
Cardiac Acetylation in Metabolic Diseases

 , , and
, , and 
Abstract
:1. Introduction
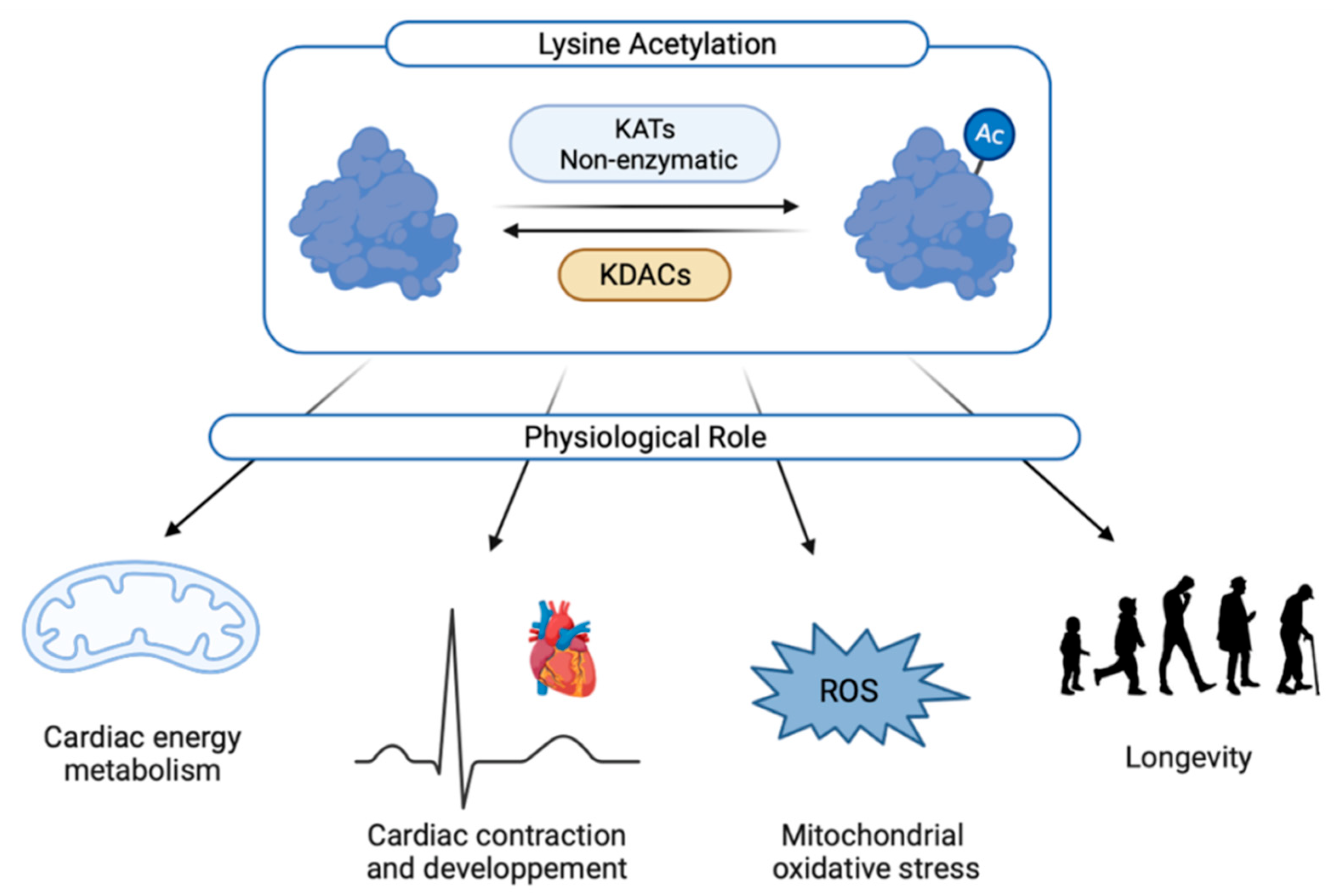
2. Cardiac Acetylation
2.1. Regulation of Lysine Acetylation
2.2. Physiological Roles of Lysine Acetylation
2.2.1. Heart Development and Cardiac Ageing
2.2.2. Cardiac Contraction
2.2.3. Role of Lysine Acetylation in Cardiac Energy Metabolism and Mitochondrial Activity
2.2.4. Mitochondrial Oxidative Stress
3. Implication of Cardiac Acetylation in Metabolic Heart Disease
3.1. Cardiac Hypertrophy
3.2. Cardiac Fibrosis
3.3. Heart Failure
3.4. Obesity
3.5. Type 2 Diabetes
3.6. Pharmacological Modulation of Cardiac Acetylation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update. Circulation 2021, 143, E254–E743. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, R.W.; Vogel, H.; Schürmann, A. Genetic and epigenetic control of metabolic health. Mol. Metab. 2013, 2, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketema, E.B.; Lopaschuk, G.D. Post-translational Acetylation Control of Cardiac Energy Metabolism. Front. Cardiovasc. Med. 2021, 8, 723996. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic Analysis of Lysine Acetylation Sites in Rat Tissues Reveals Organ Specificity and Subcellular Patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Hosp, F.; Lassowskat, I.; Santoro, V.; De Vleesschauwer, D.; Fliegner, D.; Redestig, H.; Mann, M.; Christian, S.; Hannah, M.A.; Finkemeier, I. Lysine acetylation in mitochondria: From inventory to function. Mitochondrion 2017, 33, 58–71. [Google Scholar] [CrossRef]
- Thapa, D.; Zhang, M.; Manning, J.R.; Guimarães, D.A.; Stoner, M.W.; Lai, Y.; Shiva, S.; Scott, I. Loss of GCN5L1 in cardiac cells limits mitochondrial respiratory capacity under hyperglycemic conditions. Physiol. Rep. 2019, 7, e14054. [Google Scholar] [CrossRef] [Green Version]
- Thapa, D.; Zhang, M.; Manning, J.R.; Guimarães, D.A.; Stoner, M.W.; O’Doherty, R.M.; Shiva, S.; Scott, I. Acetylation of mitochondrial proteins by GCN5L1 promotes enhanced fatty acid oxidation in the heart. Am. J. Physiol. Circ. Physiol. 2017, 313, H265–H274. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, A.; Alrob, O.A.; Zhang, L.; Wagg, C.S.; Altamimi, T.; Rawat, S.; Rebeyka, I.M.; Kantor, P.F.; Lopaschuk, G.D. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am. J. Physiol. Circ. Physiol. 2016, 311, H347–H363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Alrob, O.A.; Sankaralingam, S.; Ma, C.; Wagg, C.S.; Fillmore, N.; Jaswal, J.S.; Sack, M.N.; Lehner, R.; Gupta, M.P.; Michelakis, E.D.; et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc. Res. 2014, 103, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor γ co-activator 1 α (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Rivas, G.; Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Treviño-Saldaña, N.; Guerrero-Beltrán, C.E.; Bernal-Ramírez, J.; Torres-Quintanilla, A.; García, N.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell. Physiol. Biochem. 2019, 53, 465–479. [Google Scholar]
