly, hernias, kyphoscoliosis and corneal clouding. The occurrence of SVs is frequent in this group of pathologies, although often they are undetected and result in failed diagnosis. Indeed, several MPS-related genes undergo recurrent or private SVs as described below. Particular emphasis will be given to IDS, the causative gene of MPS II [41].
CNVs in MPS-related genes have been characterized and encompass: (i) deletion of exon 14-3′UTR, and duplication of exon 2-intron 12 in IDUA in patients affected by MPS I [42,43]; (ii) deletion of SGSH exons 1–5 in MPS IIIA patients [44]; (iii) Alu-mediated deletion of NAGLU exons 3–4 in patients with MPS IIIB/Sanfilippo type B syndrome [45,46]; (iv) heterozygous deletion of exon 15 [47] and homozygous deletion of exons 9–10 in HGSNAT in MPS IIIC or Sanfilippo type C patients [43]; (v) deletions of GNS exon 1, 2–3, 6–7, 9–14 in patients affected by MPS IIID/Sanfilippo disease type D [48,49]; (vi) deletion of multiple contiguous exons including 1–3, 2–4, 2–5, 3–14, 5–8, 9–14, 10–14, 11–12, and single exon 5 and 13 of GALNS in patients with MPS IVA/Morquio A [43,50,51,52,53]; (vii) ARSB deletion of exons 2–3, exon 4 and exon 5 MPS VI patients [54,55,56].
IDS deserves a separate discussion, which is responsible for MPS II or Hunter syndrome. The mutational spectrum associated with this gene is quite heterogeneous, ranging from point mutations to recurrent gross genomic rearrangements, occurring approximately in 10–20% of MPS II patients [41]. IDS spans about 24 kb in Xq28 and consists of nine exons. A pseudogene (IDSP1—also known as IDS2), located telomeric to the functional IDS gene, contains sequences homologous to exon 2, intron 2, exon 3 and a chimerical fragment of intron 3–intron 7, making this region prone to the occurrence of non-allelic homologous recombination events [41]. Indeed, whole IDS gene deletion (with or without the involvement of further neighboring genes) and partial IDS exon deletions have been reported in multiple studies, mainly in patients with a severe Hunter syndrome presentation [41,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. Furthermore, IDS exons duplications, IDS1/IDS2 inversions, chimeric IDS-IDS2 allele and other large complex rearrangements have been described [41,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71], as well as deletions in the promoter region in patients with a mild phenotype [72]. Recently, a case report described a girl with comorbidity of Hunter and Turner syndromes caused by a partial deletion in the long arm of chromosome X of paternal origin and a deletion of IDS inherited from the mother [73].
3.1.3. Glycogen Storage Disease
Genetic variants in GAA are responsible for Pompe disease (PD) or glycogen storage disease type II, a metabolic myopathy with a wide spectrum of clinical presentation caused by acid a-glucosidase enzyme deficiency. Depending on the residual GAA enzyme activity, the disease either develops during the first months of life as the classic infantile Pompe disease (IOPD), or later in life (childhood, adolescence or adulthood) with a milder phenotype known as late-onset Pompe disease (LOPD) [74]. Enzyme replacement therapy (ERT) should be started before symptoms appear in order to achieve optimal outcomes, thus highlighting the importance of detecting PD patients in the very early stage, and encouraging the development of newborn screening studies also with the help of NGS applications [75,76]. Multiple CNVs affecting this gene have been identified, including the most common deletion of exon 18 [77,78,79,80], and the deletion of exons 2–3 [81], exons 2–4 [80,82,83], introns 7–15 [80,84], introns 17–18 and exons 15–20 [80].
3.1.4. Glycoproteinoses
Glycoproteinoses are among the rarest and less studied LSDs and include aspartylglucosaminuria (AGA), fucosidosis (FUCA1), galactosialidosis, (CTSA), α-mannosidosis (MAN2B1), β-mannosidosis (MANBA), Schindler disease (NAGA), and sialidosis (NEU1). Being rare diseases, their genetic/genomic characterization is less detailed. Nonetheless, two large deletion (encompassing exon 4, and exons 7/8) and a 66bp duplication in exon 6 have been reported in homozygous state in FUCA1 in a patient with fucosidosis [85]. More recently, a deletion of NEU1 exon 2 was reported in a patient with type 1 sialidosis [86], as well as a heterozygous 27.5 kb deletion involving the whole coding exons of NEU1 in a girl with reduced visual acuity [87]. The power of NGS in the identification and characterization of SVs have been proved also in a case report describing a 15-year-old girl with β-Mannosidosis, revealing a complex homozygous rearrangement characterized by a partial intragenic inverted duplication of MANBA, and resulted in the mRNA skipping of multiple contiguous exons [88].
3.2. Disorders of Post-Translational Modification
Disorders of post-translational modification include multiple sulfatase deficiency and mucolipidoses (ML) II and III, and result from mutations in genes that have a role in biochemically modifying lysosomal hydrolases. In particular, mutations in GNPTAB cause both the severe type of ML (ML II alpha/beta) and the attenuated type of ML (ML III alpha/beta, or Pseudo-Hurler polydystrophy) [89]. Some genomic rearrangements altering GNPTAB structure have been described and include a duplication of exon 2 in both ML II and ML III patients [89], an Alu–Alu-mediated large homozygous genomic deletion (897 bp) encompassing GNPTAB exon 19 in a ML II a/b patient [90], and a deletion of GNPTAB exon 9 in a Chinese patient in combination with a point mutation [91].
3.3. Disorders of Integral Membrane Proteins
Disorders of integral membrane proteins include six different LSDs that, with the exception of cystinosis and Niemann–Pick type C, are rare in the general population. For five of them, both recurrent and private SVs have been reported in multiple studies.
The most common mutation causing cystinosis is a 57-kb deletion on human chromosome 17p13 that removes the majority of CTNS coding region, as well as some further adjacent genes [92]. This deletion occurs in ~60% of United States and Northern Europe patients [93,94]. A set of additional smaller SVs (from hundreds of base pairs to tens of kb) have been described in single studies (i.e., 13 kb deletion [95]; 266 bp duplication [96], deletion of exons 4–5 [97]; 10 kb deletion [98]; >1.7 kb deletion [99]).
Niemann–Pick type C is caused by mutations in either genes NPC1 (95%) or NPC2 (5%). More than 300 disease-causing mutations have been identified so far in both genes, including indel, missense, nonsense and splicing mutations [100]. Although very rare, SVs have been documented in research literature, providing insights into missing NPC1/2 mutant alleles. In particular, NPC1 whole-gene deletion has been characterized in Niemann–Pick type C patients [101,102], as well as two different larger structural variants encompassing NPC1 and flanking genes (RMC1 and part of ANKRD29 in the first one, ANKRD29 and LAMA3 in the second one) [100], and a homozygous deletion of exons 2 and 3 of NPC2 [103].
Similarly, a partial MCOLN1 gene deletion (c.1_788del) has been detected and described in patients with Mucolipidosis Type IV and identified as founder mutation in the Ashkenazi Jewish population [104], while a homozygous 94 bp deletion and a homozygous deletion of exons 8–9 in SLC17A5 were found in a child and in a prenatal hydrops fetalis with infantile free sialic acid storage disease, respectively [105,106].
Of particular interest is the Xq24 chromosomal region harboring LAMP2, whose mutations cause Danon disease. Several large deletions that alter the LAMP2 exon copy number have been described in male patients with Danon disease and involve repetitive sequence motifs, such as Alu-mediated or TA-rich repeat elements [107]. Recent detailed investigations highlighted a spectrum of peculiar SVs in female patients, such as heterozygous multi-exon LAMP2 deletions and de-novo Alu-mediated Xq24 rearrangement deleting the distal part of CUL4B and the complete sequences of LAMP2, ATP1B4, TMEM255A and ZBTB33 genes [108,109]. In addition, a tandem and likely Alu-mediated duplication of exons 4–5 in the LAMP2 gene has been identified in two brothers with typical Danon disease phenotype and was mosaically distributed in the somatic tissues of their clinically asymptomatic mother [110].
3.4. Neuronal Ceroid-Lipofuscinoses
The neuronal ceroid-lipofuscinoses are a group of neurodegenerative disorders, mostly of childhood onset, characterized by progressive neuroretinal symptomatology, progressive accumulation of auto-fluorescing waxy lipopigments (ceroid-lipofuscin) within the brain, cerebral atrophy, epilepsy, dementia and early death. These disorders were initially classified on the basis of age at onset and consisted of four major groups: infantile, late infantile, juvenile and adult. More recently, the classification was updated to include the genetic causative gene as well as the age of onset [1]. Currently, a total of 14 genes have been identified as the monogenic causes of NCLs [2].
Ceroid lipofuscinosis, neuronal type 3, or Batten disease, is the most prevalent form and is associated with biallelic mutations in CLN3, characterized by a common founder 1.02-kb intragenic deletion mutation (removing exons 7 and 8) occurring in homozygosity in 76% of patients, and compound heterozygosity in an additional 22% of patients [111,112].
