
Haematological Drugs Affecting Lipid Metabolism and Vascular Health


Abstract
1. Introduction
2. Tyrosine Kinase Inhibitors
3. Janus Associated Kinase (JAK) Inhibitors
4. PEG-Asparaginase
5. Calcineurin Inhibitors
6. Mammalian Target of Rapamycin Inhibitors
7. All-Trans Retinoic Acid
8. Corticosteroids
9. Anti-CD20
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LDL | Low-density lipoproteins |
| LDL-C | Low-density lipoprotein cholesterol |
| ASCVD | Atherosclerotic cardiovascular disease |
| VLDL | Very low density lipoproteins |
| HDL | High-density lipoproteins |
| HDL-C | High-density lipoprotein cholesterol |
| TG | Triglycerides |
| ART | Antiretroviral therapy |
| HIV | Human immunodeficiency virus |
| PIs | Protease inhibitors |
| NRTIs | Nucleoside reverse-transcriptase inhibitors |
| ALL | Acute lymphoblastic leukaemia |
| CML | Chronic myeloid leukaemia |
| TKIs | Tyrosine kinase inhibitors |
| CV | Cardiovascular |
| PAD | Peripheral arterial disease |
| ABI | Ankle brachial index |
| ApoE | Apolipoprotein E |
| JAK1 | Janus kinase 1 |
| TEK | Tyrosine kinase, endothelial |
| ICAM-1 | Intercellular adhesion molecule-1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| LPL | Lipoprotein lipase |
| PCSK-9 | Proprotein convertase subtilisin–kexin type 9 |
| GvHD | Graft-versus-host disease |
| JAK2/STAT3 | Janus kinase2/signal transducer and activator transcription3 |
| PEG-ASP | Pegylated asparaginase |
| ApoCIII | Apolipoprotein CIII |
| SNP | Single nucleotide polymorphisms |
| ApoA5 | Apolipoprotein A5 |
| CNIs | Calcineurin inhibitors |
| ApoB100 | Apolipoprotein B100 |
| sdLDL | Small dense low-density lipoproteins |
| HMG-CoA | Hydroxymethylglutaryl-CoA |
| circRNA | Circular RNA |
| miR | MicroRNA |
| SREBP | Sterol regulatory element-binding protein |
| Akt | Protein kinase B |
| mTOR | Mammalian target of rapamycin |
| APS | Antiphospholipid syndrome |
| VEGF | Vascular endothelial growth factor |
| APL | Acute promyelocytic leukaemia |
| ATRA | All-trans retinoic acid |
| PML | Promyelocytic leukaemia gene from chromosome 17 |
| RAR-α | Retinoic acid receptor α gene from chromosome 15 |
| PPAR | Peroxisome proliferator-activated receptor |
| CHOP | Cyclophosphamide, doxorubicin, vincristine, prednisolone |
| CD20 | Cluster of differentiation 20 (B-lymphocyte antigen) |
References
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; de Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; Packard, C.J.; Chapman, M.J.; Borén, J.; Aguilar-Salinas, C.A.; Averna, M.; Ference, B.A.; Gaudet, D.; Hegele, R.A.; Kersten, S.; et al. Triglyceride-rich lipoproteins and their remnants: Metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur. Heart J. 2021, 42, 4791–4806. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzo, G.; Tripaldella, M.; Mallardo, V.; Morgillo, M.; Vitelli, N.; Iannuzzi, A.; Aliberti, E.; Giallauria, F.; Tramontano, A.; Carluccio, R.; et al. Lipoprotein(a) Where Do We Stand? From the Physiopathology to Innovative Terapy. Biomedicines 2021, 9, 838. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, A.; Gentile, M.; Iannuzzo, G.; Covetti, G.; Panico, C.; Mattiello, A.; La Fata, E.; D′Elia, L.; De Michele, M.; Rubba, P. Atherogenic Lipoprotein Subfractions and Carotid Atherosclerosis in Menopausal Women. Angiology 2018, 69, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Tziomalos, K.; Athyros, V.G.; Karagiannis, A.; Mikhailidis, D.P. Dyslipidemia Induced by Drugs Used for the Prevention and Treatment of Vascular Diseases. Open Cardiovasc. Med. J. 2011, 5, 85–89. [Google Scholar] [CrossRef]
- Mantel-Teeuwisse, A.K.; Kloosterman, J.M.; der Zee, A.H.M.; Klungel, O.H.; Porsius, A.J.; de Boer, A. Drug-Induced lipid changes: A review of the unintended effects of some commonly used drugs on serum lipid levels. Drug Saf. 2001, 24, 443–456. [Google Scholar] [CrossRef]
- Ott, S.M.; Lacroix, A.Z.; Ichikawa, L.E.; Scholes, D.; Barlow, W.E. Effect of Low-Dose Thiazide Diuretics on Plasma Lipids: Results from a Double-Blind, Randomized Clinical Trial in Older Men and Women. J. Am. Geriatr. Soc. 2003, 51, 340–347. [Google Scholar] [CrossRef]
- Giugliano, D.; Acampora, R.; Marfella, R.; De Rosa, N.; Ziccardi, P.; Ragone, R.; De Angelis, L.; D′Onofrio, F. Metabolic and Cardiovascular Effects of Carvedilol and Atenolol in Non-Insulin-Dependent Diabetes Mellitus and Hypertension. Ann. Intern. Med. 1997, 126, 955–959. [Google Scholar] [CrossRef]