- Ghosh, T.K.; Aparicio-Sánchez, J.J.; Buxton, S.; Ketley, A.; Mohamed, T.; Rutland, C.S.; Loughna, S.; Brook, J.D. Acetylation of TBX5 by KAT2B and KAT2A regulates heart and limb development. J. Mol. Cell. Cardiol. 2018, 114, 185–198. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Aparicio-Sánchez, J.J.; Buxton, S.; Brook, J.D. HDAC4 and 5 repression of TBX5 is relieved by protein kinase D1. Sci. Rep. 2019, 9, 17992. [Google Scholar] [CrossRef]
- Lin, Z.; Guo, H.; Cao, Y.; Zohrabian, S.; Zhou, P.; Ma, Q.; VanDusen, N.; Guo, Y.; Zhang, J.; Stevens, S.M.; et al. Acetylation of VGLL4 Regulates Hippo-YAP Signaling and Postnatal Cardiac Growth. Dev. Cell 2016, 39, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, S.; Izumiya, Y.; Araki, S.; Nakamura, T.; Kimura, Y.; Hanatani, S.; Yamada, T.; Ishida, T.; Yamamoto, M.; Onoue, Y.; et al. Cardiomyocyte Sirt (Sirtuin) 7 Ameliorates Stress-Induced Cardiac Hypertrophy by Interacting with and Deacetylating GATA4. Hypertension 2020, 75, 98–108. [Google Scholar] [CrossRef]
- Shimizu, S.; Sunagawa, Y.; Hajika, N.; Yorimitsu, N.; Katanasaka, Y.; Funamoto, M.; Miyazaki, Y.; Sari, N.; Shimizu, K.; Hasegawa, K.; et al. Multimerization of the GATA4 transcription factor regulates transcriptional activity and cardiomyocyte hypertrophic response. Int. J. Biol. Sci. 2022, 18, 1079–1095. [Google Scholar] [CrossRef]
- Peng, C.; Luo, X.; Li, S.; Sun, H. Phenylephrine-induced cardiac hypertrophy is attenuated by a histone acetylase inhibitor anacardic acid in mice. Mol. Biosyst. 2017, 13, 714–724. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, Z.; Alibin, C.P.; Acosta, C.; Anderson, H.D. Liganded Peroxisome Proliferator-Activated Receptors (PPARs) Preserve Nuclear Histone Deacetylase 5 Levels in Endothelin-Treated Sprague-Dawley Rat Cardiac Myocytes. PLoS ONE 2014, 9, e115258. [Google Scholar] [CrossRef] [Green Version]
- Mao, Q.; Wu, S.; Peng, C.; Peng, B.; Luo, X.; Huang, L.; Zhang, H. Interactions between the ERK1/2 signaling pathway and PCAF play a key role in PE-induced cardiomyocyte hypertrophy. Mol. Med. Rep. 2021, 24, 636. [Google Scholar] [CrossRef]
- Winnik, S.; Gaul, D.S.; Siciliani, G.; Lohmann, C.; Pasterk, L.; Calatayud, N.; Weber, J.; Eriksson, U.; Auwerx, J.; van Tits, L.J.; et al. Mild endothelial dysfunction in Sirt3 knockout mice fed a high-cholesterol diet: Protective role of a novel C/EBP-β-dependent feedback regulation of SOD2. Basic Res. Cardiol. 2016, 111, 33. [Google Scholar] [CrossRef] [Green Version]
- Peugnet, V.; Chwastyniak, M.; Mulder, P.; Lancel, S.; Bultot, L.; Fourny, N.; Renguet, E.; Bugger, H.; Beseme, O.; Loyens, A.; et al. Mitochondrial-Targeted Therapies Require Mitophagy to Prevent Oxidative Stress Induced by SOD2 Inactivation in Hypertrophied Cardiomyocytes. Antioxidants 2022, 11, 723. [Google Scholar] [CrossRef]
- Leng, Y.; Wu, Y.; Lei, S.; Zhou, B.; Qiu, Z.; Wang, K.; Xia, Z. Inhibition of HDAC6 Activity Alleviates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats: Potential Role of Peroxiredoxin 1 Acetylation and Redox Regulation. Oxid. Med. Cell. Longev. 2018, 2018, 9494052. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.-J.; Cui, J.; Lin, Q.; Chen, X.-Y.; Zhang, J.; Gao, E.-H.; Wei, B.; Zhao, W. Protection of the enhanced Nrf2 deacetylation and its downstream transcriptional activity by SIRT1 in myocardial ischemia/reperfusion injury. Int. J. Cardiol. 2021, 342, 82–93. [Google Scholar] [CrossRef]
