
Developing New Treatment Options for Castration-Resistant Prostate Cancer and Recurrent Disease

 , , , and
, , , and
Abstract
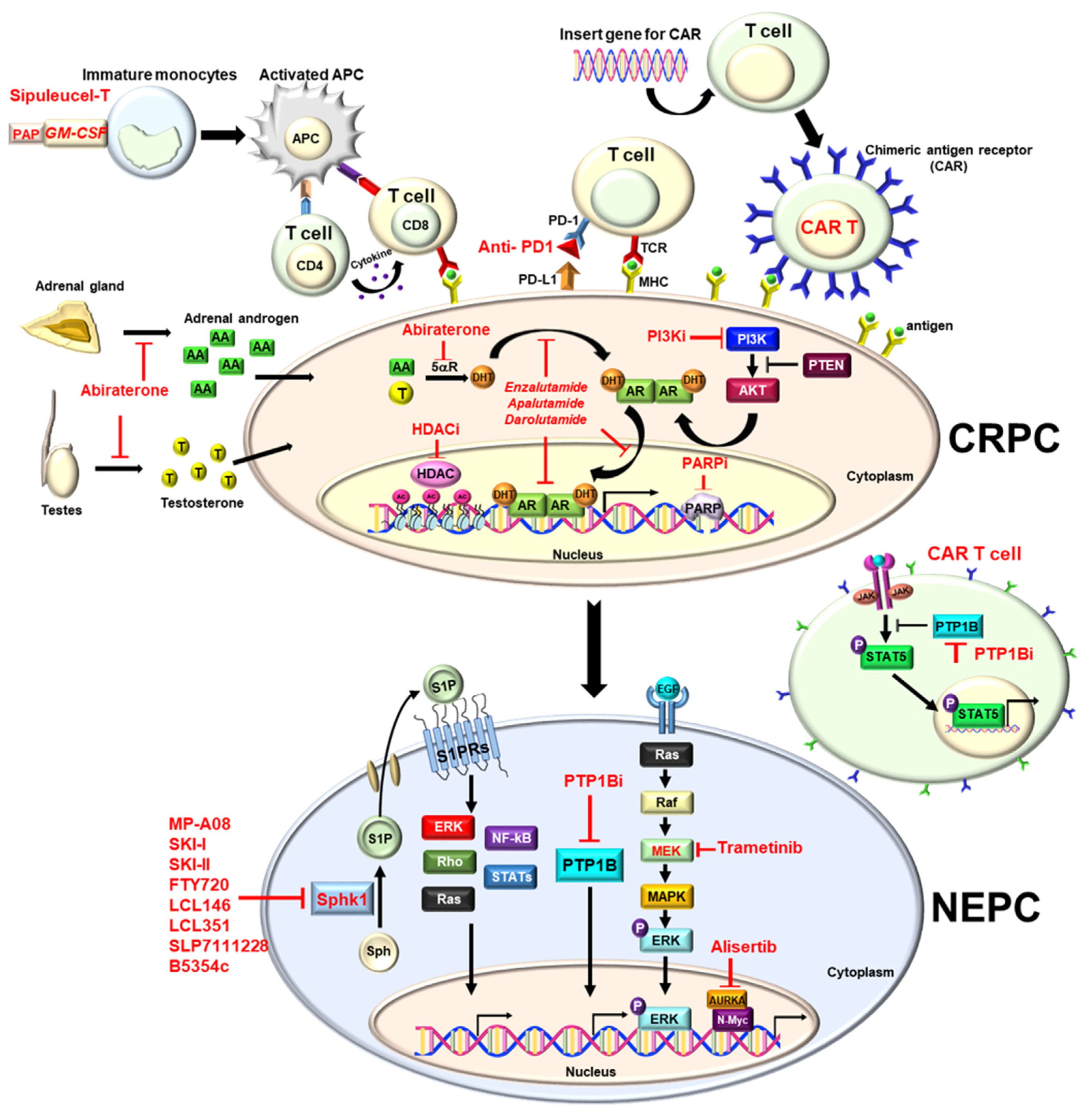
:1. Introduction
2. Hormone Therapy
2.1. Enzalutamide
2.2. Apalutamide
2.3. Darolutamide
2.4. Abiraterone
3. AR Splice Variant-7 (AR-V7) Inhibitors
4. PROTAC (Proteolysis Targeting Chimera) Degrader
5. Radiotherapy
5.1. Bone Target Agent-RADIUM-223 (Ra-223)
5.2. 177Lu-PSMA
6. Immune Therapy
6.1. GM-CSF
6.2. PD1 Inhibitor (Pembrolizumab)
7. Chimeric Antigen Receptor (CAR) T-Cells
8. Targeted Therapies
8.1. Polyadenosine Diphosphate Ribose Polymerase (PARP) Inhibitors
8.2. Olaparib
8.3. Rucaparib
8.4. Niraparib
8.5. Talazoparib
9. Phosphatidylinositol-3-Kinase (PI3K)/AKT Inhibitor
10. Aurora Kinase Inhibitor
Alisertib
11. MEK Inhibitor
Trametinib
12. Histone Deacetylase Inhibitors (HDAC Inhibitors)
13. SPHK1/S1PRs Inhibitors
14. PTP1B Inhibitors
15. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Gravis, G. Systemic treatment for metastatic prostate cancer. Asian J. Urol. 2019, 6, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Virgo, K.S.; Basch, E.; Loblaw, D.A.; Oliver, T.K.; Rumble, R.B.; Carducci, M.A.; Nordquist, L.; Taplin, M.E.; Winquist, E.; Singer, E.A. Second-Line Hormonal Therapy for Men with Chemotherapy-Naïve, Castration-Resistant Prostate Cancer: American Society of Clinical Oncology Provisional Clinical Opinion. J. Clin. Oncol. 2017, 35, 1952–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritch, C.R.; Cookson, M.S. Advances in the management of castration resistant prostate cancer. Bmj 2016, 355, i4405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conteduca, V.; Aieta, M.; Amadori, D.; De Giorgi, U. Neuroendocrine differentiation in prostate cancer: Current and emerging therapy strategies. Crit. Rev. Oncol. Hematol. 2014, 92, 11–24. [Google Scholar] [CrossRef]
- Santoni, M.; Conti, A.; Burattini, L.; Berardi, R.; Scarpelli, M.; Cheng, L.; Lopez-Beltran, A.; Cascinu, S.; Montironi, R. Neuroendocrine differentiation in prostate cancer: Novel morphological insights and future therapeutic perspectives. Biochim. Biophys. Acta 2014, 1846, 630–637. [Google Scholar] [CrossRef]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285. [Google Scholar]
- Grigore, A.D.; Ben-Jacob, E.; Farach-Carson, M.C. Prostate cancer and neuroendocrine differentiation: More neuronal, less endocrine? Front. Oncol. 2015, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grobholz, R.; Griebe, M.; Sauer, C.G.; Michel, M.S.; Trojan, L.; Bleyl, U. Influence of neuroendocrine tumor cells on proliferation in prostatic carcinoma. Hum. Pathol. 2005, 36, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidegger, I.; Brandt, M.P.; Heck, M.M. Treatment of non-metastatic castration resistant prostate cancer in 2020: What is the best? Urol. Oncol. 2020, 38, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Scher, H.I.; Miller, K.; Basch, E.; Sternberg, C.N.; Cella, D.; Forer, D.; Hirmand, M.; de Bono, J.S. Effect of enzalutamide on time to first skeletal-related event, pain, and quality of life in men with castration-resistant prostate cancer: Results from the randomised, phase 3 AFFIRM trial. Lancet Oncol. 2014, 15, 1147–1156. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Evans, C.P.; Kim, C.S.; Kimura, G.; et al. Enzalutamide in Men with Chemotherapy-naïve Metastatic Castration-resistant Prostate Cancer: Extended Analysis of the Phase 3 PREVAIL Study. Eur. Urol. 2017, 71, 151–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy with Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juárez Soto, Á.; Merseburger, A.S.; Özgüroğlu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- A Study of Rucaparib Versus Physician’s Choice of Therapy in Patients with Metastatic Castration-Resistant Prostate Cancer and Homologous Recombination Gene Deficiency (TRITON3). Available online: https://clinicaltrials.gov/ct2/show/NCT02975934?term=TRITON3&draw=2&rank=1 (accessed on 15 July 2022).