- BGaulin, B.D.; Markowitz, J.S.; Caley, C.F.; Nesbitt, L.A.; Dufresne, R.L. Clozapine-Associated Elevation in Serum Triglycerides. Am. J. Psychiatry 1999, 156, 1270–1272. [Google Scholar] [CrossRef]
- Araujo, S.; Bañón, S.; Machuca, I.; Moreno, A.; Pérez-Elías, M.J.; Casado, J.L. Prevalence of insulin resistance and risk of diabetes mellitus in HIV-infected patients receiving current antiretroviral drugs. Eur. J. Endocrinol. 2014, 171, 545–554. [Google Scholar] [CrossRef]
- Lee, D. HIV: How to manage dyslipidaemia in HIV. Drugs Context 2022, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, E. The Relationship Between Hematological Malignancy and Lipid Profile. Medeni. Med. J. 2021, 36, 146–151. [Google Scholar] [PubMed]
- Patni, N.; Li, X.; Adams-Huet, B.; Garg, A. The prevalence and etiology of extreme hypertriglyceridemia in children: Data from a tertiary children′s hospital. J. Clin. Lipidol. 2018, 12, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Mauro, M.J. Lifelong TKI therapy: How to manage cardiovascular and other risks. Hematology 2021, 2021, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Gottardi, M.; Manzato, E.; Gherlinzoni, F. Imatinib and hyperlipidemia. N. Engl. J. Med. 2005, 353, 2722–2723. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Hughes, T.P.; Larson, R.A.; Kim, D.-W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Boquimpani, C.; Pasquini, R.; Clark, R.E.; et al. Long-term outcomes with frontline nilotinib versus imatinib in newly diagnosed chronic myeloid leukemia in chronic phase: ENESTnd 10-year analysis. Leukemia 2021, 35, 440–453. [Google Scholar] [CrossRef]
- Le Coutre, P.; Rea, D.; Abruzzese, E.; Dombret, H.; Trawinska, M.M.; Herndlhofer, S.; Dörken, B.; Valent, P. Severe Peripheral Arterial Disease During Nilotinib Therapy. JNCI J. Natl. Cancer Inst. 2011, 103, 1347–1348. [Google Scholar] [CrossRef]
- Bondon-Guitton, E.; Combret, S.; Pérault-Pochat, M.C.; Stève-Dumont, M.; Bagheri, H.; Huguet, F.; Despas, F.; Pathak, A.; Montastruc, J.L. Cardiovascular risk profile of patients with peripheral arterial occlusive disease during nilotinib therapy. Target. Oncol. 2016, 11, 549–552. [Google Scholar] [CrossRef]
- Kim, T.D.; Rea, D.; Schwarz, M.; Grille, P.; Nicolini, F.E.; Rosti, G.; Levato, L.; Giles, F.J.; Dombret, H.; Mirault, T.; et al. Peripheral artery occlusive disease in chronic phase chronic myeloid leukemia patients treated with nilotinib or imatinib. Leukemia 2013, 27, 1316–1321. [Google Scholar] [CrossRef]
- Tefferi, A.; Letendre, L. Nilotinib treatment-associated peripheral artery disease and sudden death: Yet another reason to stick to imatinib as front-line therapy for chronic myelogenous leukemia. Am. J. Hematol. 2011, 86, 610–611. [Google Scholar] [CrossRef]
- Hadzijusufovic, E.; Albrecht-Schgoer, K.; Huber, K.; Hoermann, G.; Grebien, F.; Eisenwort, G.; Schgoer, W.; Herndlhofer, S.; Kaun, C.; Theurl, M.; et al. Nilotinib-induced vasculopathy: Identification of vascular endothelial cells as a primary target site. Leukemia 2017, 31, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J. Nilotinib-associated vascular events. Clin. Lymphoma Myeloma Leuk. 2012, 12, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Rea, D.; Mirault, T.; Cluzeau, T.; Gautier, J.-F.; Guilhot, F.; Dombret, H.; Messas, E. Early onset hypercholesterolemia induced by the 2nd-generation tyrosine kinase inhibitor nilotinib in patients with chronic phase-chronic myeloid leukemia. Haematologica 2014, 99, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Caocci, G.; Mulas, O.; Capodanno, I.; Bonifacio, M.; Annunziata, M.; Galimberti, S.; Luciano, L.; Tiribelli, M.; Martino, B.; Castagnetti, F.; et al. Low-density lipoprotein (LDL) levels and risk of arterial occlusive events in chronic myeloid leukemia patients treated with nilotinib. Ann. Hematol. 2021, 100, 2005–2014. [Google Scholar] [CrossRef]
- Abumiya, M.; Akamine, Y.; Sato, S.; Takahashi, S.; Yoshioka, T.; Kameoka, Y.; Takahashi, N.; Miura, M. Effects of proprotein convertase subtilisin/kexin type 9 and nilotinib plasma concentrations on nilotinib-induced hypercholesterolaemia in patients with chronic myeloid leukaemia. J. Clin. Pharm. Ther. 2021, 46, 382–387. [Google Scholar] [CrossRef]