Intragenic deletions encompassing exon 4 and a large pathogenic deletion at 13q21.33-q31.1 were described for CLN5, whose mutations are responsible of a late-infantile form of NCLs, originally described in the Finnish and Northern European populations [113,114].
Single allele deletions that complicate genetic diagnosis have also been reported for CLN8. The first de novo terminal deletion of the short arm of chromosome 8p23.3 was described in a Irish patient [115], followed by a Turkish family carrying a 2.6 kb CLN8 intragenic deletion [116]. More recently, three unrelated patients (from Argentina and Britain), each carrying large deletions encompassing the 37 kb CLN8 gene, were characterized by microarray analysis [117].
3.5. Disorders of LROs
The most notable LRO disorders include the multiple variants of Hermansky–Pudlak (HPS) disease, the Griscelli and the Chédiak–Higashi syndromes, which are all characterized by hypopigmentation (owing to a melanosome defect) and prolonged bleeding (owing to a platelet δ granule defect). Of the nine HPS subtypes, the first one was identified in a genetic isolate of central Puerto Rico and is associated to mutations in HPS1 gene [118]. A single hemizygous patient carrying a large deletion (13,966-bp) involving HPS1 and an adjacent gene, C10ORF33, has been reported to date [119].
The gene responsible for Hermansky–Pudlak type 2, AP3B1, is located on the long arm of chromosome 5 (5q14.1) and encodes the β3A subunit of the AP-3 complex [120]. Few cases of SVs in this genomic location are described in the literature. The first one, characterized in 2006, consists of a homozygous deletion of AP3B1 exon 15 (8168 bp), which completely abrogates the proper assembly and the stability of the AP-3 complex [121]. More recently, exon–exon analysis revealed a deletion of single exon 14 and exons 10–25 in two patients with Hermansky–Pudlak type 2 [120]. A consanguineous female infant with reduced pigmentation, neutropenia and recurrent infections was shown to carry a homozygous pericentric inversion inv(5)(p15.1q14.1) that likely disrupts AP3B1 at the breakpoint, thus providing a novel pathogenic mechanism for this disease [122].
4. Advantages and Drawbacks of NGS Targeted Panel Usage for the Detection of SVs in LSD-Related Genes
NGS technologies currently represent powerful tools for the characterization of SVs in human genomes by providing faster, cheaper and higher-resolution testing options [123]. Data produced by these platforms allow both the identification of point mutations, indels and SVs in a single experimental workflow and guarantee greater coverage, higher resolution, more accurate copy number estimation and more precise breakpoint detection [123,124].
Multiple algorithms have been developed to detect SVs based on different features that can be extracted from NGS data and are based on indirect inferences, such as paired-end mapping, splits read, de novo assembly and depth of coverage (DoC), for which we refer the readers to more detailed works [125,126,127,128]. Very briefly, in paired-end sequencing, DNA fragments are assumed to have a specific insert-size distribution. Tools based on the paired-end mapping approach identify CNVs by detecting fragments with a discordance between mapped paired-reads whose distances differ significantly from the predetermined average insert size. The split read approach uses reads from pair-end sequencing in which only one read of the pair is reliably mapped and the other either partially or completely fails to map to the genome. In de novo assembly, DNA fragments are reconstructed from short reads by assembling overlapping reads, and the assembled fragments are then compared to the reference genome to identify regions with different copy numbers. Finally, the DoC approach is based on the assumption that the depth of sequence coverage at any target is generally correlated with the initial copy number of the corresponding region [125,126]. Sensitivity as well as specificity is highly dependent on the SVs detection algorithms used and the estimations of the false-negative and false positive rates greatly vary [125,126]. Unfortunately, there is currently no single informatic method able to identify the full range of structural DNA variations, and multiple complementary SVs callers are required for robust variant detection.
Despite most of the SVs callers having been developed for whole-exome sequencing (WES), recent efforts were implemented to adapt these analysis workflows into adequate strategies for custom or commercial targeted gene panels (tNGS), which offer greater mean coverage of clinically relevant genes at minor cost, and allow more accurate and sensitive detection of disease-related small CNVs encompassing one or more contiguous genes and exons [125]. In the case of LSDs-related genes, for example, specific procedures for library preparation and data analysis are currently adopted when the tNGS panel targets unstable genomic regions such as for GBA and IDS described above, characterized by highly homologous regions or locus with pseudogenes in proximity [9,125]. Indeed, these similarities complicate the alignment of reads corresponding to these regions, significantly affecting data output and must be taken into account in data analysis.
Specific user-friendly web-applications, adapted for custom-designed tNGS panels and dedicated to CNVs detection, have been recently developed to perform the analysis of SNVs, CNVs and indels in a single integrated workflow. In Figure 1 and Figure 2, we show examples of CNVs detection by using tNGS with two panels we have previously used to scan the coding regions of genes implicated in LSDs [5,129]. Deep sequencing data were analyzed with the user-friendly web-application Ion Reporter Software developed by Ion Torrent, which relies on normalized read coverage (DoC) across amplicons to predict also the copy number state, corrected for GC bias and compared with a baseline coverage constructed by the user from control samples with known ploidy.
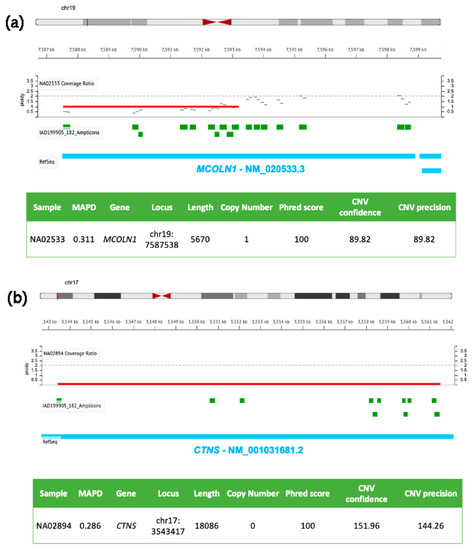
Figure 1.
Molecular details of CNVs detected in MCOLN1 and CTNS by NGS. DNA samples (NA02533 in (a) and NA02894 in (b)) were isolated from clinically diagnosed donor subjects obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research. Targeted NGS analysis was performed by using the tNGS panel previously described [126] and analyzed with the Ion Reporter Software (default parameters for germline CNVs, Median of the Absolute values of all Pairwise Differences < 0.4, CNV Confidence > 20, CNV precision > 10).
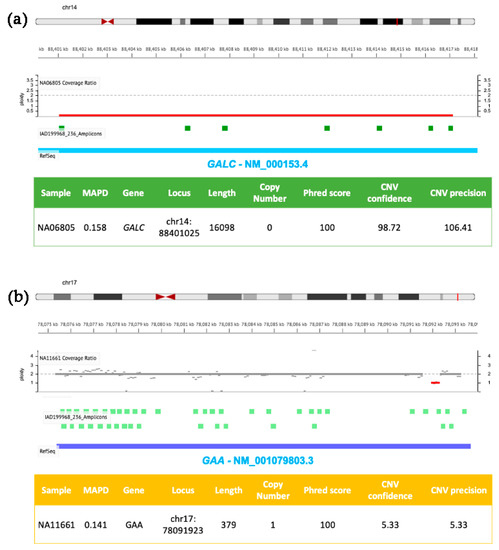
Figure 2.
Molecular details of CNVs detected in GALC and GAA by NGS. DNA samples (NA06805 in (a) and NA11661 in (b)) were isolated from clinically diagnosed donor subjects obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research. Targeted NGS analysis was performed by using the tNGS panel previously described [5] and analyzed with the Ion Reporter Software (default parameters for germline CNVs were as described in Figure 1 with the exception of panel b, where CNV Confidence > 5 and CNV precision > 5).
Figure 1 shows two genomic losses in MCOLN1 (panel a) and CTNS (panel b) detected in two DNA samples from girls affected by Mucolipidosis IV and nephropathic cystinosis, respectively. The first CNV (panel a) is a ~6 kb heterozygous deletion (ploidy state 1) spanning from exon 1 to exon 7 of MCOLN1 gene (the same patient carried also a A>G transition in the acceptor splice site of intron 3 in the other allele, rs104886461); the second panel (b) shows a homozygous ~18 kb deletion (ploidy state 0) of CTNS encompassing exons 1–10, responsible for the pathology.
Figure 2 shows two further genomic losses involving GALC (panel a) and GAA (panel b) exons present in DNA samples from patients affected by Krabbe and Pompe diseases, respectively. Sequencing reads were generated by using a custom developed NGS panel targeting six LSDs-related genes, previously described in [5]. The first CNV (panel a) is a homozygous deletion of ~16 kb in GALC (ploidy state 0) resulting in the loss of exons 11–17, successfully identified by the Ion Reporter Software developed by Ion Torrent. The second CNV (panel b) consists of a small heterozygous single exon (exon 18) deletion of GAA (ploidy state 1), characterized by low confidence and low sensitivity values (these two parameters are sensible to CNV size). The same patient also carried a T>G transversion in an acceptor site in the second allele (rs38683423). Using the confidence and sensitivity values recommended by the Ion Reporter Software, the CNV shown in Figure 2, panel b, would be filtered out, and may determine its exclusion by an unconfident user. Therefore, it is particularly important to pay attention to applying stringent filters, and the analysis of previously characterized positive controls is strongly recommended in order to evaluate both performances and limits of targeted custom NGS panels and correlated bioinformatic analysis.