- Ding, M.; Lei, J.; Han, H.; Li, W.; Qu, Y.; Fu, E.; Fu, F.; Wang, X. SIRT1 protects against myocardial ischemia–reperfusion injury via activating eNOS in diabetic rats. Cardiovasc. Diabetol. 2015, 14, 143. [Google Scholar] [CrossRef] [Green Version]
- Landim-Vieira, M.; Childers, M.C.; Wacker, A.L.; Garcia, M.R.; He, H.; Singh, R.; Brundage, E.A.; Johnston, J.R.; Whitson, B.A.; Chase, P.B.; et al. Post-translational modification patterns on β-myosin heavy chain are altered in ischemic and nonischemic human hearts. eLife 2022, 11, e74919. [Google Scholar] [CrossRef]
- Samant, S.A.; Pillai, V.B.; Sundaresan, N.R.; Shroff, S.G.; Gupta, M.P. Histone Deacetylase 3 (HDAC3)-dependent Reversible Lysine Acetylation of Cardiac Myosin Heavy Chain Isoforms Modulates Their Enzymatic and Motor Activity. J. Biol. Chem. 2015, 290, 15559–15569. [Google Scholar] [CrossRef] [Green Version]
- Loescher, C.M.; Hobbach, A.J.; Linke, W.A. Titin (TTN): From molecule to modifications, mechanics, and medical significance. Cardiovasc. Res. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Warren, C.M.; Li, J.; McKinsey, T.A.; Russell, B. Myofibril growth during cardiac hypertrophy is regulated through dual phosphorylation and acetylation of the actin capping protein CapZ. Cell. Signal. 2016, 28, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Schmidt, W.; Fritz, K.S.; Jeong, M.Y.; Cammarato, A.; Foster, D.B.; Biesiadecki, B.J.; McKinsey, T.A.; Woulfe, K.C. Site-specific acetyl-mimetic modification of cardiac troponin I modulates myofilament relaxation and calcium sensitivity. J. Mol. Cell. Cardiol. 2020, 139, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD Blocks Cardiac Hypertrophic Response via Activation of the SIRT3-LKB1-AMP-activated Kinase Pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Chen, X.-F.; Wang, N.-Y.; Wang, X.-M.; Liang, S.-T.; Zheng, W.; Lu, Y.-B.; Zhao, X.; Hao, D.-L.; Zhang, Z.-Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Hu, L.; Yang, H.; Gao, C.; Zeng, K.; Yu, M.; Feng, J.; Qiu, J.; Liu, C.; Fu, F.; et al. Reduction of SIRT1 blunts the protective effects of ischemic post-conditioning in diabetic mice by impairing the Akt signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Jeong, A.; Kwon, D.-H.; Lee, Y.-U.; Kim, Y.-K.; Ahn, Y.; Kook, T.; Park, W.-J.; Kook, H. P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. Int. J. Mol. Sci. 2021, 22, 9944. [Google Scholar] [CrossRef] [PubMed]