- Sadiq, S.; Owen, E.; Foster, T.; Knight, K.; Wang, L.; Pirmohamed, M.; Clark, R.E.; Pushpakom, S. Nilotinib-induced metabolic dysfunction: Insights from a translational study using in vitro adipocyte models and patient cohorts. Leukemia 2019, 33, 1810–1814. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; Le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A Phase 2 Trial of Ponatinib in Philadelphia Chromosome–Positive Leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef]
- Chan, O.; Talati, C.; Isenalumhe, L.; Shams, S.; Nodzon, L.; Fradley, M.; Sweet, K.; Pinilla-Ibarz, J. Side-effects profile and outcomes of ponatinib in the treatment of chronic myeloid leukemia. Blood Adv. 2020, 4, 530–538. [Google Scholar] [CrossRef]
- Caocci, G.; Mulas, O.; Capodanno, I.; Abruzzese, E.; Iurlo, A.; Luciano, L.; Albano, F.; Annunziata, M.; Tiribelli, M.; Bonifacio, M.; et al. Low low-density lipoprotein (LDL), cholesterol and triglycerides plasma levels are associated with reduced risk of arterial occlusive events in chronic myeloid leukemia patients treated with ponatinib in the real-life. A Campus CML study. Blood Cancer J. 2020, 10, 66. [Google Scholar] [CrossRef]
- Zeng, P.; Schmaier, A. Ponatinib and other CML Tyrosine Kinase Inhibitors in Thrombosis. Int. J. Mol. Sci. 2020, 21, 6556. [Google Scholar] [CrossRef]
- Breccia, M.; Alimena, G.; Baccarani, M.; Bocchia, M.; Di Raimondo, F.; Passerini, C.G.; Gozzini, A.; Morra, E.; Pane, F.; Pregno, P.; et al. Current management of CML patients: Summary of the Italian Consensus Meeting held in Rome, April 11–12, 2013. Crit. Rev. Oncol. 2014, 90, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, A.; Rubba, P.; Gentile, M.; Mallardo, V.; Calcaterra, I.; Bresciani, A.; Covetti, G.; Cuomo, G.; Merone, P.; Di Lorenzo, A.; et al. Carotid Atherosclerosis, Ultrasound and Lipoproteins. Biomedicines 2021, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Verstovsek, S. Molecular Pathways: JAK/STAT Pathway: Mutations, Inhibitors, and Resistance. Clin. Cancer Res. 2013, 19, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Verstovsek, S.; Gupta, V.; Mascarenhas, J.O.; Atallah, E.; Burn, T.; Sun, W.; Sandor, V.; Gotlib, J. Effects of Ruxolitinib Treatment on Metabolic and Nutritional Parameters in Patients With Myelofibrosis From COMFORT-I. Clin. Lymphoma Myeloma Leuk. 2014, 15, 214–221.e1. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.P.; Brunstein, C.G.; Holtan, S.G. Life-Threatening Hypertriglyceridemia in a Patient on Ruxolitinib and Sirolimus for Chronic Graft-versus-Host Disease. Case Rep. Transplant. 2018, 2018, 4539757. [Google Scholar] [CrossRef] [PubMed]
- Bauters, T.; Bordon, V.; Laureys, G.; Dhooge, C. Combined use of ruxolitinib and sirolimus: Increased monitoring of triglycerides required. Bone Marrow Transplant. 2019, 54, 1372–1373. [Google Scholar] [CrossRef]
- Mollé, N.; Krichevsky, S.; Kermani, P.; Silver, R.T.; Ritchie, E.; Scandura, J.M. Ruxolitinib can cause weight gain by blocking leptin signaling in the brain via JAK2/STAT3. Blood 2020, 135, 1062–1066. [Google Scholar] [CrossRef]
- Derman, B.A.; Streck, M.; Wynne, J.; Christ, T.N.; Curran, E.; Stock, W.; Knoebel, R.W. Efficacy and toxicity of reduced vs. standard dose pegylated asparaginase in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia. Leuk. Lymphoma 2020, 61, 614–622. [Google Scholar] [CrossRef]
- Parsons, S.K.; Skapek, S.X.; Neufeld, E.J.; Kuhlman, C.; Young, M.L.; Donnelly, M.; Brunzell, J.D.; Otvos, J.D.; Sallan, S.E.; Rifai, N. Asparaginase-associated lipid abnormalities in children with acute lymphoblastic leukemia. Blood 1997, 89, 1886–1895. [Google Scholar] [CrossRef]
- Hoogerbrugge, N.; Jansen, H.; Hoogerbrugge, P.M. Transient hyperlipidemia during treatment of ALL with L-asparaginase is related to decreased lipoprotein lipase activity. Leukemia 1997, 11, 1377–1379. [Google Scholar] [CrossRef][Green Version]
- Cremer, P.; Lakomek, M.; Beck, W.; Prindull, G. The effect of L-asparaginase on lipid metabolism during induction chemotherapy of childhood lymphoblastic leukaemia. Eur. J. Pediatr. 1988, 147, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.H.; Pieters, R.; de Groot-Kruseman, H.A.; Hop, W.C.; Boos, J.; Tissing, W.J.; van der Sluis, I.M. The toxicity of very prolonged courses of PEGasparaginase or Erwinia asparaginase in relation to asparaginase activity, with a special focus on dyslipidemia. Haematologica 2014, 99, 1716–1721. [Google Scholar] [CrossRef]