Along with the technical criticisms related to adequate SVs calling and visualization, the interpretation of their clinical relevance still remains an open current challenge, especially for CNVs. Indeed, the speed at which novel genetic variants are identified by NGS applications is far greater than our ability to interpret data and assign them pathogenicity significance. Therefore, determining what a non-recurrent or a de novo SV actually means for an individual’s health and what is its clinical meaning remains a very difficult task, which commonly generates discrepancies in inter-laboratory practices [130].
Although there are no generally established rules for analysis, interpretation and classification of VUS (Variants of Uncertain Significance) and de novo SVs, some attention should be emphasized before reporting a SV in a diagnostic report. For example, it is important to correlate the identified SV with the downstream metabolic activity, to assess the impact of the variant in multiple mRNAs transcripts and protein generated, to verify family segregation in order to understand if the family history of the disease is consistent with de novo inheritance, to investigate the cis or trans position of the SV compared to a second mutation. It is fundamental to query dedicated public databases (GnomAD, DGV, ClinVar, Decipher) and verify if the identified variant overlaps with other items already described (pathogenic, likely pathogenic, benign, likely benign), to evaluate the frequency in local population and to use a proper prediction tool for SV interpretation (e.g., SV interpreter) [131,132].
It should be kept in mind that, when targeted NGS panels are used, it is not possible to resolve the precise SV configuration and breakpoints; therefore, it could be necessary to use other molecular approaches, such as aCGH, long-read genome sequencing and gene expression analysis to investigate pathogenicity and characterize the possible mechanisms of complex SVs formation [133]. Moreover, given the constantly evolving landscape of SVs, it could be useful to include, in both the policies of the lab and in the informed consent signed by patients, the possibility of re-interpreting and updating the VUS and recontact patients [134].
Another important point concerns the emergence of newborn screening (NBS) programs for LSDs, which are taking place in the form of pilot studies or regional/national initiatives worldwide, and are mainly based on biochemical assays evaluating enzymatic activity or substrates accumulation (i.e., digital microfluidic fluorometry and tandem mass spectrometry) [2,5]. In the era of NBS programs, the search for confirmatory diagnosis on an asymptomatic neonate with a positive finding from a biochemical enzyme test is a great challenge and genotyping is becoming a primary tool, in which even the analysis of SVs should not be underestimated.
5. Conclusions
Conventional diagnostic procedures are able to identify and diagnose most individuals with LSDs. However, a number of cases remain unexplained for difficulties in detecting CNVs or other complex rearrangements altering gene expression and enzymatic function. Numerous case-reports have highlighted the advantages obtained by the use of NGS for the diagnostic validation of LSDs, especially for the detection of SVs in cases for which conventional procedures determine insufficient evidence. The clinical interpretation of complex non-recurrent SVs is still limited and sharing phenotypic and molecular information about patients is essential to define SVs’ clinical relevance and impact on the disease. Additional efforts are ongoing and should be implemented by the scientific community to standardize NGS procedures and data evaluation for introducing this type of analysis in clinical diagnostics of LSDs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines10081836/s1, Table S1: Structural variants altering LSDs-related genes. References [135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161] are cited in the supplementary materials.
Author Contributions
Conceptualization, V.L.C. and S.C.; methodology, V.L.C.; validation, V.L.C.; formal analysis, V.L.C.; investigation, V.L.C.; data curation, V.L.C.; writing—original draft preparation, V.L.C.; writing—review and editing, V.L.C. and S.C.; supervision, S.C.; project administration, S.C.; funding acquisition, S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the joint project between IRIB-CNR and SANOFI “Early diagnosis of some lysosomal diseases: analysis of the clinical utility and diagnostic validity of genomic techniques for their molecular diagnosis. Assessments of the implications of the inclusion of lysosomal diseases in the context of a national neonatal screening program” (project no. 2018/9848).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors gratefully acknowledge Cristina Calì, Alfia Corsino, Maria Patrizia D’Angelo and Francesco Marino for administrative and technical support.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- La Cognata, V.; Guarnaccia, M.; Polizzi, A.; Ruggieri, M.; Cavallaro, S. Highlights on Genomics Applications for Lysosomal Storage Diseases. Cells 2020, 9, 1902. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Guarnaccia, M.; Morello, G.; Ruggieri, M.; Polizzi, A.; Cavallaro, S. Design and Validation of a Custom NGS Panel Targeting a Set of Lysosomal Storage Diseases Candidate for NBS Applications. Int. J. Mol. Sci. 2021, 22, 10064. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.V.F.; Rojas Malaga, D.; Kubaski, F.; Fischinger Moura de Souza, C.; de Oliveira Poswar, F.; Baldo, G.; Giugliani, R. Precision Medicine for Lysosomal Disorders. Biomolecules 2020, 10, 1110. [Google Scholar] [CrossRef]
- Malaga, D.R.; Brusius-Facchin, A.C.; Siebert, M.; Pasqualim, G.; Saraiva-Pereira, M.L.; Souza, C.F.M.; Schwartz, I.V.D.; Matte, U.; Giugliani, R. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 2019, 42, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, A.; D’Avanzo, F.; Bertoldi, L.; Zampieri, G.; Feltrin, E.; De Pascale, F.; Rampazzo, A.; Forzan, M.; Valle, G.; Tomanin, R. Setup and Validation of a Targeted Next-Generation Sequencing Approach for the Diagnosis of Lysosomal Storage Disorders. J. Mol. Diagn. 2020, 22, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Cattarossi, S.; Bembi, B.; Dardis, A. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. J. Mol. Diagn. 2017, 19, 733–741. [Google Scholar] [CrossRef]
- Zampieri, S.; Cattarossi, S.; Pavan, E.; Barbato, A.; Fiumara, A.; Peruzzo, P.; Scarpa, M.; Ciana, G.; Dardis, A. Accurate Molecular Diagnosis of Gaucher Disease Using Clinical Exome Sequencing as a First-Tier Test. Int. J. Mol. Sci. 2021, 22, 5538. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Roberts, R.; Mercer, T.R.; Xu, J.; Sedlazeck, F.J.; Tong, W. Towards accurate and reliable resolution of structural variants for clinical diagnosis. Genome Biol. 2022, 23, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef]
- Lupski, J.R. 2018 Victor A. McKusick Leadership Award: Molecular Mechanisms for Genomic and Chromosomal Rearrangements. Am. J. Hum. Genet. 2019, 104, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Komlosi, K.; Gyenesei, A.; Bene, J. Editorial: Copy Number Variation in Rare Disorders. Front. Genet. 2022, 13, 898059. [Google Scholar] [CrossRef]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.M.; McLysaght, A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 2017, 8, 14366. [Google Scholar] [CrossRef] [PubMed]
- Harel, T.; Lupski, J.R. Genomic disorders 20 years on-mechanisms for clinical manifestations. Clin. Genet. 2018, 93, 439–449. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Morello, G.; D’Agata, V.; Cavallaro, S. Copy number variability in Parkinson’s disease: Assembling the puzzle through a systems biology approach. Hum. Genet. 2017, 136, 13–37. [Google Scholar] [CrossRef]