- Moilanen, A.M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykänen, P.S.; Törmäkangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, 12007. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Hussain, M.; Saad, F.; Fizazi, K.; Sternberg, C.N.; Crawford, E.D.; Kopyltsov, E.; Park, C.H.; Alekseev, B.; Montesa-Pino, Á.; et al. Darolutamide and Survival in Metastatic, Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2022, 386, 1132–1142. [Google Scholar] [CrossRef]
- Barrie, S.E.; Haynes, B.P.; Potter, G.A.; Chan, F.C.; Goddard, P.M.; Dowsett, M.; Jarman, M. Biochemistry and pharmacokinetics of potent non-steroidal cytochrome P450(17alpha) inhibitors. J. Steroid. Biochem. Mol. Biol. 1997, 60, 347–351. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, A.; Belldegrun, A. Novel therapeutic strategies for castration resistant prostate cancer: Inhibition of persistent androgen production and androgen receptor mediated signaling. J. Urol. 2011, 185, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Naseer, F.; Ahmad, T.; Kousar, K.; Anjum, S. Advanced Therapeutic Options for Treatment of Metastatic Castration Resistant Prostatic Adenocarcinoma. Front. Pharmacol. 2021, 12, 728054. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.E.; Eedunuri, V.K.; Chew, S.A.; Zimmermann, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Blattner, M.; Lee, D.J.; O’Reilly, C.; Park, K.; MacDonald, T.Y.; Khani, F.; Turner, K.R.; Chiu, Y.L.; Wild, P.J.; Dolgalev, I.; et al. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia 2014, 16, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Nakazawa, M.; Fang, M.; Marshall, C.H.; Lotan, T.L.; Isaacsson Velho, P.; Antonarakis, E.S. Clinical and genomic features of SPOP-mutant prostate cancer. Prostate 2022, 82, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, A.I.; Jim, H.S.L.; Gonzalez, B.D.; Small, B.J.; Gilvary, D.; Breen, E.C.; Bower, J.E.; Fishman, M.; Zachariah, B.; Jacobsen, P.B. Systemic inflammation and symptomatology in patients with prostate cancer treated with androgen deprivation therapy: Preliminary findings. Cancer 2021, 127, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Sciarra, A.; Gentilucci, A.; Salciccia, S.; Pierella, F.; Del Bianco, F.; Gentile, V.; Silvestri, I.; Cattarino, S. Prognostic value of inflammation in prostate cancer progression and response to therapeutic: A critical review. J. Inflamm. 2016, 13, 35. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.P.; Li, J.; Tewari, A.K. Inflammation and prostate cancer: The role of interleukin 6 (IL-6). BJU Int. 2014, 113, 986–992. [Google Scholar] [CrossRef]
- Del Re, M.; Conteduca, V.; Crucitta, S.; Gurioli, G.; Casadei, C.; Restante, G.; Schepisi, G.; Lolli, C.; Cucchiara, F.; Danesi, R.; et al. Androgen receptor gain in circulating free DNA and splicing variant 7 in exosomes predict clinical outcome in CRPC patients treated with abiraterone and enzalutamide. Prostate Cancer Prostatic. Dis. 2021, 24, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hille, C.; Gorges, T.M.; Riethdorf, S.; Mazel, M.; Steuber, T.; Amsberg, G.V.; König, F.; Peine, S.; Alix-Panabières, C.; Pantel, K. Detection of Androgen Receptor Variant 7 (ARV7) mRNA Levels in EpCAM-Enriched CTC Fractions for Monitoring Response to Androgen Targeting Therapies in Prostate Cancer. Cells 2019, 8, 1067. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Armstrong, C.; Zhu, Y.; Lou, W.; Gao, A.C. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget 2016, 7, 32210–32220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georget, V.; Térouanne, B.; Nicolas, J.C.; Sultan, C. Mechanism of antiandrogen action: Key role of hsp90 in conformational change and transcriptional activity of the androgen receptor. Biochemistry 2002, 41, 11824–11831. [Google Scholar] [CrossRef]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraldeschi, R.; Welti, J.; Powers, M.V.; Yuan, W.; Smyth, T.; Seed, G.; Riisnaes, R.; Hedayat, S.; Wang, H.; Crespo, M.; et al. Second-Generation HSP90 Inhibitor Onalespib Blocks mRNA Splicing of Androgen Receptor Variant 7 in Prostate Cancer Cells. Cancer Res. 2016, 76, 2731–2742. [Google Scholar] [CrossRef] [Green Version]