- Finch, E.R.; Smith, C.A.; Yang, W.; Liu, Y.; Kornegay, N.M.; Panetta, J.C.; Crews, K.R.; Molinelli, A.R.; Cheng, C.; Pei, D.; et al. Asparaginase formulation impacts hypertriglyceridemia during therapy for acute lymphoblastic leukemia. Pediatr. Blood Cancer 2019, 67, e28040. [Google Scholar] [CrossRef] [PubMed]
- Ridola, V.; Buonuomo, P.S.; Maurizi, P.; Putzulu, R.; Annunziata, M.L.; Pietrini, D.; Riccardi, R. Severe acute hypertriglyceridemia during acute lymphoblastic leukemia induction successfully treated with plasmapheresis. Pediatr. Blood Cancer 2008, 50, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, A.; Annunziata, M.; Fortunato, G.; Giacobbe, C.; Palma, D.; Bresciani, A.; Aliberti, E.; Iannuzzo, G. Case Report: Genetic Analysis of PEG-Asparaginase Induced Severe Hypertriglyceridemia in an Adult With Acute Lymphoblastic Leukaemia. Front. Genet. 2022, 13, 832890. [Google Scholar] [CrossRef]
- Liu, A.Y.; Nabel, C.S.; Finkelman, B.S.; Ruth, J.R.; Kurzrock, R.; van Rhee, F.; Krymskaya, V.P.; Kelleher, D.; Rubenstein, A.H.; Fajgenbaum, D.C. Idiopathic multicentric Castleman′s disease: A systematic literature review. Lancet Haematol 2016, 3, e163–e175. [Google Scholar] [CrossRef]
- Storb, R.; Deeg, H.J.; Whitehead, J.; Appelbaum, F.; Beatty, P.; Bensinger, W.; Buckner, C.D.; Clift, R.; Doney, K.; Farewell, V.; et al. Methotrexate and Cyclosporine Compared with Cyclosporine Alone for Prophylaxis of Acute Graft versus Host Disease after Marrow Transplantation for Leukemia. N. Engl. J. Med. 1986, 314, 729–735. [Google Scholar] [CrossRef]
- Ratanatharathorn, V.; Nash, R.A.; Przepiorka, D.; Devine, S.M.; Klein, J.L.; Weisdorf, D.; Fay, J.W.; Nademanee, A.; Antin, J.H.; Christiansen, N.P.; et al. Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft-versus-host disease prophylaxis after HLA-identical sibling bone marrow transplantation. Blood 1998, 92, 2303–2314. [Google Scholar]
- Paviglianiti, A.; Labopin, M.; Blaise, D.; Socié, G.; Bulabois, C.E.; Lioure, B.; Ceballos, P.; Blau, I.W.; Guillerm, G.; Maertens, J. Comparison of mycophenolate mofetil and calcineurin inhibitor versus calcineurin inhibitor-based graft-versus-host-disease prophylaxis for matched unrelated donor transplant in acute myeloid leukemia. A study from the ALWP of the EBMT. Bone Marrow Transpl. 2021, 56, 1077–1085. [Google Scholar] [CrossRef]
- Kockx, M.; Glaros, E.; Leung, B.; Ng, T.W.; Berbée, J.F.; Deswaerte, V.; Nawara, D.; Quinn, C.; Rye, K.-A.; Jessup, W.; et al. Low-Density Lipoprotein Receptor–Dependent and Low-Density Lipoprotein Receptor–Independent Mechanisms of Cyclosporin A–Induced Dyslipidemia. Arter. Thromb. Vasc. Biol. 2016, 36, 1338–1349. [Google Scholar] [CrossRef]
- Taylor, D.O.; Barr, M.L.; Radovancevic, B.; Renlund, D.G.; Jr, R.M.M.; Smart, F.W.; Tolman, D.E.; Frazier, O.; Young, J.B.; VanVeldhuisen, P. A randomized, multicenter comparison of tacrolimus and cyclosporine immunosuppressive regimens in cardiac transplantation: Decreased hyperlipidemia and hypertension with tacrolimus. J. Heart Lung Transplant. 1999, 18, 336–345. [Google Scholar] [CrossRef]
- Zimmermann, A.; Zobeley, C.; Weber, M.; Lang, H.; Galle, P.R.; Zimmermann, T. Changes in lipid and carbohydrate metabolism under mTOR- and calcineurin-based immunosuppressive regimen in adult patients after liver transplantation. Eur. J. Intern. Med. 2016, 29, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Kuster, G.M.; Drexel, H.; Bleisch, J.A.; Rentsch, K.; Pei, P.; Binswanger, U.; Amann, F.W. Relation of cyclosporine blood levels to adverse effects on lipoproteins. Transplantation 1994, 57, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Palming, J.; Rizell, M.; Aureliano, M.; Carvalho, E.; Svensson, M.K.; Eriksson, J.W. The immunosuppressive agents rapamycin, cyclosporin A and tacrolimus increase lipolysis, inhibit lipid storage and alter expression of genes involved in lipid metabolism in human adipose tissue. Mol. Cell. Endocrinol. 2013, 365, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, Y.; Ferrari, L.; Souidi, M.; Batt, A.M.; Lutton, C.; Siest, G.; Visvikis, S. Compared effect of immunosuppressive drugs cyclosporine A and rapamycin on cholesterol homeostasis key enzymes CYP27A1 and HMG-CoA reductase. Basic Clin. Pharmacol. Toxicol. 2007, 100, 392–397. [Google Scholar] [CrossRef]