- Lupski, J.R.; Stankiewicz, P. Genomic disorders: Molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005, 1, e49. [Google Scholar] [CrossRef]
- Alves, M.Q.; Le Trionnaire, E.; Ribeiro, I.; Carpentier, S.; Harzer, K.; Levade, T.; Ribeiro, M.G. Molecular basis of acid ceramidase deficiency in a neonatal form of Farber disease: Identification of the first large deletion in ASAH1 gene. Mol. Genet. Metab. 2013, 109, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Eng, B.; Heshka, T.; Tarnopolsky, M.A.; Nakamura, L.M.; Nowaczyk, M.J.; Waye, J.S. Infantile metachromatic leukodystrophy (MLD) in a compound heterozygote for the c.459 + 1G > A mutation and a complete deletion of the ARSA gene. Am. J. Med. Genet. A 2004, 128, 95–97. [Google Scholar] [CrossRef]
- Zhao, S.; Zhan, X.; Wang, Y.; Ye, J.; Han, L.; Qiu, W.; Gao, X.; Gu, X.; Zhang, H. Large-scale study of clinical and biochemical characteristics of Chinese patients diagnosed with Krabbe disease. Clin. Genet. 2018, 93, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Gaignard, P.; Fagart, J.; Niemir, N.; Puech, J.P.; Azouguene, E.; Dussau, J.; Caillaud, C. Characterization of seven novel mutations on the HEXB gene in French Sandhoff patients. Gene 2013, 512, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Cattarossi, S.; Oller Ramirez, A.M.; Rosano, C.; Lourenco, C.M.; Passon, N.; Moroni, I.; Uziel, G.; Pettinari, A.; Stanzial, F.; et al. Sequence and copy number analyses of HEXB gene in patients affected by Sandhoff disease: Functional characterization of 9 novel sequence variants. PLoS ONE 2012, 7, e41516. [Google Scholar] [CrossRef] [PubMed]
- Sobek, A.K.; Evers, C.; Dekomien, G. Integrated multiplex ligation dependent probe amplification (MLPA) assays for the detection of alterations in the HEXB, GM2A and SMARCAL1 genes to support the diagnosis of Morbus Sandhoff, M. Tay-Sachs variant AB and Schimke immuno-osseous dysplasia in humans. Mol. Cell. Probes 2013, 27, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, D.; Gupta, S.K.; Sarma, A.S.; Ranganath, P.; Jain, S.J.; Sheth, J.; Mistri, M.; Gupta, N.; Kabra, M.; Phadke, S.R.; et al. Functional characterization of novel variants in SMPD1 in Indian patients with acid sphingomyelinase deficiency. Hum. Mutat. 2021, 42, 1336–1350. [Google Scholar] [CrossRef]
- Amico, G.; Grossi, S.; Vijzelaar, R.; Lanza, F.; Mazzotti, R.; Corsolini, F.; Ketema, M.; Filocamo, M. MLPA-based approach for initial and simultaneous detection of GBA deletions and recombinant alleles in patients affected by Gaucher Disease. Mol. Genet. Metab. 2016, 119, 329–337. [Google Scholar] [CrossRef]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Torralba, M.A.; Alfonso, P.; Pérez-Calvo, J.I.; Cenarro, A.; Pastores, G.M.; Giraldo, P.; Civeira, F.; Pocoví, M. High Prevalence of the 55-bp Deletion (c.1263del55) in Exon 9 of the Glucocerebrosidase Gene Causing Misdiagnosis (for Homozygous N370S (c.1226A > G) Mutation) in Spanish Gaucher Disease Patients. Blood Cells Mol. Dis. 2002, 29, 35–40. [Google Scholar] [CrossRef]
- Cozar, M.; Bembi, B.; Dominissini, S.; Zampieri, S.; Vilageliu, L.; Grinberg, D.; Dardis, A. Molecular characterization of a new deletion of the GBA1 gene due to an inter Alu recombination event. Mol. Genet. Metab. 2011, 102, 226–228. [Google Scholar] [CrossRef]
- Filocamo, M.; Mazzotti, R.; Stroppiano, M.; Seri, M.; Giona, F.; Parenti, G.; Regis, S.; Corsolini, F.; Zoboli, S.; Gatti, R. Analysis of the glucocerebrosidase gene and mutation profile in 144 Italian gaucher patients. Hum. Mutat. 2002, 20, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Velayati, A.; Knight, M.A.; Stubblefield, B.K.; Sidransky, E.; Tayebi, N. Identification of Recombinant Alleles Using Quantitative Real-Time PCR. J. Mol. Diagn. 2011, 13, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Farr, M.; Ferreira, S.; Al-Dilaimi, A.; Bogeholz, S.; Goesmann, A.; Kalinowski, J.; Knabbe, C.; Faber, L.; Oliveira, J.P.; Rudolph, V. Fabry disease: Detection of Alu-mediated exon duplication by NGS. Mol. Cell. Probes 2019, 45, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, A.; Centra, M.; Prattichizzo, C.; Gigante, M.; De Fabritiis, M.; Giancaspro, V.; Petrarulo, F.; Santacroce, R.; Margaglione, M.; Gesualdo, L.; et al. Identification of GLA gene deletions in Fabry patients by Multiplex Ligation-dependent Probe Amplification (MLPA). Mol. Genet. Metab. 2008, 94, 382–385. [Google Scholar] [CrossRef]
- Bernstein, H.S.; Bishop, D.F.; Astrin, K.H.; Kornreich, R.; Eng, C.M.; Sakuraba, H.; Desnick, R.J. Fabry disease: Six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J. Clin. Investig. 1989, 83, 1390–1399. [Google Scholar] [CrossRef]
- Kornreich, R.; Bishop, D.F.; Desnick, R.J. Alpha-galactosidase A gene rearrangements causing Fabry disease. Identification of short direct repeats at breakpoints in an Alu-rich gene. J. Biol. Chem. 1990, 265, 9319–9326. [Google Scholar] [CrossRef]
- Eng, C.M.; Resnick-Silverman, L.A.; Niehaus, D.J.; Astrin, K.H.; Desnick, R.J. Nature and frequency of mutations in the alpha-galactosidase A gene that cause Fabry disease. Am. J. Hum. Genet. 1993, 53, 1186–1197. [Google Scholar]
- Nowak, A.; Murik, O.; Mann, T.; Zeevi, D.A.; Altarescu, G. Detection of single nucleotide and copy number variants in the Fabry disease-associated GLA gene using nanopore sequencing. Sci. Rep. 2021, 11, 22372. [Google Scholar] [CrossRef]
- Alabbas, F.; Elyamany, G.; Alanzi, T.; Ali, T.B.; Albatniji, F.; Alfaraidi, H. Wolman’s disease presenting with secondary hemophagocytic lymphohistiocytosis: A case report from Saudi Arabia and literature review. BMC Pediatr. 2021, 21, 72. [Google Scholar] [CrossRef]
- Brusius-Facchin, A.C.; Schwartz, I.V.; Zimmer, C.; Ribeiro, M.G.; Acosta, A.X.; Horovitz, D.; Monlleo, I.L.; Fontes, M.I.; Fett-Conte, A.; Sobrinho, R.P.; et al. Mucopolysaccharidosis type II: Identification of 30 novel mutations among Latin American patients. Mol. Genet. Metab. 2014, 111, 133–138. [Google Scholar] [CrossRef]
- Jahic, A.; Gunther, S.; Muschol, N.; Fossoy Stadheim, B.; Braaten, O.; Kjensli Hyldebrandt, H.; Kuiper, G.A.; Tylee, K.; Wijburg, F.A.; Beetz, C. “Missing mutations” in MPS I: Identification of two novel copy number variations by an IDUA-specific in house MLPA assay. Mol. Genet. Genomic Med. 2019, 7, e00615. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.R.; Rafati, M.; Shadnoush, M.; Pourbabaee, S.; Aghighi, M.; Mirab Samiee, S.; Kermanchi, J.; Alaei, M.R.; Salehpour, S.; Amirkashani, D.; et al. Molecular characterization of a large cohort of mucopolysaccharidosis patients: Iran Mucopolysaccharidosis RE-diagnosis study (IMPRESsion). Hum. Mutat. 2022, 43, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Ouesleti, S.; Brunel, V.; Ben Turkia, H.; Dranguet, H.; Miled, A.; Miladi, N.; Ben Dridi, M.F.; Lavoinne, A.; Saugier-Veber, P.; Bekri, S. Molecular characterization of MPS IIIA, MPS IIIB and MPS IIIC in Tunisian patients. Clin. Chim. Acta 2011, 412, 2326–2331. [Google Scholar] [CrossRef] [PubMed]
- Champion, K.J.; Basehore, M.J.; Wood, T.; Destree, A.; Vannuffel, P.; Maystadt, I. Identification and characterization of a novel homozygous deletion in the alpha-N-acetylglucosaminidase gene in a patient with Sanfilippo type B syndrome (mucopolysaccharidosis IIIB). Mol. Genet. Metab. 2010, 100, 51–56. [Google Scholar] [CrossRef]
- Ozkinay, F.; Emecen, D.A.; Kose, M.; Isik, E.; Bozaci, A.E.; Canda, E.; Tuysuz, B.; Zubarioglu, T.; Atik, T.; Onay, H. Clinical and genetic features of 13 patients with mucopolysaccarhidosis type IIIB: Description of two novel NAGLU gene mutations. Mol. Genet. Metab. Rep. 2021, 27, 100732. [Google Scholar] [CrossRef]