- Potjewyd, F.; Turner, A.W.; Beri, J.; Rectenwald, J.M.; Norris-Drouin, J.L.; Cholensky, S.H.; Margolis, D.M.; Pearce, K.H.; Herring, L.E.; James, L.I. Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem. Biol. 2020, 27, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Crews, C.M. Waste disposal-An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, S.M.; Dong, J.; Xu, Z.Y.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Huang, W.; Zheng, X.; Liyan, C.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef] [PubMed]
- PROTAC Shrinks Mutated Prostate Tumors. Cancer Discov. 2022, 12, Of2. [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef] [PubMed]
- Ledet, E.M.; Lilly, M.B.; Sonpavde, G.; Lin, E.; Nussenzveig, R.H.; Barata, P.C.; Yandell, M.; Nagy, R.J.; Kiedrowski, L.; Agarwal, N.; et al. Comprehensive Analysis of AR Alterations in Circulating Tumor DNA from Patients with Advanced Prostate Cancer. Oncologist 2020, 25, 327–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Lewington, V.J. Bone-seeking radionuclides for therapy. J. Nucl. Med. 2005, 46 (Suppl. S1), 38s–47s. [Google Scholar]
- Henriksen, G.; Breistøl, K.; Bruland, S.Ø.; Fodstad, Ø.; Larsen, R.H. Significant antitumor effect from bone-seeking, alpha-particle-emitting (223)Ra demonstrated in an experimental skeletal metastases model. Cancer Res. 2002, 62, 3120–3125. [Google Scholar]
- Hoskin, P.; Sartor, O.; O’Sullivan, J.M.; Johannessen, D.C.; Helle, S.I.; Logue, J.; Bottomley, D.; Nilsson, S.; Vogelzang, N.J.; Fang, F.; et al. Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: A prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. Lancet Oncol. 2014, 15, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, A.K.; Anilkumar, G.; Christiansen, J.J. Is prostate-specific membrane antigen a multifunctional protein? Am. J. Physiol. Cell Physiol. 2005, 288, C975–C981. [Google Scholar] [CrossRef] [Green Version]
- Bostwick, D.G.; Pacelli, A.; Blute, M.; Roche, P.; Murphy, G.P. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: A study of 184 cases. Cancer 1998, 82, 2256–2261. [Google Scholar] [CrossRef]
- Wright, G.L., Jr.; Grob, B.M.; Haley, C.; Grossman, K.; Newhall, K.; Petrylak, D.; Troyer, J.; Konchuba, A.; Schellhammer, P.F.; Moriarty, R. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology 1996, 48, 326–334. [Google Scholar] [CrossRef]
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. S10), S13–S18. [Google Scholar] [PubMed]
- Liu, H.; Rajasekaran, A.K.; Moy, P.; Xia, Y.; Kim, S.; Navarro, V.; Rahmati, R.; Bander, N.H. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998, 58, 4055–4060. [Google Scholar] [PubMed]
- Emmett, L.; Willowson, K.; Violet, J.; Shin, J.; Blanksby, A.; Lee, J. Lutetium (177) PSMA radionuclide therapy for men with prostate cancer: A review of the current literature and discussion of practical aspects of therapy. J. Med. Radiat. Sci. 2017, 64, 52–60. [Google Scholar] [CrossRef]
- Caroli, P.; Sandler, I.; Matteucci, F.; De Giorgi, U.; Uccelli, L.; Celli, M.; Foca, F.; Barone, D.; Romeo, A.; Sarnelli, A.; et al. 68Ga-PSMA PET/CT in patients with recurrent prostate cancer after radical treatment: Prospective results in 314 patients. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2035–2044. [Google Scholar] [CrossRef]