- de Groen, P.C. Cyclosporine, low-density lipoprotein, and cholesterol. Mayo Clin. Proc. 1998, 63, 1012–1021. [Google Scholar] [CrossRef]
- Derfler, K.; Hayde, M.; Heinz, G.; Hirschl, M.M.; Steger, G.; Hauser, A.-C.; Balcke, P.; Widhalm, K. Decreased postheparin lipolytic activity in renal transplant recipients with cyclosporin A. Kidney Int. 1991, 40, 720–727. [Google Scholar] [CrossRef][Green Version]
- Ichimaru, N.; Takahara, S.; Kokado, Y.; Wang, J.D.; Hatori, M.; Kameoka, H.; Inoue, T.; Okuyama, A. Changes in lipid metabolism and effect of simvastatin in renal transplant recipients induced by cyclosporine or tacrolimus. Atherosclerosis 2001, 158, 417–423. [Google Scholar] [CrossRef]
- Kimak, E.; Solski, J.; Baranowicz-Gąszczyk, I.; Książek, A. A Long-Term Study of Dyslipidemia and Dyslipoproteinemia in Stable Post-Renal Transplant Patients. Ren. Fail. 2006, 28, 483–486. [Google Scholar] [CrossRef]
- Tur, M.; Garrigue, V.; Vela, C.; Dupuy, A.M.; Descomps, B.; Cristol, J.; Mourad, G. Apolipoprotein CIII is upregulated by anticalcineurins and rapamycin: Implications in transplantation-induced dyslipidemia. Transplant. Proc. 2000, 32, 2783–2784. [Google Scholar] [CrossRef]
- Neto, A.M.; Bovi, T.; Righetto, C.; Fiore, A.; Lot, L.; Perales, S.; De Ataide, E.; Boin, I. Clinical Profile of Patients With Diabetes Mellitus and Liver Transplantation: Results After a Multidisciplinary Team Intervention. Transplant. Proc. 2018, 50, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, K.; Wei, R.; Fan, G.; Cai, X.; Xu, L.; Cen, B.; Wang, J.; Xie, H.; Zheng, S.; et al. The circFASN/miR-33a pathway participates in tacrolimus-induced dysregulation of hepatic triglyceride homeostasis. Signal Transduct. Target. Ther. 2020, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- ARodriguez-Rodriguez, A.E.; Porrini, E.; Torres, A. Beta-Cell Dysfunction Induced by Tacrolimus: A Way to Explain Type 2 Diabetes? Int. J. Mol. Sci. 2021, 22, 10311. [Google Scholar] [CrossRef] [PubMed]
- Fazal, M.A.; Idrees, M.K.; Akhtar, S.F. Dyslipidaemia among renal transplant recipients: Cyclosporine versus tacrolimus. J. Pak. Med. Assoc. 2014, 64, 496–499. [Google Scholar] [PubMed]
- Holdaas, H.; Julian, D. The use of statins after solid organ transplantation. Nephrol. Dial. Transplant. 2002, 17, 1537. [Google Scholar] [CrossRef]
- Moore, R.; Hernandez, D.; Valantine, H. Calcineurin Inhibitors and Post-Transplant Hyperlipidaemias. Drug Saf. 2001, 24, 755–766. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, X.; Cassady, K.; Zou, Z.; Yang, S.; Wang, Z.; Zhang, X. The Role of mTOR Inhibitors in Hematologic Disease: From Bench to Bedside. Front. Oncol. 2021, 10, 611690. [Google Scholar] [CrossRef]
- Platzbecker, U.; Haase, M.; Herbst, R.; Hänel, A.; Voigtmann, K.; Thiede, C.; Mohr, B.; Schleyer, E.; Leopold, T.; Orth, M.; et al. Activity of sirolimus in patients with myelodysplastic syndrome—Results of a pilot study. Br. J. Haematol. 2005, 128, 625–630. [Google Scholar] [CrossRef]
- Bartalucci, N.; Guglielmelli, P.; Vannucchi, A.M. Rationale for Targeting the PI3K/Akt/mTOR Pathway in Myeloproliferative Neoplasms. Clin. Lymphoma Myeloma Leuk. 2013, 13, S307–S309. [Google Scholar] [CrossRef]
- Kalfa, T.A. Warm antibody autoimmune hemolytic anemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 690–697. [Google Scholar] [CrossRef]
- Armand, P.; Gannamaneni, S.; Kim, H.T.; Cutler, C.S.; Ho, V.T.; Koreth, J.; Alyea, E.P.; LaCasce, A.S.; Jacobsen, E.D.; Fisher, D.C.; et al. Improved Survival in Lymphoma Patients Receiving Sirolimus for Graft-Versus-Host Disease Prophylaxis After Allogeneic Hematopoietic Stem-Cell Transplantation With Reduced-Intensity Conditioning. J. Clin. Oncol. 2008, 26, 5767–5774. [Google Scholar] [CrossRef] [PubMed]
- Sandmaier, B.M.; Kornblit, B.; Storer, B.E.; Olesen, G.; Maris, M.B.; Langston, A.A.; Gutman, J.A.; Petersen, S.L.; Chauncey, T.R.; Bethge, W.A.; et al. Addition of sirolimus to standard cyclosporine plus mycophenolate mofetil-based graft-versus-host disease prophylaxis for patients after unrelated non-myeloablative haemopoietic stem cell transplantation: A multicentre, randomised, phase 3 trial. Lancet Haematol. 2019, 6, e409–e418. [Google Scholar] [CrossRef]