- Sudrie-Arnaud, B.; Snanoudj, S.; Dabaj, I.; Dranguet, H.; Abily-Donval, L.; Lebas, A.; Vezain, M.; Heron, B.; Marie, I.; Duval-Arnould, M.; et al. Next-Generation Molecular Investigations in Lysosomal Diseases: Clinical Integration of a Comprehensive Targeted Panel. Diagnostics 2021, 11, 294. [Google Scholar] [CrossRef]
- Beesley, C.E.; Concolino, D.; Filocamo, M.; Winchester, B.G.; Strisciuglio, P. Identification and characterisation of an 8.7 kb deletion and a novel nonsense mutation in two Italian families with Sanfilippo syndrome type D (mucopolysaccharidosis IIID). Mol. Genet. Metab. 2007, 90, 77–80. [Google Scholar] [CrossRef]
- Valstar, M.J.; Bertoli-Avella, A.M.; Wessels, M.W.; Ruijter, G.J.; de Graaf, B.; Olmer, R.; Elfferich, P.; Neijs, S.; Kariminejad, R.; Suheyl Ezgu, F.; et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum. Mutat. 2010, 31, E1348–E1360. [Google Scholar] [CrossRef]
- Caciotti, A.; Tonin, R.; Rigoldi, M.; Ferri, L.; Catarzi, S.; Cavicchi, C.; Procopio, E.; Donati, M.A.; Ficcadenti, A.; Fiumara, A.; et al. Optimizing the molecular diagnosis of GALNS: Novel methods to define and characterize Morquio-A syndrome-associated mutations. Hum. Mutat. 2015, 36, 357–368. [Google Scholar] [CrossRef]
- Hori, T.; Tomatsu, S.; Nakashima, Y.; Uchiyama, A.; Fukuda, S.; Sukegawa, K.; Shimozawa, N.; Suzuki, Y.; Kondo, N.; Horiuchi, T.; et al. Mucopolysaccharidosis type IVA: Common double deletion in the N-acetylgalactosamine-6-sulfatase gene (GALNS). Genomics 1995, 26, 535–542. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Nishioka, T.; Gutierrez, M.A.; Pena, O.M.; Tranda Firescu, G.G.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 2005, 26, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Tomatsu, S.; Masuno, M.; Ogawa, T.; Yamagishi, A.; Rezvi, G.M.; Sukegawa, K.; Shimozawa, N.; Suzuki, Y.; Kondo, N.; et al. Mucopolysaccharidosis IVA: Submicroscopic deletion of 16q24.3 and a novel R386C mutation of N-acetylgalactosamine-6-sulfate sulfatase gene in a classical Morquio disease. Hum. Mutat. 1996, 7, 123–134. [Google Scholar] [CrossRef]
- Ittiwut, C.; Boonbuamas, S.; Srichomthong, C.; Ittiwut, R.; Suphapeetiporn, K.; Shotelersuk, V. Novel Mutations, Including a Large Deletion in the ARSB Gene, Causing Mucopolysaccharidosis Type VI. Genet. Test. Mol. Biomark. 2017, 21, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.D.; Ke, Y.Y.; Chou, I.C.; Wang, C.H.; Tsai, F.J. Deletion of exon 4 in the N-acetylgalactosamine-4-sulfatase gene in a Taiwanese patient with mucopolysaccharidosis type VI. Tohoku J. Exp. Med. 2015, 235, 267–273. [Google Scholar] [CrossRef][Green Version]
- Villani, G.R.; Grosso, M.; Pontarelli, G.; Chierchia, A.; Sessa, R.; Sibilio, M.; Parenti, G.; Di Natale, P. Large deletion involving exon 5 of the arylsulfatase B gene caused apparent homozygosity in a mucopolysaccharidosis type VI patient. Genet. Test. Mol. Biomark. 2010, 14, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Verma, G.; Saxena, D.; Kabra, M.; Gupta, N.; Mandal, K.; Moirangthem, A.; Sheth, J.; Puri, R.D.; Bijarnia-Mahay, S.; et al. Genotype-phenotype spectrum of 130 unrelated Indian families with Mucopolysaccharidosis type II. Eur. J. Med. Genet. 2022, 65, 104447. [Google Scholar] [CrossRef]
- Alcantara-Ortigoza, M.A.; Garcia-de Teresa, B.; Gonzalez-Del Angel, A.; Berumen, J.; Guardado-Estrada, M.; Fernandez-Hernandez, L.; Navarrete-Martinez, J.I.; Maza-Morales, M.; Rius-Dominguez, R. Wide allelic heterogeneity with predominance of large IDS gene complex rearrangements in a sample of Mexican patients with Hunter syndrome. Clin. Genet. 2016, 89, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Mangas, M.; Prata, M.J.; Ribeiro, G.; Lopes, L.; Ribeiro, H.; Pinto-Basto, J.; Lima, M.R.; Lacerda, L. Molecular characterization of Portuguese patients with mucopolysaccharidosis type II shows evidence that the IDS gene is prone to splicing mutations. J. Inherit. Metab. Dis. 2006, 29, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, M.L.; Dahl, N.; Malmgren, H.; Kleijer, W.J.; Tonnesen, T.; Carlberg, B.M.; Pettersson, U. Inversion of the IDS gene resulting from recombination with IDS-related sequences is a common cause of the Hunter syndrome. Hum. Mol. Genet. 1995, 4, 615–621. [Google Scholar] [CrossRef]
- Bunge, S.; Rathmann, M.; Steglich, C.; Bondeson, M.L.; Tylki-Szymanska, A.; Popowska, E.; Gal, A. Homologous nonallelic recombinations between the iduronate-sulfatase gene and pseudogene cause various intragenic deletions and inversions in patients with mucopolysaccharidosis type II. Eur. J. Hum. Genet. 1998, 6, 492–500. [Google Scholar] [CrossRef]
- Chkioua, L.; Grissa, O.; Leban, N.; Gribaa, M.; Boudabous, H.; Turkia, H.B.; Ferchichi, S.; Tebib, N.; Laradi, S. The mutational spectrum of hunter syndrome reveals correlation between biochemical and clinical profiles in Tunisian patients. BMC Med. Genet. 2020, 21, 111. [Google Scholar] [CrossRef]
- Filocamo, M.; Bonuccelli, G.; Corsolini, F.; Mazzotti, R.; Cusano, R.; Gatti, R. Molecular analysis of 40 Italian patients with mucopolysaccharidosis type II: New mutations in the iduronate-2-sulfatase (IDS) gene. Hum. Mutat. 2001, 18, 164–165. [Google Scholar] [CrossRef]
- Galvis, J.; Gonzalez, J.; Uribe, A.; Velasco, H. Deep Genotyping of the IDS Gene in Colombian Patients with Hunter Syndrome. JIMD Rep. 2015, 19, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Hartog, C.; Fryer, A.; Upadhyaya, M. Mutation analysis of iduronate-2-sulphatase gene in 24 patients with Hunter syndrome: Characterisation of 6 novel mutations. Mutation in brief no. 249. Online. Hum. Mutat. 1999, 14, 87. [Google Scholar] [CrossRef]
- Kosuga, M.; Mashima, R.; Hirakiyama, A.; Fuji, N.; Kumagai, T.; Seo, J.H.; Nikaido, M.; Saito, S.; Ohno, K.; Sakuraba, H.; et al. Molecular diagnosis of 65 families with mucopolysaccharidosis type II (Hunter syndrome) characterized by 16 novel mutations in the IDS gene: Genetic, pathological, and structural studies on iduronate-2-sulfatase. Mol. Genet. Metab. 2016, 118, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Oshima, J.; Lee, J.A.; Breman, A.M.; Fernandes, P.H.; Babovic-Vuksanovic, D.; Ward, P.A.; Wolfe, L.A.; Eng, C.M.; Del Gaudio, D. LCR-initiated rearrangements at the IDS locus, completed with Alu-mediated recombination or non-homologous end joining. J. Hum. Genet. 2011, 56, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Uttarilli, A.; Ranganath, P.; Matta, D.; Md Nurul Jain, J.; Prasad, K.; Babu, A.S.; Girisha, K.M.; Verma, I.C.; Phadke, S.R.; Mandal, K.; et al. Identification and characterization of 20 novel pathogenic variants in 60 unrelated Indian patients with mucopolysaccharidoses type I and type II. Clin. Genet. 2016, 90, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, A.; Tomanin, R.; Rampazzo, A.; Rigon, C.; Gasparotto, N.; Cassina, M.; Clementi, M.; Scarpa, M. A Hunter Patient with a Severe Phenotype Reveals Two Large Deletions and Two Duplications Extending 1.2 Mb Distally to IDS Locus. JIMD Rep. 2014, 17, 13–21. [Google Scholar] [CrossRef]
- Jezela-Stanek, A.; Pokora, P.; Mlynek, M.; Smyk, M.; Ziemkiewicz, K.; Rozdzynska-Swiatkowska, A.; Tylki-Szymanska, A. Diverse clinical outcome of Hunter syndrome in patients with chromosomal aberration encompassing entire and partial IDS deletions: What is important for early diagnosis and counseling? Clin. Dysmorphol. 2021, 30, 76–82. [Google Scholar] [CrossRef]