- Hernando Polo, S.; Moreno Muñoz, D.; Rosero Rodríguez, A.C.; Silva Ruiz, J.; Rosero Rodríguez, D.I.; Couñago, F. Changing the History of Prostate Cancer with New Targeted Therapies. Biomedicines 2021, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Bander, N.H.; Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J. Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J. Clin. Oncol. 2005, 23, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.; Vallabhajosula, S.; Christos, P.; Akhtar, N.H.; Osborne, J.; Goldsmith, S.J.; Larson, S.; Taskar, N.P.; et al. Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 5182–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagawa, S.T.; Vallabhajosula, S.; Christos, P.J.; Jhanwar, Y.S.; Batra, J.S.; Lam, L.; Osborne, J.; Beltran, H.; Molina, A.M.; Goldsmith, S.J.; et al. Phase 1/2 study of fractionated dose lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 ((177) Lu-J591) for metastatic castration-resistant prostate cancer. Cancer 2019, 125, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Niaz, M.J.; Batra, J.S.; Walsh, R.D.; Ramirez-Fort, M.K.; Vallabhajosula, S.; Jhanwar, Y.S.; Molina, A.M.; Nanus, D.M.; Osborne, J.R.; Bander, N.H.; et al. Pilot Study of Hyperfractionated Dosing of Lutetium-177-Labeled Antiprostate-Specific Membrane Antigen Monoclonal Antibody J591 ((177) Lu-J591) for Metastatic Castration-Resistant Prostate Cancer. Oncologist 2020, 25, e477–e895. [Google Scholar] [CrossRef] [Green Version]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [(177)Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Yadav, M.P.; Ballal, S.; Sahoo, R.K.; Dwivedi, S.N.; Bal, C. Radioligand Therapy With (177)Lu-PSMA for Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis. AJR Am. J. Roentgenol. 2019, 213, 275–285. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Ng, S.; et al. [(177)Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): A randomised, open-label, phase 2 trial. Lancet 2021, 397, 797–804. [Google Scholar] [CrossRef]
- Quinn, D.I.; Shore, N.D.; Egawa, S.; Gerritsen, W.R.; Fizazi, K. Immunotherapy for castration-resistant prostate cancer: Progress and new paradigms. Urol. Oncol. 2015, 33, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.X.; Chan, S.; Kwek, S.; Lewis, J.; Dao, V.; Zhang, L.; Cooperberg, M.R.; Ryan, C.J.; Lin, A.M.; Friedlander, T.W.; et al. Systemic GM-CSF Recruits Effector T Cells into the Tumor Microenvironment in Localized Prostate Cancer. Cancer Immunol. Res. 2016, 4, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Glode, L.M.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Gulley, J.L.; Pico-Navarro, C. Revised Overall Survival Analysis of a Phase II, Randomized, Double-Blind, Controlled Study of PROSTVAC in Men with Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Fay, E.K.; Graff, J.N. Immunotherapy in Prostate Cancer. Cancers 2020, 12, 1752. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate. Cancer Prostatic. Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Bruno, T.C.; Maris, C.H.; Xu, L.; Thoburn, C.J.; DeMarzo, A.M.; Meeker, A.K.; Isaacs, W.B.; Drake, C.G. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin. Cancer Res. 2008, 14, 3254–3261. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, E.; Srinivas, S.; Antonarakis, E.S.; Armstrong, A.J.; Bekelman, J.E.; Cheng, H.; D’Amico, A.V.; Davis, B.J.; Desai, N.; Dorff, T.; et al. NCCN Guidelines Insights: Prostate Cancer, Version 1.2021. J. Natl. Compr. Canc. Netw. 2021, 19, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.T.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019, 25, 7035–7045. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barata, P.; Agarwal, N.; Nussenzveig, R.; Gerendash, B.; Jaeger, E.; Hatton, W.; Ledet, E.; Lewis, B.; Layton, J.; Babiker, H.; et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J. Immunother. Cancer 2020, 8, e001065. [Google Scholar] [CrossRef]