- Kaplan, B.; Qazi, Y.; Wellen, J.R. Strategies for the management of adverse events associated with mTOR inhibitors. Transplant. Rev. 2014, 28, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Sivendran, S.; Agarwal, N.; Gartrell, B.; Ying, J.; Boucher, K.M.; Choueiri, T.K.; Sonpavde, G.; Oh, W.K.; Galsky, M.D. Metabolic complications with the use of mTOR inhibitors for cancer therapy. Cancer Treat. Rev. 2014, 40, 190–196. [Google Scholar] [CrossRef]
- Morviducci, L.; Rota, F.; Rizza, L.; Di Giacinto, P.; Ramponi, S.; Nardone, M.; Tubili, C.; Lenzi, A.; Zuppi, P.; Baldelli, R. Everolimus is a new anti-cancer molecule: Metabolic side effects as lipid disorders and hyperglycemia. Diabetes Res. Clin. Pract. 2018, 143, 428–431. [Google Scholar] [CrossRef]
- Badiou, S.; Cristol, J.-P.; Mourad, G. Dyslipidemia following kidney transplantation: Diagnosis and treatment. Curr. Diabetes Rep. 2009, 9, 305–311. [Google Scholar] [CrossRef]
- Houde, V.P.; Brûlé, S.; Festuccia, W.T.; Blanchard, P.-G.; Bellmann, K.; Deshaies, Y.; Marette, A. Chronic Rapamycin Treatment Causes Glucose Intolerance and Hyperlipidemia by Upregulating Hepatic Gluconeogenesis and Impairing Lipid Deposition in Adipose Tissue. Diabetes 2010, 59, 1338–1348. [Google Scholar] [CrossRef]
- Liu, Q.-Y.; Nambi, P. Sirolimus upregulates aP2 expression in human monocytes and macrophages. Transplant. Proc. 2004, 36, 3229–3231. [Google Scholar] [CrossRef]
- Hoogeveen, R.; Ballantyne, C.M.; Pownall, H.; Opekun, A.R.; Hachey, D.L.; Jaffe, J.S.; Oppermann, S.; Kahan, B.D.; Morrisett, J.D. Effect of sirolimus on the metabolism of apob100- containing lipoproteins in renal transplant patients1. Transplantation 2001, 72, 1244–1250. [Google Scholar] [CrossRef]
- Colonna, V.D.G.; Pavanello, C.; Rusconi, F.; Sartore-Bianchi, A.; Siena, S.; Castelnuovo, S.; Sirtori, C.R.; Mombelli, G. Lipid-lowering therapy of everolimus-related severe hypertriglyceridaemia in a pancreatic neuroendocrine tumour (pNET). J. Clin. Pharm. Ther. 2018, 43, 114–116. [Google Scholar] [CrossRef]
- Iannuzzo, G.; Cuomo, G.; Di Lorenzo, A.; Tripaldella, M.; Mallardo, V.; Idelson, P.I.; Sagnelli, C.; Sica, A.; Creta, M.; Baltar, J.; et al. Dyslipidemia in Transplant Patients: Which Therapy? J. Clin. Med. 2022, 11, 4080. [Google Scholar] [CrossRef] [PubMed]
- Arachchillage, D.R.; Laffan, M. Pathogenesis and management of antiphospholipid syndrome. Br. J. Haematol. 2017, 178, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Neeff, H.; Thomusch, O.; Strate, T.; Tittelbach-Helmrich, D.; Hopt, U.T.; von Dobschuetz, E. Everolimus Improves Microcirculatory Derangements in Experimental Postischemic Pancreatitis Modulating the Expression of Vascular Endothelial Growth Factor, Interleukin 6, and Toll-Like Receptor 4. Pancreas 2015, 44, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Haffer, H.; Wang, P.; Richter, M.; Chun, F.K.H.; Kornberger, A.; Beiras-Fernandez, A. Influence of an Early Application of Mammalian Target of Rapamycin Inhibitors Everolimus and Sirolimus on Acute Vascular Inflammatory Responses After Ischemia-Reperfusion Injury. Exp. Clin. Transplant. 2021, 19, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, J.; Qualtieri, J.; Head, D.; Savani, B.N.; Reddy, N. High prevalence of obesity in acute promyelocytic leukemia (APL): Implications for differentiating agents in APL and metabolic syndrome. Ther. Adv. Hematol. 2011, 2, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Marsden, J. Lipid metabolism and retinoid therapy. Pharmacol. Ther. 1989, 40, 55–65. [Google Scholar] [CrossRef]
- Staels, B. Regulation of lipid and lipoprotein metabolism by retinoids. J. Am. Acad. Dermatol. 2001, 45, S158–S167. [Google Scholar] [CrossRef]
- JSun, J.; Lou, Y.; Zhu, J.; Shen, H.; Zhou, D.; Zhu, L.; Yang, X.; Xie, M.; Li, L.; Huang, X.; et al. Hypertriglyceridemia in Newly Diagnosed Acute Promyelocytic Leukemia. Front. Oncol. 2020, 10, 577796. [Google Scholar]
- Kanamaru, A.; Takemoto, Y.; Tanimoto, M.; Murakami, H.; Asou, N.; Kobayashi, T.; Kuriyama, K.; Ohmoto, E.; Sakamaki, H.; Tsubaki, K. All-trans retinoic acid for the treatment of newly diagnosed acute promyelocytic leukemia. Japan Adult Leukemia Study Group. Blood 1995, 85, 1202–1206. [Google Scholar] [CrossRef]