- Bugyei-Twum, A.; Ford, C.; Civitarese, R.; Seegobin, J.; Advani, S.L.; Desjardins, J.-F.; Kabir, G.; Zhang, Y.; Mitchell, M.; Switzer, J.; et al. Sirtuin 1 activation attenuates cardiac fibrosis in a rodent pressure overload model by modifying Smad2/3 transactivation. Cardiovasc. Res. 2018, 114, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zeng, Y.; Yang, X.; Wu, Y.; Zhang, S.; Huang, S.; Zhong, Y.; Chen, M. Resveratrol ameliorates myocardial fibrosis by regulating Sirt1/Smad3 deacetylation pathway in rat model with dilated cardiomyopathy. BMC Cardiovasc. Disord. 2022, 22, 17. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, B.; Lin, Q.; Xu, W.; Zuo, W.; Li, J.; Liu, N.; Tu, T.; Zhang, B.; Xiao, Y.; et al. Nicotinamide mononucleotide attenuates isoproterenol-induced cardiac fibrosis by regulating oxidative stress and Smad3 acetylation. Life Sci. 2021, 274, 119299. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Mitochondrial Acetylation and Diseases of Aging. J. Aging Res. 2011, 2011, 234875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Chernaya, V.; Johnson, C.; Yang, W.Y.; Cueto, R.; Sha, X.; Zhang, Y.; Qin, X.; Sun, J.; Choi, E.T.; et al. Metabolic Diseases Downregulate the Majority of Histone Modification Enzymes, Making a Few Upregulated Enzymes Novel Therapeutic Targets—“Sand Out and Gold Stays”. J. Cardiovasc. Transl. Res. 2016, 9, 49–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, P.; Feng, X.; Zhang, X.; Huang, X.; Liu, S.; Lu, X.; Li, J.; You, J.; Lu, J.; Li, Z.; et al. SIRT6 suppresses phenylephrine-induced cardiomyocyte hypertrophy though inhibiting p300. J. Pharmacol. Sci. 2016, 132, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, Y.; Ren, J. Acetylation in cardiovascular diseases: Molecular mechanisms and clinical implications. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165836. [Google Scholar] [CrossRef]
- Pane, L.S.; Fulcoli, F.G.; Cirino, A.; Altomonte, A.; Ferrentino, R.; Bilio, M.; Baldini, A. Tbx1 represses Mef2c gene expression and is correlated with histone 3 deacetylation of the anterior heart field enhancer. Dis. Model. Mech. 2018, 11, dmm029967. [Google Scholar] [CrossRef] [Green Version]
- Bühler, A.; Gahr, B.M.; Park, D.-D.; Bertozzi, A.; Boos, A.; Dalvoy, M.; Pott, A.; Oswald, F.; Kovall, R.A.; Kühn, B.; et al. Histone deacetylase 1 controls cardiomyocyte proliferation during embryonic heart development and cardiac regeneration in zebrafish. PLOS Genet. 2021, 17, e1009890. [Google Scholar] [CrossRef]
- Cencioni, C.; Spallotta, F.; Mai, A.; Martelli, F.; Farsetti, A.; Zeiher, A.M.; Gaetano, C. Sirtuin function in aging heart and vessels. J. Mol. Cell. Cardiol. 2015, 83, 55–61. [Google Scholar] [CrossRef]
- Watroba, M.; Szukiewicz, D. Sirtuins at the Service of Healthy Longevity. Front. Physiol. 2021, 12, 724506. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Schwer, B.; Alt, F.W.; Mostoslavsky, R. SIRT6 in DNA repair, metabolism and ageing. J. Intern. Med. 2008, 263, 128–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M. Longevity genes, cardiac ageing, and the pathogenesis of cardiomyopathy: Implications for understanding the effects of current and future treatments for heart failure. Eur. Heart J. 2020, 41, 3856–3861. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Conti, S.; Perico, L.; Corna, D.; Cerullo, D.; Zentilin, L.; Zoja, C.; Perna, A.; Lionetti, V.; et al. Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxid. Redox Signal. 