- Shia, J.; Stadler, Z.; Weiser, M.R.; Rentz, M.; Gonen, M.; Tang, L.H.; Vakiani, E.; Katabi, N.; Xiong, X.; Markowitz, A.J.; et al. Immunohistochemical staining for DNA mismatch repair proteins in intestinal tract carcinoma: How reliable are biopsy samples? Am. J. Surg. Pathol. 2011, 35, 447–454. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Author Correction: Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95, Erratum in Nature 2019, 571, E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Schepisi, G.; Cursano, M.C.; Casadei, C.; Menna, C.; Altavilla, A.; Lolli, C.; Cerchione, C.; Paganelli, G.; Santini, D.; Tonini, G.; et al. CAR-T cell therapy: A potential new strategy against prostate cancer. J. Immunother. Cancer 2019, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.P.J.; Thomas, P.B.; Risbridger, G.P.; Taylor, R.; Azad, A.; Hofman, M.S.; Williams, E.D.; Vela, I. Chimeric Antigen Receptor T-Cell Therapy in Metastatic Castrate-Resistant Prostate Cancer. Cancers 2022, 14, 503. [Google Scholar] [CrossRef] [PubMed]
- Amini, L.; Silbert, S.K.; Maude, S.L.; Nastoupil, L.J.; Ramos, C.A.; Brentjens, R.J.; Sauter, C.S.; Shah, N.N.; Abou-El-Enein, M. Preparing for CAR T cell therapy: Patient selection, bridging therapies and lymphodepletion. Nat. Rev. Clin. Oncol 2022, 19, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Hupe, M.C.; Philippi, C.; Roth, D.; Kümpers, C.; Ribbat-Idel, J.; Becker, F.; Joerg, V.; Duensing, S.; Lubczyk, V.H.; Kirfel, J.; et al. Expression of Prostate-Specific Membrane Antigen (PSMA) on Biopsies Is an Independent Risk Stratifier of Prostate Cancer Patients at Time of Initial Diagnosis. Front. Oncol. 2018, 8, 623. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Slovin, S. Adoptive Transfer of Autologous T Cells Targeted to Prostate Specific Membrane Antigen (PSMA) for the Treatment of Castrate Metastatic Prostate Cancer (CMPC). Available online: https://clinicaltrials.gov/ct2/show/NCT01140373?term=NCT01140373&draw=2&rank=1 (accessed on 15 July 2022).
- Slovin, S.F.; Dorff, T.B.; Falchook, G.S.; Wei, X.X.; Gao, X.; McKay, R.R.; Oh, D.Y.; Wibmer, A.G.; Spear, M.A.; McCaigue, J.; et al. Phase 1 Study of P-PSMA-101 CAR-T Cells in Patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC). Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2022.40.6_suppl.098 (accessed on 15 July 2022).
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation and Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Helfand, B.T.; Carneiro, B.A.; Qin, W.; Yang, X.J.; Lee, C.; Zhang, W.; Giles, F.J.; Cristofanilli, M.; Kuzel, T.M. Efficacy Against Human Prostate Cancer by Prostate-specific Membrane Antigen-specific, Transforming Growth Factor-β Insensitive Genetically Targeted CD8(+) T-cells Derived from Patients with Metastatic Castrate-resistant Disease. Eur. Urol. 2018, 73, 648–652. [Google Scholar] [CrossRef] [PubMed]
- A Study of CART-PSMA-TGFβRDN in Patients with Metastatic Castration Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04227275?term=NCT04227275&draw=2&rank=1 (accessed on 15 July 2022).
- Anurathapan, U.; Chan, R.C.; Hindi, H.F.; Mucharla, R.; Bajgain, P.; Hayes, B.C.; Fisher, W.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther. 2014, 22, 623–633. [Google Scholar] [CrossRef] [Green Version]
- PSCA-CAR T Cells in Treating Patients With PSCA+ Metastatic Castration Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03873805?term=NCT03873805&draw=2&rank=1 (accessed on 15 July 2022).
- Safety and Activity Study of PSCA-Targeted CAR-T Cells (BPX-601) in Subjects with Selected Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02744287?term=NCT02744287&draw=2&rank=1 (accessed on 15 July 2022).
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu. Rev. Pathol. 2009, 4, 461–487. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell Biol. 2007, 27, 5597–5605. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef]
- PROpel: Phase III Trial of Olaparib (ola) and Abiraterone (abi) Versus Placebo (pbo) and abi as First-Line (1L) Therapy for Patients (pts) with Metastatic Castration-Resistant Prostate Cancer (mCRPC). Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2022.40.6_suppl.011 (accessed on 5 July 2022).