- Kalisz, M.; Chmielowska, M.; Martyńska, L.; Domańska, A.; Bik, W.; Litwiniuk, A. All-trans-retinoic acid ameliorates atherosclerosis, promotes perivascular adipose tissue browning, and increases adiponectin production in Apo-E mice. Sci. Rep. 2021, 11, 4451. [Google Scholar] [CrossRef]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor beta/delta and retinoic acid receptor. Mol. Cell Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, G.; Scandali, V.M.; Trementino, L.; Cardinaletti, M.; Appolloni, G.; Boscaro, M. Pathophysiology of Dyslipidemia in Cushing’s Syndrome. Neuroendocrinology 2010, 92, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Jefferys, D.B.; Lessof, M.H.; Mattock, M.B. Corticosteroid treatment, serum lipids and coronary artery disease. Postgrad. Med. J. 1980, 56, 491–493. [Google Scholar] [CrossRef] [PubMed]
- El-Shaboury, A.H.; Hayes, T.M. Hyperlipidaemia in Asthmatic Patients Receiving Long-term Steroid Therapy. BMJ 1973, 2, 85–86. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chow, E.J.; Pihoker, C.; Hunt, K.; Wilkinson, K.; Friedman, D.L. Obesity and hypertension among children after treatment for acute lymphoblastic leukemia. Cancer 2007, 110, 2313–2320. [Google Scholar] [CrossRef]
- Geer, E.B.; Islam, J. Buettner, Mechanisms of glucocorticoid-induced insulin resistance: Focus on adipose tissue function and lipid metabolism. Endocrinol. Metab. Clin. N. Am. 2014, 43, 75–102. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Nikkilä, E.A.; Pelkonen, R.; Sane, T. Plasma lipoproteins, lipolytic enzymes, and very low density lipoprotein triglyceride turnover in Cushing′s syndrome. J. Clin. Endocrinol. Metab. 1983, 57, 619–626. [Google Scholar] [CrossRef]
- Benucci, M.; Saviola, G.; Manfredi, M.; Sarzi-Puttini, P.; Atzeni, F. Factors correlated with improvement of endothelial dysfunction during rituximab therapy in patients with rheumatoid arthritis. Biol. Targets Ther. 2013, 7, 69–75. [Google Scholar] [CrossRef][Green Version]
- Novikova, D.S.; Popkova, T.V.; Lukina, G.V.; Luchikhina, E.L.; Karateev, D.E.; Volkov, A.V.; Novikov, A.A.; Aleksandrova, E.N.; Nasonov, E.L. The Effects of Rituximab on Lipids, Arterial Stiffness and Carotid Intima-Media Thickness in Rheumatoid Arthritis. J. Korean Med. Sci. 2016, 31, 202–207. [Google Scholar] [CrossRef]
- Zhao, T.X.; Aetesam-Ur-Rahman, M.; Sage, A.P.; Victor, S.; Kurian, R.; Fielding, S.; Ait-Oufella, H.; Chiu, Y.-D.; Binder, C.J.; Mckie, M.; et al. Rituximab in patients with acute ST-elevation myocardial infarction: An experimental medicine safety study. Cardiovasc. Res. 2022, 118, 872–882. [Google Scholar] [CrossRef]
- Béliard, S.; Di Filippo, M.; Kaplanski, G.; Valéro, R. Highly efficacious, long-term, triglyceride lowering with rituximab therapy in a patient with autoimmune hypertriglyceridemia. J. Clin. Lipidol. 2018, 12, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-K.; Chung, J.-S.; Lee, G.-W.; Cho, S.-H.; Hong, J.; Shin, D.-Y.; Shin, H.-J. Statin use has negative clinical impact on non-germinal center in patients with diffuse large B cell lymphoma in rituximab era. Leuk. Res. 2015, 39, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Gouni, S.; Strati, P.; Toruner, G.; Aradhya, A.; Landgraf, R.; Bilbao, D.; Vega, F.; Agarwal, N.K. Statins enhance the chemosensitivity of R-CHOP in diffuse large B-cell lymphoma. Leuk. Lymphoma 2022, 63, 1302–1313. [Google Scholar] [CrossRef]
- Spinelli, G.; Felipe, C.; Park, S.; Mandia-Sampaio, E.; Tedesco-Silva, H.; Medina-Pestana, J. Lipid Profile Changes During the First Year After Kidney Transplantation: Risk Factors and Influence of the Immunosuppressive Drug Regimen. Transplant. Proc. 2011, 43, 3730–3737. [Google Scholar] [CrossRef] [PubMed]
- Bonet, M.L.; Ribot, J.; Palou, A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Popkova, T.V.; Nasonov, E.L. The Effect of Anti-B-cell Therapy on the Development of Atherosclerosis in Patients with Rheumatoid Arthritis. Curr. Pharm. Des. 2012, 18, 1512–1518. [Google Scholar]


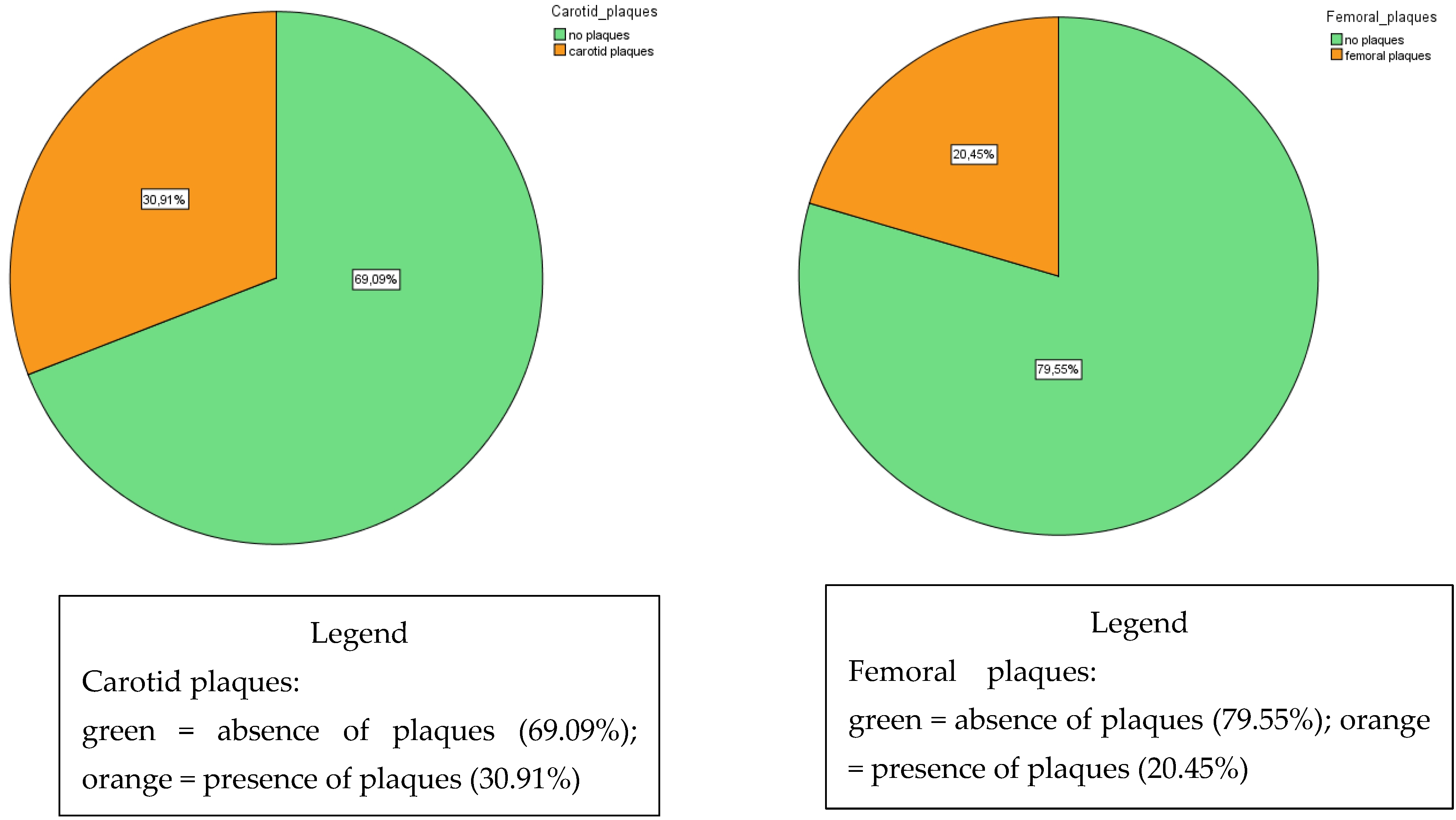{kind=link}
| Mean ± SD | Range | |
|---|---|---|
| Age (years) | 51.1 ± 13.6 | 25–79 |
| Sex (M/F) | 51% | |
| Systolic arterial blood pressure (mmHg) | 129 ± 18 | 106–180 |
| Diastolic arterial blood pressure (mmHg) | 82 ± 6 | 70–90 |
| Ankle–brachial pressure index (Right) | 1.14 ± 0.12 | 0.92–1.42 |