- Dvorakova, L.; Vlaskova, H.; Sarajlija, A.; Ramadza, D.P.; Poupetova, H.; Hruba, E.; Hlavata, A.; Bzduch, V.; Peskova, K.; Storkanova, G.; et al. Genotype-phenotype correlation in 44 Czech, Slovak, Croatian and Serbian patients with mucopolysaccharidosis type II. Clin. Genet. 2017, 91, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Brusius-Facchin, A.C.; Abrahao, L.; Schwartz, I.V.; Lourenco, C.M.; Santos, E.S.; Zanetti, A.; Tomanin, R.; Scarpa, M.; Giugliani, R.; Leistner-Segal, S. Extension of the molecular analysis to the promoter region of the iduronate 2-sulfatase gene reveals genomic alterations in mucopolysaccharidosis type II patients with normal coding sequence. Gene 2013, 526, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina, A.N.; Voskoboeva, E.Y.; Zakharova, E.Y.; Nikolaeva, E.A.; Kanivets, I.V.; Kolotii, A.D.; Baydakova, G.V.; Kharabadze, M.N.; Kuramagomedova, R.G.; Melnikova, N.V. Case report: A rare case of Hunter syndrome (type II mucopolysaccharidosis) in a girl. BMC Med. Genet. 2019, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Vanherpe, P.; Fieuws, S.; D’Hondt, A.; Bleyenheuft, C.; Demaerel, P.; De Bleecker, J.; Van den Bergh, P.; Baets, J.; Remiche, G.; Verhoeven, K.; et al. Late-onset Pompe disease (LOPD) in Belgium: Clinical characteristics and outcome measures. Orphanet J. Rare Dis. 2020, 15, 83. [Google Scholar] [CrossRef]
- Sawada, T.; Kido, J.; Nakamura, K. Newborn Screening for Pompe Disease. Int. J. Neonatal Screen. 2020, 6, 31. [Google Scholar] [CrossRef]
- Tang, H.; Feuchtbaum, L.; Sciortino, S.; Matteson, J.; Mathur, D.; Bishop, T.; Olney, R.S. The First Year Experience of Newborn Screening for Pompe Disease in California. Int. J. Neonatal Screen. 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Vanderkraan, M.; Kroos, M.A.; Joosse, M.; Bijvoet, A.G.A.; Verbeet, M.P.; Kleijer, W.J.; Reuser, A.J.J. Deletion of Exon 18 Is a Frequent Mutation in Glycogen Storage Disease Type II. Biochem. Biophys. Res. Commun. 1994, 203, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Vorgerd, M.; Burwinkel, B.; Reichmann, H.; Malin, J.-P.; Kilimann, M.W. Adult-onset glycogen storage disease type II: Phenotypic and allelic heterogeneity in German patients. Neurogenetics 1998, 1, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Semplicini, C.; Letard, P.; De Antonio, M.; Taouagh, N.; Perniconi, B.; Bouhour, F.; Echaniz-Laguna, A.; Orlikowski, D.; Sacconi, S.; Salort-Campana, E.; et al. Late-onset Pompe disease in France: Molecular features and epidemiology from a nationwide study. J. Inherit. Metab. Dis. 2018, 41, 937–946. [Google Scholar] [CrossRef]
- Mori, M.; Haskell, G.; Kazi, Z.; Zhu, X.; DeArmey, S.M.; Goldstein, J.L.; Bali, D.; Rehder, C.; Cirulli, E.T.; Kishnani, P.S. Sensitivity of whole exome sequencing in detecting infantile- and late-onset Pompe disease. Mol. Genet. Metab. 2017, 122, 189–197. [Google Scholar] [CrossRef]
- Bali, D.S.; Goldstein, J.L.; Banugaria, S.; Dai, J.; Mackey, J.; Rehder, C.; Kishnani, P.S. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: Lessons learned from 10 years of clinical laboratory testing experience. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.; Pomponio, R.J.; van Vliet, L.; Palmer, R.E.; Phipps, M.; Van der Helm, R.; Halley, D.; Reuser, A. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum. Mutat. 2008, 29, E13–E26. [Google Scholar] [CrossRef]
- McCready, M.E.; Carson, N.L.; Chakraborty, P.; Clarke, J.T.R.; Callahan, J.W.; Skomorowski, M.A.; Chan, A.K.J.; Bamforth, F.; Casey, R.; Rupar, C.A.; et al. Development of a clinical assay for detection of GAA mutations and characterization of the GAA mutation spectrum in a Canadian cohort of individuals with glycogen storage disease, type II. Mol. Genet. Metab. 2007, 92, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Huie, M.L.; Anyane-Yeboa, K.; Guzman, E.; Hirschhorn, R. Homozygosity for Multiple Contiguous Single-Nucleotide Polymorphisms as an Indicator of Large Heterozygous Deletions: Identification of a Novel Heterozygous 8-kb Intragenic Deletion (IVS7–19 to IVS15–17) in a Patient with Glycogen Storage Disease Type II. Am. J. Hum. Genet. 2002, 70, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.J.; Seo, H.C.; Coucke, P.; Tonlorenzi, R.; O’Brien, J.S. Spectrum of mutations in fucosidosis. Eur. J. Hum. Genet. 1999, 7, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.P.; Lee, N.C.; Lin, C.H. Clinical and electrophysiological characteristics of a type 1 sialidosis patient with a novel deletion mutation in NEU1 gene. J. Formos. Med. Assoc. 2020, 119, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Q. Heterozygous structural variation mimicking homozygous missense mutations in NEU1 associated with presenting clinical signs in eyes alone. Ophthalmic Genet. 2020, 41, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, M.; Smeland, M.F.; Lindgren, J.; Sikora, P.; Riise Stensland, H.M.F.; Asin-Cayuela, J. beta-Mannosidosis caused by a novel homozygous intragenic inverted duplication in MANBA. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003954. [Google Scholar] [CrossRef]
- Otomo, T.; Muramatsu, T.; Yorifuji, T.; Okuyama, T.; Nakabayashi, H.; Fukao, T.; Ohura, T.; Yoshino, M.; Tanaka, A.; Okamoto, N.; et al. Mucolipidosis II and III alpha/beta: Mutation analysis of 40 Japanese patients showed genotype-phenotype correlation. J. Hum. Genet. 2009, 54, 145–151. [Google Scholar] [CrossRef]
- Coutinho, M.F.; da Silva Santos, L.; Lacerda, L.; Quental, S.; Wibrand, F.; Lund, A.M.; Johansen, K.B.; Prata, M.J.; Alves, S. Alu-Alu Recombination Underlying the First Large Genomic Deletion in GlcNAc-Phosphotransferase Alpha/Beta (GNPTAB) Gene in a MLII Alpha/Beta Patient. JIMD Rep. 2012, 4, 117–124. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, J.; Qiu, W.J.; Han, L.S.; Gao, X.L.; Liang, L.L.; Gu, X.F.; Zhang, H.W. Identification of predominant GNPTAB gene mutations in Eastern Chinese patients with mucolipidosis II/III and a prenatal diagnosis of mucolipidosis II. Acta Pharmacol. Sin. 2019, 40, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Freed, K.A.; Blangero, J.; Howard, T.; Johnson, M.P.; Curran, J.E.; Garcia, Y.R.; Lan, H.C.; Abboud, H.E.; Moses, E.K. The 57 kb deletion in cystinosis patients extends into TRPV1 causing dysregulation of transcription in peripheral blood mononuclear cells. J. Med. Genet. 2011, 48, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Kiehntopf, M.; Schickel, J.; Gonne, B.; Koch, H.G.; Superti-Furga, A.; Steinmann, B.; Deufel, T.; Harms, E. Analysis of the CTNS gene in patients of German and Swiss origin with nephropathic cystinosis. Hum. Mutat. 2002, 20, 237. [Google Scholar] [CrossRef] [PubMed]
- Bendavid, C.; Kleta, R.; Long, R.; Ouspenskaia, M.; Muenke, M.; Haddad, B.R.; Gahl, W.A. FISH diagnosis of the common 57-kb deletion in CTNS causing cystinosis. Hum. Genet. 2004, 115, 510–514. [Google Scholar] [CrossRef]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- Taranta, A.; Wilmer, M.J.; van den Heuvel, L.P.; Bencivenga, P.; Bellomo, F.; Levtchenko, E.N.; Emma, F. Analysis of CTNS gene transcripts in nephropathic cystinosis. Pediatr. Nephrol. 2010, 25, 1263–1267. [Google Scholar] [CrossRef][Green Version]
- Anikster, Y.; Shotelersuk, V.; Gahl, W.A. CTNS mutations in patients with cystinosis. Hum. Mutat. 1999, 14, 454–458. [Google Scholar] [CrossRef]
- Topaloglu, R.; Vilboux, T.; Coskun, T.; Ozaltin, F.; Tinloy, B.; Gunay-Aygun, M.; Bakkaloglu, A.; Besbas, N.; van den Heuvel, L.; Kleta, R.; et al. Genetic basis of cystinosis in Turkish patients: A single-center experience. Pediatr. Nephrol. 2012, 27, 115–121. [Google Scholar] [CrossRef]
- Kiehntopf, M.; Varga, R.E.; Koch, H.G.; Beetz, C. A homemade MLPA assay detects known CTNS mutations and identifies a novel deletion in a previously unresolved cystinosis family. Gene 2012, 495, 89–92. [Google Scholar] [CrossRef]
- Rodriguez-Pascau, L.; Toma, C.; Macias-Vidal, J.; Cozar, M.; Cormand, B.; Lykopoulou, L.; Coll, M.J.; Grinberg, D.; Vilageliu, L. Characterisation of two deletions involving NPC1 and flanking genes in Niemann-Pick type C disease patients. Mol. Genet. Metab. 2012, 107, 716–720. [Google Scholar] [CrossRef]