2019, 31, 1255–1271. [Google Scholar] [CrossRef] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic Instability and Aging-like Phenotype in the Absence of Mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 Is a Stress-Responsive Deacetylase in Cardiomyocytes That Protects Cells from Stress-Mediated Cell Death by Deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-H.; Major, J.L.; Liebner, T.; Hourani, Z.; Travers, J.G.; Wennersten, S.A.; Haefner, K.R.; Cavasin, M.A.; Wilson, C.E.; Jeong, M.Y.; et al. HDAC6 modulates myofibril stiffness and diastolic function of the heart. J. Clin. Investig. 2022, 132, e148333. [Google Scholar] [CrossRef]
- Pougovkina, O.; te Brinke, H.; Ofman, R.; van Cruchten, A.G.; Kulik, W.; Wanders, R.J.A.; Houten, S.M.; de Boer, V.C.J. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 2014, 23, 3513–3522. [Google Scholar] [CrossRef] [Green Version]
- Tomczyk, M.M.; Cheung, K.G.; Xiang, B.; Tamanna, N.; Fonseca Teixeira, A.L.; Agarwal, P.; Kereliuk, S.M.; Spicer, V.; Lin, L.; Treberg, J.; et al. Mitochondrial Sirtuin-3 (SIRT3) Prevents Doxorubicin-Induced Dilated Cardiomyopathy by Modulating Protein Acetylation and Oxidative Stress. Circ. Heart Fail. 2022, 15, e008547. [Google Scholar] [CrossRef]
- Maissan, P.; Mooij, E.J.; Barberis, M. Sirtuins-mediated system-level regulation of mammalian tissues at the interface between metabolism and cell cycle: A systematic review. Biology 2021, 10, 194. [Google Scholar] [CrossRef]
- Khan, D.; Sarikhani, M.; Dasgupta, S.; Maniyadath, B.; Pandit, A.S.; Mishra, S.; Ahamed, F.; Dubey, A.; Fathma, N.; Atreya, H.S.; et al. SIRT6 deacetylase transcriptionally regulates glucose metabolism in heart. J. Cell. Physiol. 2018, 233, 5478–5489. [Google Scholar] [CrossRef]
- Khan, D.; Ara, T.; Ravi, V.; Rajagopal, R.; Tandon, H.; Parvathy, J.; Gonzalez, E.A.; Asirvatham-Jeyaraj, N.; Krishna, S.; Mishra, S.; et al. SIRT6 transcriptionally regulates fatty acid transport by suppressing PPARγ. Cell Rep. 2021, 35, 109190. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1525–1534. [Google Scholar] [CrossRef]
- Hou, Y.-S.; Wang, J.-Z.; Shi, S.; Han, Y.; Zhang, Y.; Zhi, J.-X.; Xu, C.; Li, F.-F.; Wang, G.-Y.; Liu, S.-L. Identification of epigenetic factor KAT2B gene variants for possible roles in congenital heart diseases. Biosci. Rep. 2020, 40, BSR20191779. [Google Scholar] [CrossRef] [Green Version]
- Sunagawa, Y.; Katayama, A.; Funamoto, M.; Shimizu, K.; Shimizu, S.; Sari, N.; Katanasaka, Y.; Miyazaki, Y.; Hosomi, R.; Hasegawa, K.; et al. The polyunsaturated fatty acids, EPA and DHA, ameliorate myocardial infarction-induced heart failure by inhibiting p300-HAT activity in rats. J. Nutr. Biochem. 2022, 106, 109031. [Google Scholar] [CrossRef]
- Li, N.; Zhou, H.; Ma, Z.G.; Zhu, J.X.; Liu, C.; Song, P.; Kong, C.Y.; Wu, H.M.; Deng, W.; Tang, Q.Z. Geniposide alleviates isoproterenol-induced cardiac fibrosis partially via SIRT1 activation in vivo and in vitro. Front. Pharmacol. 2018, 9, 854. [Google Scholar] [CrossRef]