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015, 4, e09207. [Google Scholar] [CrossRef]
- Phase 3 MAGNITUDE Study: First Results of Niraparib (NIRA) with Abiraterone Acetate and Prednisone (AAP) as First-Line Therapy in Patients (pts) with Metastatic Castration-Resistant Prostate Cancer (mCRPC) with and without Homologous Recombination Repair (HRR) Gene Alterations. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2022.40.6_suppl.012 (accessed on 15 July 2022).
- Talazoparib in Combination with Belinostat for Metastatic Breast Cancer, Metastatic Castration Resistant Prostate Cancer, and Metastatic Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04703920?term=NCT+04703920&draw=2&rank=1 (accessed on 15 July 2022).
- Cuzick, J.; Yang, Z.H.; Fisher, G.; Tikishvili, E.; Stone, S.; Lanchbury, J.S.; Camacho, N.; Merson, S.; Brewer, D.; Cooper, C.S.; et al. Prognostic value of PTEN loss in men with conservatively managed localised prostate cancer. Br. J. Cancer 2013, 108, 2582–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.; Grignard, G.; Margue, C.; Dippel, W.; Capesius, C.; Mossong, J.; Nathan, M.; Giacchi, S.; Scheiden, R.; Kieffer, N. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int. J. Cancer 2007, 120, 1284–1292. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinonen, K.A.; Saramäki, O.R.; Furusato, B.; Kimura, T.; Takahashi, H.; Egawa, S.; Suzuki, H.; Keiger, K.; Ho Hahm, S.; Isaacs, W.B.; et al. Loss of PTEN is associated with aggressive behavior in ERG-positive prostate cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2333–2344. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Fu, W.; Li, P.; Nicosia, S.V.; Jenster, G.; Zhang, X.; Bai, W. FoxO1 mediates PTEN suppression of androgen receptor N- and C-terminal interactions and coactivator recruitment. Mol. Endocrinol. 2009, 23, 213–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraldeschi, R.; Nava Rodrigues, D.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Billalabeitia, E.; Seitzer, N.; Song, S.J.; Song, M.S.; Patnaik, A.; Liu, X.S.; Epping, M.T.; Papa, A.; Hobbs, R.M.; Chen, M.; et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 2014, 4, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.N.; Higano, C.S.; Hahn, N.M.; Taylor, M.H.; Zhang, B.; Zhou, X.; Venkatakrishnan, K.; Leonard, E.J.; Sarantopoulos, J. Open-label, multicenter, phase 1 study of alisertib (MLN8237), an aurora A kinase inhibitor, with docetaxel in patients with solid tumors. Cancer 2016, 122, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase a Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Patel, S.A.; Sama, A.R.; Hoffman-Censits, J.H.; Kennedy, B.; Kilpatrick, D.; Ye, Z.; Yang, H.; Mu, Z.; Leiby, B.; et al. A Phase I/II Study of the Investigational Drug Alisertib in Combination with Abiraterone and Prednisone for Patients With Metastatic Castration-Resistant Prostate Cancer Progressing on Abiraterone. Oncologist 2016, 21, 1296–1297e. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicken, H.; Hensley, P.J.; Kyprianou, N. Prostate tumor neuroendocrine differentiation via EMT: The road less traveled. Asian J. Urol. 2019, 6, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Lo, U.G.; Lee, C.F.; Lee, M.S.; Hsieh, J.T. The Role and Mechanism of Epithelial-to-Mesenchymal Transition in Prostate Cancer Progression. Int. J. Mol. Sci. 2017, 18, 2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate. Cancer Prostatic. Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef]
- Ducasse, M.; Brown, M.A. Epigenetic aberrations and cancer. Mol. Cancer 2006, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Han, J.W. Targeting epigenetics for cancer therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottet, D.; Castronovo, V. Histone deacetylases: Target enzymes for cancer therapy. Clin. Exp. Metastasis 2008, 25, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; North, B.; Van Mellaert, F.; de Leval, J.; Verdin, E.; Castronovo, V. Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. Eur. J. Histochem. 2004, 48, 273–290. [Google Scholar]