- Bauer, P.; Knoblich, R.; Bauer, C.; Finckh, U.; Hufen, A.; Kropp, J.; Braun, S.; Kustermann-Kuhn, B.; Schmidt, D.; Harzer, K.; et al. NPC1: Complete genomic sequence, mutation analysis, and characterization of haplotypes. Hum. Mutat. 2002, 19, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Macias-Vidal, J.; Rodriguez-Pascau, L.; Sanchez-Olle, G.; Lluch, M.; Vilageliu, L.; Grinberg, D.; Coll, M.J.; Spanish, N.P.C.W.G. Molecular analysis of 30 Niemann-Pick type C patients from Spain. Clin. Genet. 2011, 80, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, M.; Prasada, L.H.; Bhowmik, A.D.; Trujillano, D.; Shukla, A.; Chakraborti, S.; Kandaswamy, K.K.; Rolfs, A.; Kamath, N.; Dalal, A.; et al. Homozygous deletion of exons 2 and 3 of NPC2 associated with Niemann-Pick disease type C. Am. J. Med. Genet. A 2016, 170, 2486–2489. [Google Scholar] [CrossRef] [PubMed]
- Bach, G.; Webb, M.B.; Bargal, R.; Zeigler, M.; Ekstein, J. The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of 3 novel MCOLN1 mutations. Hum. Mutat. 2005, 26, 591. [Google Scholar] [CrossRef] [PubMed]
- Zigman, T.; Petkovic Ramadza, D.; Lusic, M.; Zekusic, M.; Ninkovic, D.; Gardijan, D.; Potocki, K.; Omerza, L.; Beljan, L.; Zarkovic, K.; et al. Hypogammaglobulinemia and imaging features in a patient with infantile free sialic acid storage disease (ISSD) and a novel mutation in the SLC17A5 gene. J. Pediatr. Endocrinol. Metab. 2018, 31, 1155–1159. [Google Scholar] [CrossRef]
- Hasnain, A.; Burnett, S.; Agatep, R.; Spriggs, E.; Chodirker, B.; Mhanni, A.A.A. Prenatal hydrops fetalis associated with infantile free sialic acid storage disease due to a novel homozygous deletion in the SLC17A5 gene. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006106. [Google Scholar] [CrossRef]
- Yang, Z.; Funke, B.H.; Cripe, L.H.; Vick, G.W., 3rd; Mancini-Dinardo, D.; Pena, L.S.; Kanter, R.J.; Wong, B.; Westerfield, B.H.; Varela, J.J.; et al. LAMP2 microdeletions in patients with Danon disease. Circ. Cardiovasc. Genet. 2010, 3, 129–137. [Google Scholar] [CrossRef][Green Version]
- Majer, F.; Piherova, L.; Reboun, M.; Stara, V.; Pelak, O.; Norambuena, P.; Stranecky, V.; Krebsova, A.; Vlaskova, H.; Dvorakova, L.; et al. LAMP2 exon-copy number variations in Danon disease heterozygote female probands: Infrequent or underdetected? Am. J. Med. Genet. A 2018, 176, 2430–2434. [Google Scholar] [CrossRef]
- Majer, F.; Kousal, B.; Dusek, P.; Piherova, L.; Reboun, M.; Mihalova, R.; Gurka, J.; Krebsova, A.; Vlaskova, H.; Dvorakova, L.; et al. Alu-mediated Xq24 deletion encompassing CUL4B, LAMP2, ATP1B4, TMEM255A, and ZBTB33 genes causes Danon disease in a female patient. Am. J. Med. Genet. A 2020, 182, 219–223. [Google Scholar] [CrossRef]
- Majer, F.; Pelak, O.; Kalina, T.; Vlaskova, H.; Dvorakova, L.; Honzik, T.; Palecek, T.; Kuchynka, P.; Masek, M.; Zeman, J.; et al. Mosaic tissue distribution of the tandem duplication of LAMP2 exons 4 and 5 demonstrates the limits of Danon disease cellular and molecular diagnostics. J. Inherit. Metab. Dis. 2014, 37, 117–124. [Google Scholar] [CrossRef]
- Ku, C.A.; Hull, S.; Arno, G.; Vincent, A.; Carss, K.; Kayton, R.; Weeks, D.; Anderson, G.W.; Geraets, R.; Parker, C.; et al. Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol. 2017, 135, 749–760. [Google Scholar] [CrossRef] [PubMed]
- De los Reyes, E.; Dyken, P.R.; Phillips, P.; Brodsky, M.; Bates, S.; Glasier, C.; Mrak, R.E. Profound infantile neuroretinal dysfunction in a heterozygote for the CLN3 genetic defect. J. Child Neurol. 2004, 19, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Mullen, T.E.; Kiely, R.; Min, J.; Feng, X.; Cao, Y.; O’Malley, L.; Shen, Y.; Chu-Shore, C.; Mole, S.E.; et al. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology 2010, 74, 565–571. [Google Scholar] [CrossRef]
- Li, W.; Fan, X.; Zhang, Y.; Huang, L.; Jiang, T.; Qin, Z.; Su, J.; Luo, J.; Yi, S.; Zhang, S.; et al. A novel pathogenic frameshift variant unmasked by a large de novo deletion at 13q21.33-q31.1 in a Chinese patient with neuronal ceroid lipofuscinosis type 5. BMC Med. Genet. 2020, 21, 100. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.M.; O’HIci, B.; Anderson, G.; Nestor, T.; Lynch, S.A.; King, M.D. Variant late-infantile neuronal ceroid lipofuscinosis due to a novel heterozygous CLN8 mutation and de novo 8p23.3 deletion. Clin. Genet. 2012, 81, 602–604. [Google Scholar] [CrossRef]
- Reinhardt, K.; Grapp, M.; Schlachter, K.; Bruck, W.; Gartner, J.; Steinfeld, R. Novel CLN8 mutations confirm the clinical and ethnic diversity of late infantile neuronal ceroid lipofuscinosis. Clin. Genet. 2010, 77, 79–85. [Google Scholar] [CrossRef]
- Beesley, C.; Guerreiro, R.J.; Bras, J.T.; Williams, R.E.; Taratuto, A.L.; Eltze, C.; Mole, S.E. CLN8 disease caused by large genomic deletions. Mol. Genet. Genomic Med. 2017, 5, 85–91. [Google Scholar] [CrossRef]
- Anikster, Y.; Huizing, M.; White, J.; Shevchenko, Y.O.; Fitzpatrick, D.L.; Touchman, J.W.; Compton, J.G.; Bale, S.J.; Swank, R.T.; Gahl, W.A.; et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat. Genet. 2001, 28, 376–380. [Google Scholar] [CrossRef]
- Griffin, A.E.; Cobb, B.R.; Anderson, P.D.; Claassen, D.A.; Helip-Wooley, A.; Huizing, M.; Gahl, W.A. Detection of hemizygosity in Hermansky-Pudlak syndrome by quantitative real-time PCR. Clin. Genet. 2005, 68, 23–30. [Google Scholar] [CrossRef]
- Alizadeh, Z.; Nabilou, S.; Mazinani, M.; Tajik, S.; Hamidieh, A.A.; Houshmand, M.; Fazlollahi, M.R.; Pourpak, Z. Partial albinism and immunodeficiency in patients with Hermansky-Pudlak Type II: Introducing 2 novel mutations. Scand. J. Immunol. 2021, 93, e12966. [Google Scholar] [CrossRef]
- Jung, J.; Bohn, G.; Allroth, A.; Boztug, K.; Brandes, G.; Sandrock, I.; Schaffer, A.A.; Rathinam, C.; Kollner, I.; Beger, C.; et al. Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood 2006, 108, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Murden, S.L.; Brooks, C.; Maloney, V.; Manning, R.A.; Gilmour, K.C.; Bharadwaj, V.; de la Fuente, J.; Chakravorty, S.; Mumford, A.D. Disruption of AP3B1 by a chromosome 5 inversion: A new disease mechanism in Hermansky-Pudlak syndrome type 2. BMC Med. Genet. 2013, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Larson, D.E.; Chen, K.; Ding, L.; Wilson, R.K. Massively parallel sequencing approaches for characterization of structural variation. Methods Mol. Biol. 2012, 838, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.M.; Quinlan, A.R. Detection and interpretation of genomic structural variation in mammals. Methods Mol. Biol. 2012, 838, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Roca, I.; Gonzalez-Castro, L.; Fernandez, H.; Couce, M.L.; Fernandez-Marmiesse, A. Free-access copy-number variant detection tools for targeted next-generation sequencing data. Mutat. Res. Rev. Mutat. Res. 2019, 779, 114–125. [Google Scholar] [CrossRef]
- Gong, T.; Hayes, V.M.; Chan, E.K.F. Detection of somatic structural variants from short-read next-generation sequencing data. Brief. Bioinform. 2021, 22, bbaa056. [Google Scholar] [CrossRef] [PubMed]
- Abel, H.J.; Duncavage, E.J. Detection of structural DNA variation from next generation sequencing data: A review of informatic approaches. Cancer Genet. 2013, 206, 432–440. [Google Scholar] [CrossRef]
- Hayes, M. Computational Analysis of Structural Variation in Cancer Genomes. Methods Mol. Biol. 2019, 1878, 65–83. [Google Scholar] [CrossRef]
- La Cognata, V.; Cavallaro, S. A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases. Genes 2021, 12, 1750. [Google Scholar] [CrossRef]
- Zhang, K.; Lin, G.; Han, D.; Han, Y.; Peng, R.; Li, J. Adaptation of ACMG-ClinGen Technical Standards for Copy Number Variant Interpretation Concordance. Front. Genet. 2022, 13, 829728. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Fino, J.; Marques, B.; Dong, Z.; David, D. SVInterpreter: A Comprehensive Topologically Associated Domain-Based Clinical Outcome Prediction Tool for Balanced and Unbalanced Structural Variants. Front. Genet. 2021, 12, 757170. [Google Scholar] [CrossRef] [PubMed]