| Ankle–brachial pressure index (Left) | 1.14 ± 0.17 | 0.67–1.54 |
| Carotid intima–media thickness (right) (mm) | 1.36 ± 0.7 | 0.6–3.0 |
| Carotid intima–media thickness (left) (mm) | 1.23 ± 0.7 | 0.7–4.0 |
| Cardiac ejection fraction (%) | 60.7 ± 3.5 | 55–65 |
| Smokers (%) | 60% No 27% Ex 13% Yes | |
| Hypertensive (%) | 29% | |
| Diabetic (%) | 12% |
| Name of the Drug | Haematological Diseases in Which They Are Used | Mechanism of Action | Main Effects on Lipids or Vasculopathy | Pathogenesis | Treatment (Lifestyle Modifications Are Always Encouraged) |
|---|---|---|---|---|---|
| Nilotinib | LMC | Second-generation tyrosine kinase inhibitor | ↑ LDL-C [23] ↑↑ Vascular disease (PAD) [17,18,19,20] | Not fully understood prothrombotic and antiangiogenic effects | -Statins -Vasoactive drugs -Nilotinib discontinuation |
| Ponatinib | LMC | Third-generation tyrosine kinase inhibitor | ↑ Vascular disease (PAD) [27] | Not fully understood prothrombotic effects | -Statins -Vasoactive drugs -Ponatinib discontinuation |
| Ruxolitinib | Idiopathic myelofibrosis | JAK1/2 inhibitor | ↑↑ TG (together with sirolimus) [35,36] | Dysregulation of the leptin receptor [37] | In severe hyperTG: -Ruxolitinib discontinuation -Plasma exchange |
| PEG-asparaginase | LLA | Depletion of amino acid L-asparagine | ↑↑ TG (together with corticosteroids) [44,45] | ↑ VLDL [39] ↓ LPL [40] | -Omega 3 -Fibrates In severe hyperTG: -PEG-ASP discontinuation -Plasma exchange |
| Cyclosporine | Prophylaxis of GvHD after haematopoietic stem-cell transplantation, Castleman disease | Calcineurin inhibitor | ↑ LDL [51] ↑TG [51] ↑ sdLDL [60] | ↑ Insulin resistance [54] ↑ Cholesterol synthesis [55] ↓ Clearance VLDL [57] ↑ Apo CIII [60] ↓ LPL [58,59] | -Fluvastatin, pravastatin -fibrates |
| Tacrolimus | Prophylaxis of GvHD after haematopoietic stem-cell transplantation | Calcineurin inhibitor | ↑ LDL [104] ↑ TG [104] | ↑ Insulin resistance [61] ↓ Akt phosphorylation [63] ↓ Circ-RNA [62] | -Fluvastatin, pravastatin -fibrates |
| Sirolimus | Prophylaxis of GvHD after haematopoietic stem-cell transplantation; myelodysplastic syndrome; autoimmune haemolytic anaemia | mTOR inhibitor | ↑ TG [60,73,74] ↑ VLDL [79] ↑ sd LDL [52] ↑ LDL [73,74] | ↑ Apo CIII [60] ↑ Fatty acid binding protein [78] ↑ Gluconeogenesis [77] ↓ LPL activity [75] ↓ Clearance LDL [75,76] | -Fenofibrate -Statins (↑ risk of rhabdomyolysis) -Omega-3-acid ethyl esters |
| Everolimus | Prophylaxis of GvHD after haematopoietic stem-cell transplantation | mTOR inhibitor | ↑ TG [73,75] ↑ VLDL [80] ↑ sd LDL [52] ↑ LDL [75] | ↑ Apo CIII [80] ↓ LPL activity [75,80] ↓ Clearance LDL [75,80] | -Fenofibrate -Statins (↑ risk of rhabdomyolysis) -Omega-3-acid ethyl esters |
| ATRA | APL | Antineoplastic agent | ↑↑ TG [89] | ↑ Hepatic TG synthesis [105] ↑ apo CIII [87] | -Omega-3-acid ethyl esters -Fibrates -ATRA withdrawal |
| Corticosteroids | Used in conjunction with other agents in multiple haematological diseases | Anti-inflammatory steroid | ↑ TG [95] ↑= LDL [93,94] | ↑ Insulin resistance [96] ↑ VLDL [97] | -Statins -Fibrates |
| Rituximab | Non-Hodgkin lymphomas | Anti-CD20 | ↓ Carotid IMT [98,99] ↓ TG [101] | Suppression of active B2 cells [106] | Not necessary |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrella, A.; Iannuzzi, A.; Annunziata, M.; Covetti, G.; Cavallaro, R.; Aliberti, E.; Tortori, E.; Iannuzzo, G. Haematological Drugs Affecting Lipid Metabolism and Vascular Health. Biomedicines 2022, 10, 1935. https://doi.org/10.3390/biomedicines10081935
Parrella A, Iannuzzi A, Annunziata M, Covetti G, Cavallaro R, Aliberti E, Tortori E, Iannuzzo G. Haematological Drugs Affecting Lipid Metabolism and Vascular Health. Biomedicines. 2022; 10(8):1935. https://doi.org/10.3390/biomedicines10081935
Chicago/Turabian StyleParrella, Antonio, Arcangelo Iannuzzi, Mario Annunziata, Giuseppe Covetti, Raimondo Cavallaro, Emilio Aliberti, Elena Tortori, and Gabriella Iannuzzo. 2022. "Haematological Drugs Affecting Lipid Metabolism and Vascular Health" Biomedicines 10, no. 8: 1935. https://doi.org/10.3390/biomedicines10081935
APA StyleParrella, A., Iannuzzi, A., Annunziata, M., Covetti, G., Cavallaro, R., Aliberti, E., Tortori, E., & Iannuzzo, G. (2022). Haematological Drugs Affecting Lipid Metabolism and Vascular Health. Biomedicines, 10(8), 1935. https://doi.org/10.3390/biomedicines10081935