- Lin, J.; Wang, C.; Kelly, W.K. Targeting epigenetics for the treatment of prostate cancer: Recent progress and future directions. Semin. Oncol. 2013, 40, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, Y.H.; Li, Y.J.; Cohen, A.; Li, Z.; Squires, J.; Zhang, W.; Chen, X.F.; Zhang, M.; Huang, J.T. Potential therapeutic effect of epigenetic therapy on treatment-induced neuroendocrine prostate cancer. Asian J. Androl. 2017, 19, 686–693. [Google Scholar] [CrossRef]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): Trial results and interleukin-6 analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 2009, 115, 5541–5549. [Google Scholar] [CrossRef] [Green Version]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Yang, Z.; Sun, Y.; Dong, H.; Ma, J. Interaction of microRNAs with sphingosine kinases, sphingosine-1 phosphate, and sphingosine-1 phosphate receptors in cancer. Discov. Oncol. 2021, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Doumerc, N.; Golzio, M.; Naymark, M.; Teissie, J.; Kohama, T.; Waxman, J.; Malavaud, B.; Cuvillier, O. Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Mol. Cancer Ther. 2008, 7, 1836–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, L.; Nunes, J.; Salunkhe, V.; Skalska, L.; Kohama, T.; Cuvillier, O.; Waxman, J.; Pchejetski, D. Sphingosine kinase 1 inhibition sensitizes hormone-resistant prostate cancer to docetaxel. Int. J. Cancer 2009, 125, 2728–2736. [Google Scholar] [CrossRef]
- Brizuela, L.; Dayon, A.; Doumerc, N.; Ader, I.; Golzio, M.; Izard, J.C.; Hara, Y.; Malavaud, B.; Cuvillier, O. The sphingosine kinase-1 survival pathway is a molecular target for the tumor-suppressive tea and wine polyphenols in prostate cancer. FASEB J. 2010, 24, 3882–3894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.O.; Kim, J.S.; Lee, M.S.; Lee, H.J. Anti-cancer effect of pristimerin by inhibition of HIF-1alpha involves the SPHK-1 pathway in hypoxic prostate cancer cells. BMC Cancer 2016, 16, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckham, T.H.; Cheng, J.C.; Lu, P.; Shao, Y.; Troyer, D.; Lance, R.; Marrison, S.T.; Norris, J.S.; Liu, X. Acid ceramidase induces sphingosine kinase 1/S1P receptor 2-mediated activation of oncogenic Akt signaling. Oncogenesis 2013, 2, e49. [Google Scholar] [CrossRef] [Green Version]
- Brizuela, L.; Ader, I.; Mazerolles, C.; Bocquet, M.; Malavaud, B.; Cuvillier, O. First evidence of sphingosine 1-phosphate lyase protein expression and activity downregulation in human neoplasm: Implication for resistance to therapeutics in prostate cancer. Mol. Cancer Ther. 2012, 11, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Akao, Y.; Banno, Y.; Nakagawa, Y.; Hasegawa, N.; Kim, T.J.; Murate, T.; Igarashi, Y.; Nozawa, Y. High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem. Biophys. Res. Commun. 2006, 342, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Bohler, T.; Brizuela, L.; Sauer, L.; Doumerc, N.; Golzio, M.; Salunkhe, V.; Teissie, J.; Malavaud, B.; Waxman, J.; et al. FTY720 (fingolimod) sensitizes prostate cancer cells to radiotherapy by inhibition of sphingosine kinase-1. Cancer Res. 2010, 70, 8651–8661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Dang, A.; Hernandez, E.; Pong, R.C.; Chen, B.; Sonavane, R.; Raj, G.; Kapur, P.; Lin, H.Y.; Wu, S.R.; et al. Activation of sphingosine kinase by lipopolysaccharide promotes prostate cancer cell invasion and metastasis via SphK1/S1PR4/matriptase. Oncogene 2019, 38, 5580–5598. [Google Scholar] [CrossRef] [PubMed]
- Malavaud, B.; Pchejetski, D.; Mazerolles, C.; de Paiva, G.R.; Calvet, C.; Doumerc, N.; Pitson, S.; Rischmann, P.; Cuvillier, O. Sphingosine kinase-1 activity and expression in human prostate cancer resection specimens. Eur. J. Cancer 2010, 46, 3417–3424. [Google Scholar] [CrossRef]
- Lee, C.F.; Chen, Y.A.; Hernandez, E.; Pong, R.C.; Ma, S.; Hofstad, M.; Kapur, P.; Zhau, H.; Chung, L.W.; Lai, C.H.; et al. The central role of Sphingosine kinase 1 in the development of neuroendocrine prostate cancer (NEPC): A new targeted therapy of NEPC. Clin. Transl. Med. 2022, 12, e695. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Tsou, R.C.; Bence, K.K. The Genetics of PTPN1 and Obesity: Insights from Mouse Models of Tissue-Specific PTP1B Deficiency. J. Obes. 2012, 2012, 926857. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef]