- Sanchis-Juan, A.; Stephens, J.; French, C.E.; Gleadall, N.; Megy, K.; Penkett, C.; Shamardina, O.; Stirrups, K.; Delon, I.; Dewhurst, E.; et al. Complex structural variants in Mendelian disorders: Identification and breakpoint resolution using short- and long-read genome sequencing. Genome. Med. 2018, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Vears, D.F.; Niemiec, E.; Howard, H.C.; Borry, P. Analysis of VUS reporting, variant reinterpretation and recontact policies in clinical genomic sequencing consent forms. Eur. J. Hum. Genet. 2018, 26, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.A.; Luzi, P.; Chen, Y.Q.; Wenger, D.A. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum. Mol. Genet. 1995, 4, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Selleri, S.; Torchiana, E.; Pareyson, D.; Lulli, L.; Bertagnolio, B.; Savoiardo, M.; Farina, L.; Carrara, F.; Filocamo, M.; Gatti, R.; et al. Deletion of exons 11-17 and novel mutations of the galactocerebrosidase gene in adult- and early-onset patients with Krabbe disease. J. Neurol. 2000, 247, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Le Coutre, P.; Demina, A.; Beutler, E.; Beck, M.; Petrides, P.E. Molecular analysis of Gaucher disease: Distribution of eight mutations and the complete gene deletion in 27 patients from Germany. Hum. Genet. 1997, 99, 816–821. [Google Scholar] [CrossRef]
- Breen, C.; Mercer, J.; Jones, S.A.; Jahic, A.; Heptinstall, L.; Tylee, K.; Newman, W.G.; Beetz, C. Maternal mosaicism for IDUA deletion clarifies recurrence risk in MPS I. Hum. Genome Var. 2016, 3, 16031. [Google Scholar] [CrossRef]
- Birot, A.M.; Bouton, O.; Froissart, R.; Maire, I.; Bozon, D. IDS gene-pseudogene exchange responsible for an intragenic deletion in a Hunter patient. Hum. Mutat. 1996, 8, 44–50. [Google Scholar] [CrossRef]
- Toscano, A.; Tsujino, S.; Vita, G.; Shanske, S.; Messina, C.; Dimauro, S. Molecular basis of muscle phosphoglycerate mutase (PGAM-M) deficiency in the Italian kindred. Muscle Nerve 1996, 19, 1134–1137. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Regis, S.; Filocamo, M.; Corsolini, F.; Caroli, F.; Gatti, R. A deletion involving exons 2-4 in the iduronate-2-sulfatase gene of a patient with intermediate Hunter syndrome. Clin. Genet. 1998, 53, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Brusius-Facchin, A.C.; De Souza, C.F.; Schwartz, I.V.; Riegel, M.; Melaragno, M.I.; Correia, P.; Moraes, L.M.; Llerena, J., Jr.; Giugliani, R.; Leistner-Segal, S. Severe phenotype in MPS II patients associated with a large deletion including contiguous genes. Am. J. Med. Genet. A 2012, 158, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Timms, K.M.; Huckett, L.E.; Belmont, J.W.; Shapira, S.K.; Gibbs, R.A. DNA deletion confined to the iduronate-2-sulfatase promoter abolishes IDS gene expression. Hum. Mutat. 1998, 11, 121–126. [Google Scholar] [CrossRef]
- Flomen, R.H.; Green, P.M.; Bentley, D.R.; Giannelli, F.; Green, E.P. Detection of point mutations and a gross deletion in six Hunter syndrome patients. Genomics 1992, 13, 543–550. [Google Scholar] [CrossRef]
- Gort, L.; Chabas, A.; Coll, M.J. Hunter disease in the Spanish population: Molecular analysis in 31 families. J. Inherit. Metab. Dis. 1998, 21, 655–661. [Google Scholar] [CrossRef]
- Karsten, S.L.; Lagerstedt, K.; Carlberg, B.M.; Kleijer, W.J.; Zaremba, J.; Van Diggelen, O.P.; Czartoryska, B.; Pettersson, U.; Bondeson, M.L. Two distinct deletions in the IDS gene and the gene W: A novel type of mutation associated with the Hunter syndrome. Genomics 1997, 43, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Karsten, S.; Voskoboeva, E.; Tishkanina, S.; Pettersson, U.; Krasnopolskaja, X.; Bondeson, M.L. Mutational spectrum of the iduronate-2-sulfatase (IDS) gene in 36 unrelated Russian MPS II patients. Hum. Genet. 1998, 103, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Ki, C.S.; Kim, C.H.; Ko, A.R.; Yook, Y.J.; Lee, S.J.; Kim, S.J.; Park, S.W.; Yeau, S.; Kwon, E.K.; et al. Identification of 11 novel mutations in 49 Korean patients with mucopolysaccharidosis type II. Clin. Genet. 2012, 81, 185–190. [Google Scholar] [CrossRef]
- Steen-Bondeson, M.L.; Dahl, N.; Tonnesen, T.; Kleijer, W.J.; Seidlitz, G.; Gustavson, K.H.; Wilson, P.J.; Morris, C.P.; Hopwood, J.J.; Pettersson, U. Molecular analysis of patients with Hunter syndrome: Implication of a region prone to structural alterations within the IDS gene. Hum. Mol. Genet. 1992, 1, 195–198. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Cooper, A.; Heptinstall, L.E.; Hatton, C.E.; Thornley, M.; Wraith, J.E. Mutation analysis in 57 unrelated patients with MPS II (Hunter’s disease). Arch. Dis. Child. 1998, 79, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Vollebregt, A.A.M.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Oussoren, E.; Plug, I.; Ruijter, G.J.; van der Ploeg, A.T.; Pijnappel, W. Genotype-phenotype relationship in mucopolysaccharidosis II: Predictive power of IDS variants for the neuronopathic phenotype. Dev. Med. Child Neurol. 2017, 59, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, Y.; Miura, A.; Yamazaki, N.; So, T.; Kosuga, M.; Yanagi, K.; Kaname, T.; Yamagata, T.; Sakuraba, H.; Okuyama, T. A cDNA analysis disclosed the discordance of genotype-phenotype correlation in a patient with attenuated MPS II and a 76-base deletion in the gene for iduronate-2-sulfatase. Mol. Genet. Metab. Rep. 2020, 25, 100692. [Google Scholar] [CrossRef] [PubMed]
- Lagerstedt, K.; Carlberg, B.M.; Karimi-Nejad, R.; Kleijer, W.J.; Bondeson, M.L. Analysis of a 43.6 kb deletion in a patient with Hunter syndrome (MPSII): Identification of a fusion transcript including sequences from the gene W and the IDS gene. Hum. Mutat. 2000, 15, 324–331. [Google Scholar] [CrossRef]
- Pollard, L.M.; Jones, J.R.; Wood, T.C. Molecular characterization of 355 mucopolysaccharidosis patients reveals 104 novel mutations. J. Inherit. Metab. Dis. 2013, 36, 179–187. [Google Scholar] [CrossRef]
- In‘t Groen, S.L.M.; de Faria, D.O.S.; Iuliano, A.; van den Hout, J.M.P.; Douben, H.; Dijkhuizen, T.; Cassiman, D.; Witters, P.; Barba Romero, M.A.; de Klein, A.; et al. Novel GAA Variants and Mosaicism in Pompe Disease Identified by Extended Analyses of Patients with an Incomplete DNA Diagnosis. Mol. Ther. Methods Clin. Dev. 2020, 17, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Wamelink, M.M.; Struys, E.A.; Jansen, E.E.; Levtchenko, E.N.; Zijlstra, F.S.; Engelke, U.; Blom, H.J.; Jakobs, C.; Wevers, R.A. Sedoheptulokinase deficiency due to a 57-kb deletion in cystinosis patients causes urinary accumulation of sedoheptulose: Elucidation of the CARKL gene. Hum. Mutat. 2008, 29, 532–536. [Google Scholar] [CrossRef]
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef] [PubMed]
- Kitzmuller, C.; Haines, R.L.; Codlin, S.; Cutler, D.F.; Mole, S.E. A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2008, 17, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Lauronen, L.; Munroe, P.B.; Jarvela, I.; Autti, T.; Mitchison, H.M.; O’Rawe, A.M.; Gardiner, R.M.; Mole, S.E.; Puranen, J.; Hakkinen, A.M.; et al. Delayed classic and protracted phenotypes of compound heterozygous juvenile neuronal ceroid lipofuscinosis. Neurology 1999, 52, 360–365. [Google Scholar] [CrossRef]
- Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium. Cell 1995, 82, 949–957. [Google Scholar] [CrossRef]
- Jarvela, I.; Autti, T.; Lamminranta, S.; Aberg, L.; Raininko, R.; Santavuori, P. Clinical and magnetic resonance imaging findings in Batten disease: Analysis of the major mutation (1.02-kb deletion). Ann. Neurol. 1997, 42, 799–802. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).