- Lu, T.M.; Tsai, J.Y.; Chen, Y.C.; Huang, C.Y.; Hsu, H.L.; Weng, C.F.; Shih, C.C.; Hsu, C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014, 21, 57. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, A.S.; El-Kott, A.F.; Eleawa, S.M.; El-Gerbed, M.S.A.; Khalifa, H.S.; El-Kenawy, A.E.; Albadrani, G.M.; Abdel-Daim, M.M. Kaempferol protects against streptozotocin-induced diabetic cardiomyopathy in rats by a hypoglycemic effect and upregulating sirt1. J. Physiol. Pharmacol. 2021, 72, 339–355. [Google Scholar]
- Qiu, L.; Xu, C.; Xia, H.; Chen, J.; Liu, H.; Jiang, H. Downregulation of P300/CBP-Associated Factor Attenuates Myocardial Ischemia-Reperfusion Injury Via Inhibiting Autophagy. Int. J. Med. Sci. 2020, 17, 1196–1206. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhan, L.; Zhou, Q.Y.; Zhang, L.L.; Chen, X.M.; Hu, X.M.; Yuan, X.C. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur. J. Pharmacol. 2015, 764, 554–561. [Google Scholar] [CrossRef]
- Turdi, S.; Li, Q.; Lopez, F.L.; Ren, J. Catalase alleviates cardiomyocyte dysfunction in diabetes: Role of Akt, Forkhead transcriptional factor and silent information regulator 2. Life Sci. 2007, 81, 895–905. [Google Scholar] [CrossRef]
- Lai, Y.C.; Tabima, D.M.; Dube, J.J.; Hughan, K.S.; Vanderpool, R.R.; Goncharov, D.A.; St. Croix, C.M.; Garcia-Ocanã, A.; Goncharova, E.A.; Tofovic, S.P.; et al. SIRT3-AMP-Activated Protein Kinase Activation by Nitrite and Metformin Improves Hyperglycemia and Normalizes Pulmonary Hypertension Associated with Heart Failure with Preserved Ejection Fraction. Circulation 2016, 133, 717–731. [Google Scholar]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Möbius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef]
- Li, L.; Zeng, H.; He, X.; Chen, J. Sirtuin 3 Alleviates Diabetic Cardiomyopathy by Regulating TIGAR and Cardiomyocyte Metabolism. J. Am. Heart Assoc. 2021, 10, e018913. [Google Scholar] [CrossRef]
- Dai, L.; Xie, Y.; Zhang, W.; Zhong, X.; Wang, M.; Jiang, H.; He, Z.; Liu, X.; Zeng, H.; Wang, H. Weighted Gene Co-Expression Network Analysis Identifies ANGPTL4 as a Key Regulator in Diabetic Cardiomyopathy via FAK/SIRT3/ROS Pathway in Cardiomyocyte. Front. Endocrinol. 2021, 12, 705154. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Gao, B.; Yang, Y.; Jia, S.-B.; Ma, X.-P.; Zhang, M.-H.; Wang, L.-J.; Ma, A.-Q.; Zhang, Q.-N. Histone deacetylase 3 suppresses the expression of SHP-1 via deacetylation of DNMT1 to promote heart failure. Life Sci. 2022, 292, 119552. [Google Scholar] [CrossRef]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.; Chen, S.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Yang, J.-J.; Shi, K.-H.; Li, J. Epigenetic factors MeCP2 and HDAC6 control α-tubulin acetylation in cardiac fibroblast proliferation and fibrosis. Inflamm. Res. 2016, 65, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Bauters, C.; Dubois, E.; Porouchani, S.; Saloux, E.; Fertin, M.; de Groote, P.; Lamblin, N.; Pinet, F. Long-term prognostic impact of left ventricular remodeling after a first myocardial infarction in modern clinical practice. PLoS ONE 2017, 12, e0188884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chen, X.; Bi, Z.; Liao, J.; Zhao, W.; Huang, W. Curcumin attenuates renal ischemia reperfusion injury via JNK pathway with the involvement of p300/CBP-mediated histone acetylation. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2021, 25, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Sunagawa, Y.; Maekawa, T.; Funamoto, M.; Shimizu, S.; Shimizu, K.; Katanasaka, Y.; Komiyama, M.; Hawke, P.; Hara, H.; et al. Ecklonia stolonifera Okamura Extract Suppresses Myocardial Infarction-Induced Left Ventricular Systolic Dysfunction by Inhibiting p300-HAT Activity. Nutrients 2022, 14, 580. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Funamoto, M.; Shimizu, K.; Shimizu, S.; Sari, N.; Katanasaka, Y.; Miyazaki, Y.; Kakeya, H.; Hasegawa, K.; Morimoto, T. Curcumin, an inhibitor of p300-hat activity, suppresses the development of hypertension-induced left ventricular hypertrophy with preserved ejection fraction in dahl rats. Nutrients 2021, 13, 2608. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Shimizu, K.; Katayama, A.; Funamoto, M.; Shimizu, K.; Nurmila, S.; Shimizu, S.; Miyazaki, Y.; Katanasaka, Y.; Hasegawa, K.; et al. Metformin suppresses phenylephrine-induced hypertrophic responses by inhibiting p300-HAT activity in cardiomyocytes. J. Pharmacol. Sci. 2021, 147, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C., Jr.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 1, e84897. [Google Scholar] [CrossRef]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602. [Google Scholar] [CrossRef] [Green Version]
- Prola, A.; Pires Da Silva, J.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2α deacetylation. Cell Death Differ. 2017, 24, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Renguet, E.; De Loof, M.; Fourny, N.; Ginion, A.; Bouzin, C.; Poüs, C.; Horman, S.; Beauloye, C.; Bultot, L.; Bertrand, L. α-Tubulin acetylation on lysine 40 controls cardiac glucose uptake. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H1032–H1043. [Google Scholar] [CrossRef]
- Gao, S.; Yang, Q.; Peng, Y.; Kong, W.; Liu, Z.; Li, Z.; Chen, J.; Bao, M.; Li, X.; Zhang, Y.; et al. SIRT6 regulates obesity-induced oxidative stress via ENDOG/SOD2 signaling in the heart. Cell Biol. Toxicol. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, Q.; Sun, X.; Qian, H.; Lyu, S.; Chen, R.; Xia, H.; Yuan, W. Sirtuin 3: Emerging therapeutic target for cardiovascular diseases. Free Radic. Biol. Med. 2022, 180, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, S.; Zhang, B.; Liu, J. SIRT3 as a potential therapeutic target for heart failure. Pharmacol. Res. 2021, 165, 105432. [Google Scholar] [CrossRef] [PubMed]
- Saiyang, X.; Deng, W.; Qizhu, T. Sirtuin 6: A potential therapeutic target for cardiovascular diseases. Pharmacol. Res. 2021, 163, 105214. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Class | Name | KAT | KDAC | Function of Acetylation in the Heart | References |
|---|---|---|---|---|---|
| Mitochondrial proteins | LCAD/SCAD | GCN5L1 | SIRT3 | Increased activity and modulated fatty acid oxidation | [5,10,11,12,13] |