- Lessard, L.; Stuible, M.; Tremblay, M.L. The two faces of PTP1B in cancer. Biochim. Biophys. Acta 2010, 1804, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Labbe, D.P.; Deblois, G.; Begin, L.R.; Hardy, S.; Mes-Masson, A.M.; Saad, F.; Trotman, L.C.; Giguere, V.; Tremblay, M.L. PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res. 2012, 72, 1529–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Wu, N.; Li, X.; Guo, C.; Li, C.; Jiang, B.; Wang, H.; Shi, D. Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway. Cell Death Dis. 2019, 10, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Cao, Y.; Zhou, X.; Wei, B.; Zhang, Y.; Liu, X. PTP1B promotes the malignancy of ovarian cancer cells in a JNK-dependent mechanism. Biochem. Biophys. Res. Commun. 2018, 503, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, E.; Das, A.M.; Swets, M.; Cao, W.; van der Woude, C.J.; Bruno, M.J.; Peppelenbosch, M.P.; Kuppen, P.J.; Ten Hagen, T.L.; Fuhler, G.M. Increased PTP1B expression and phosphatase activity in colorectal cancer results in a more invasive phenotype and worse patient outcome. Oncotarget 2016, 7, 21922–21938. [Google Scholar] [CrossRef]
- Tonks, N.K.; Muthuswamy, S.K. A brake becomes an accelerator: PTP1B--a new therapeutic target for breast cancer. Cancer Cell 2007, 11, 214–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zhang, L.; Bourne, P.A.; Reeder, J.E.; di Sant’Agnese, P.A.; Yao, J.L.; Na, Y.; Huang, J. Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differentiation of prostate cancer. Prostate 2006, 66, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Lu, K.H.; Du, X.; Zeissig, M.N.; Xu, R.; Goh, P.K.; Xirouchaki, C.E.; Hogarth, S.J.; Greatorex, S.; Sek, K.; et al. PTP1B Is an Intracellular Checkpoint that Limits T-cell and CAR T-cell Antitumor Immunity. Cancer Discov. 2022, 12, 752–773. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Drug Class | Therapy | Indication | Clinical Trial | Efficacy (Mon.) | Impact on PCa Treatment | Reference |
|---|---|---|---|---|---|---|
| 2nd-generation antiandrogen | Enzalutamide | mCRPC | AFFIRM | OS 18.4 vs. 13.6 | ENZ improves OS after chemotherapy | [14] |
| PREVAIL | OS 35.3 vs. 31.3 | ENZ improves OS in chemotherapy naïve | [15,16] | |||
| nmCRPC | PROSPER | MFS 36.6 vs. 14.7 | ENZ decreases the risk of metastasis | [17] | ||
| mCSPC | ENZEMET | 80% vs. 72% (3-year OS) | ENZ improves OS and decreases disease progression | [18] | ||
| ARCHES | rPFS NR vs. 19.0 | ENZ decreases the risk of metastatic progression | [19] | |||
| Apalutamide | nmCRPC | SPARTAN | MFS 40.5 vs. 16.2 | APA decreasse the risk of metastasis | [20] | |
| mCSPC | TITAN | 2-year OS 82.4 vs. 73.5 | APA improves OS and PFS | [21] | ||
| Darolutamide | nmCRPC | ARAMIS | MFS 40.4 vs. 18.4 | DA decreases the risk of metastasis | [22] | |
| Abiraterone | mCRPC | COA-301 | OS 15.8 vs. 11.2 | AB improves OS after chemotherapy | [23] | |
| COA-302 | OS 34.7 vs. 30.3 | AB improves OS in chemotherapy naïve | [24] | |||
| High-volume CSPC | LATITUDE | OS 53.3 vs. 36.5 | AB improves OS in high-risk CSPC | [25,26] | ||
| Radiotherapy | Radium 223 | CRPC with symptomatic bone metastasis, no visceral metastasis | ALSYMPCA | OS 14.9 vs. 11.3 | RA-223 improves OS in symptomatic bony metastatic mCRPC | [27] |
| 177Lu-PSMA 617 | PSMA-positive mCRPC and already treated with ARB and chemotherapy | VISION | OS 15.3 vs. 11.3 rPFS 8.7 vs. 3.4 | 177Lu-PSMA 617 improves OS and rPFS in PSMA-positive mCRPC | [28] | |
| Immunotherapy | Sipuleucel-T | mCRPC | IMPACT | OS 25.8 vs. 21.7 | Sipuleucel-T improves OS in mCRPC | [29] |
| PARP-I | Olaparib | mCRPC with HRR genes mutation after ENZ or AB | PROfound | OS 18.5 vs.15.1 rPFS 7.4 cs. 3.6 | Olaparib improves OS and rPFS in mCRPC with HRR gene mutation | [30] |
| Rubraca | mCRPC with BRCA genes after ARB or chemotherapy | TRITON2 (phase2) TRITON3 (phase3) | ORR 43.5% (IRR) PSA response rate 54.8% | Rubraca has the antitumor activity in mCRPC with BRCA gene mutation | [31,32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.-R.; Chen, Y.-A.; Kao, W.-H.; Lai, C.-H.; Lin, H.; Hsieh, J.-T. Developing New Treatment Options for Castration-Resistant Prostate Cancer and Recurrent Disease. Biomedicines 2022, 10, 1872. https://doi.org/10.3390/biomedicines10081872
Wang B-R, Chen Y-A, Kao W-H, Lai C-H, Lin H, Hsieh J-T. Developing New Treatment Options for Castration-Resistant Prostate Cancer and Recurrent Disease. Biomedicines. 2022; 10(8):1872. https://doi.org/10.3390/biomedicines10081872
Chicago/Turabian StyleWang, Bo-Ren, Yu-An Chen, Wei-Hsiang Kao, Chih-Ho Lai, Ho Lin, and Jer-Tsong Hsieh. 2022. "Developing New Treatment Options for Castration-Resistant Prostate Cancer and Recurrent Disease" Biomedicines 10, no. 8: 1872. https://doi.org/10.3390/biomedicines10081872