| β-HAD | GCN5L1 | SIRT3 | Increased activity and modulated fatty acid oxidation | [5,12,14] | |
| OPA1 | Unknown | SIRT3 | Decreases its activity | [15] | |
| PGC1α | Unknown | SIRT1 | Increases its expression | [16] | |
| Cyclophilin D | GCN5L1 | SIRT3 | Induces mPTP opening | [17] | |
| Transcription factors | TBX5 | KAT2A, KAT2B | HDAC4 HDAC5 | Increases transcriptional activity | [18,19] |
| VGLL4 | P300 | Unknown | Negatively regulates its binding to TEAD1 | [20] | |
| GATA4 | P300 | SIRT7 | Activates its DNA binding activity | [21,22] | |
| MEF2A | P300 KAT2B | HDAC5 | Increased hypertrophy | [23,24] | |
| MEF2C | KAT2B | HDAC5 | Increased hypertrophy | [24,25] | |
| Anti-oxidant proteins | SOD2 | Unknown | Decreases SOD2 activity | [26] | |
| SIRT3 | Increased mitochondrial oxidative stress and hypertrophy | [27] | |||
| Prx1 | Unknown | HDAC6 | Increased peroxide-reduction activity | [28] | |
| Nrf2 | Unknown | SIRT1 | Decreases its activity | [29] | |
| eNOS | Unknown | SIRT1 | Inactive form | [30] | |
| Contractile proteins | β-MHC | Unknown | HDAC6 | Impact myosin head positioning | [31,32] |
| Titin | Unknown | HDAC6 | Cardiac contraction | [33] | |
| CapZβ1 | Unknown | HDAC3/6 | Cardiac contraction | [34] | |
| TnI | Unknown | HDAC6 | Cardiac contraction | [35] | |
| Signaling pathway | LKB1 | Unknown | SIRT2 SIRT3 | Induces its activation by phosphorylation | [36,37] |
| Akt | Unknown | SIRT1 | Inhibition of Akt phosphorylation and activation | [38] | |
| SMAD2 | KAT2B | SIRT1 | Induced fibrosis | [39,40] | |
| SMAD3 | Unknown | SIRT1 | Induced fibrosis | [40,41,42] |
| KATs | KDACs | ||||
|---|---|---|---|---|---|
| Name | Heart Failure | Metabolic Diseases | Name | Heart Failure | Metabolic Diseases |
| P300 | Increase [46,69] | Unknown | SIRT1 | Decrease [70,71] | Decrease [30,38,72] |
| KAT2B | Increase [23,25,73] | Unknown | SIRT2 | Decrease [37] | Decrease [74,75] |
| GCN5L1 | Unknown | Increase in pre-diabetes [11,76] | SIRT3 | Decrease [77] | Decrease [17,78,79] |
| Decrease in diabetes [10] | SIRT6 | Decrease [46,63] | Decrease [63] | ||
| SIRT7 | Increase [21] | Unknown | |||
| HDAC3 | Increase [80] | Increase [81] | |||
| HDAC6 | Unknown | Increase [28] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubois-Deruy, E.; El Masri, Y.; Turkieh, A.; Amouyel, P.; Pinet, F.; Annicotte, J.-S. Cardiac Acetylation in Metabolic Diseases. Biomedicines 2022, 10, 1834. https://doi.org/10.3390/biomedicines10081834
Dubois-Deruy E, El Masri Y, Turkieh A, Amouyel P, Pinet F, Annicotte J-S. Cardiac Acetylation in Metabolic Diseases. Biomedicines. 2022; 10(8):1834. https://doi.org/10.3390/biomedicines10081834
Chicago/Turabian StyleDubois-Deruy, Emilie, Yara El Masri, Annie Turkieh, Philippe Amouyel, Florence Pinet, and Jean-Sébastien Annicotte. 2022. "Cardiac Acetylation in Metabolic Diseases" Biomedicines 10, no. 8: 1834. https://doi.org/10.3390/biomedicines10081834
APA StyleDubois-Deruy, E., El Masri, Y., Turkieh, A., Amouyel, P., Pinet, F., & Annicotte, J. -S. (2022). Cardiac Acetylation in Metabolic Diseases. Biomedicines, 10(8), 1834. https://doi.org/10.3390/biomedicines10081834