
Upregulated miR-18a-5p in Colony Forming Unit-Hill’s in Subclinical Cardiovascular Disease and Metformin Therapy; MERIT Study †

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Meso Scale Discovery (MSD) Assay
2.3. Flow Cytometric Evaluation of Circulating Endothelial Progenitor Cells
2.4. Culture and Quantification of CFU-Hill’s Colonies
2.5. Plasma Sample Preparation
2.6. MiRNA Expression Using Real-Time Quantitative PCR
2.7. Pathway Analysis: Ingenuity Pathway Analysis of miR-18a-5p
2.8. Statistical Analysis
3. Results
3.1. Characteristics of Studied Subjects
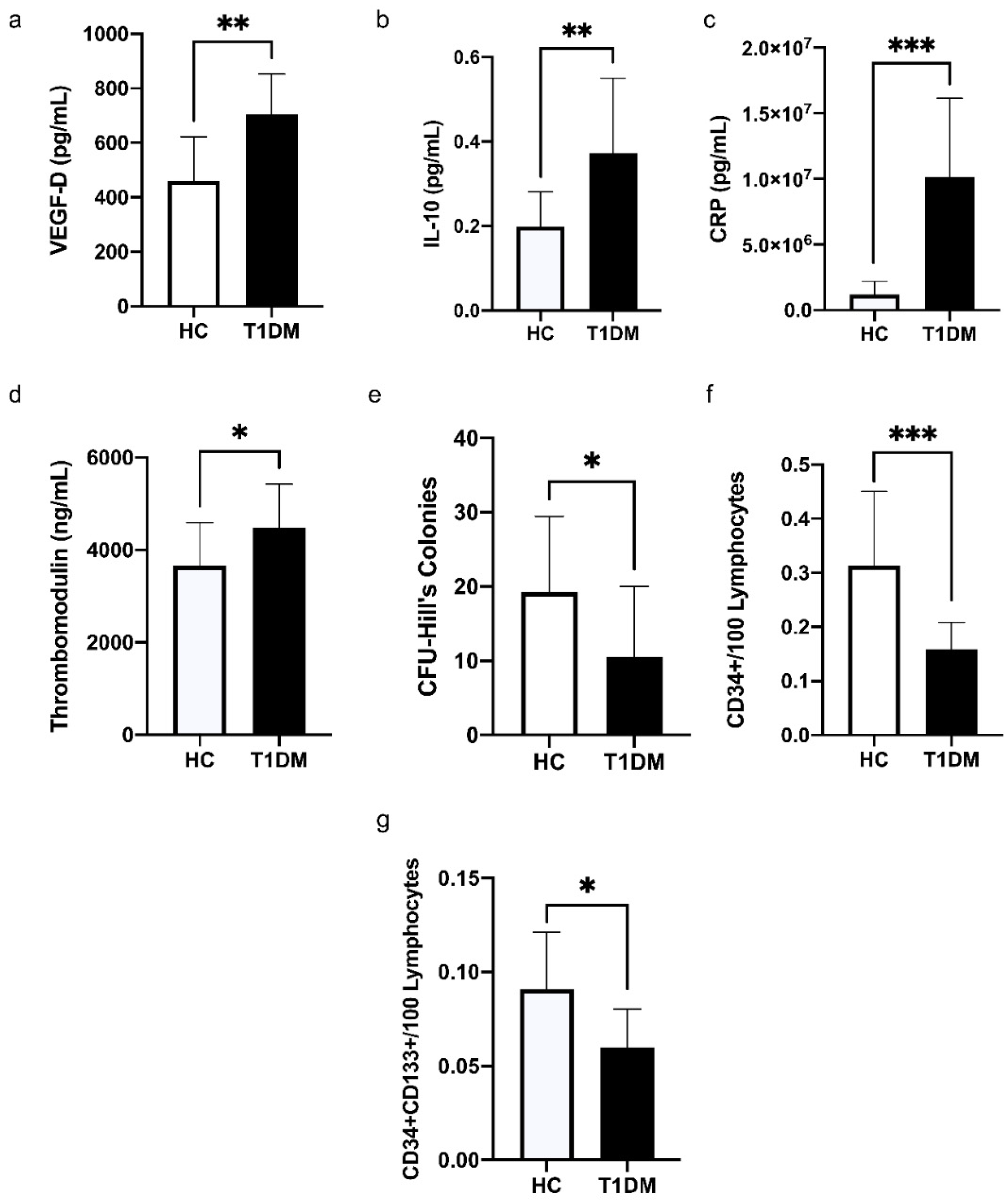
3.2. Comparison of the Levels of Cytokines, C-Reactive Protein (CRP), and Vascular Health Indices between HC and T1DM
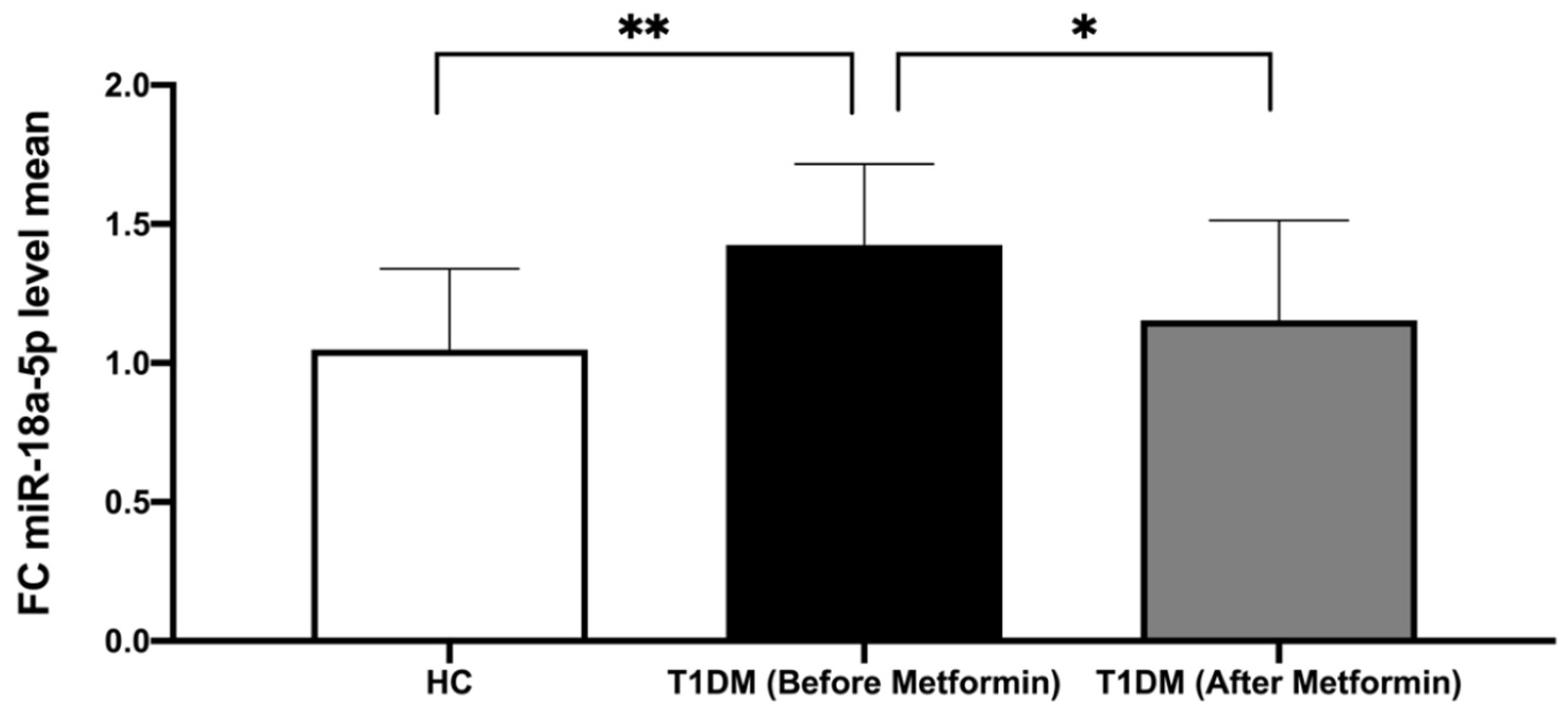
3.3. The Expression of miR-18a-5p in Study Participants
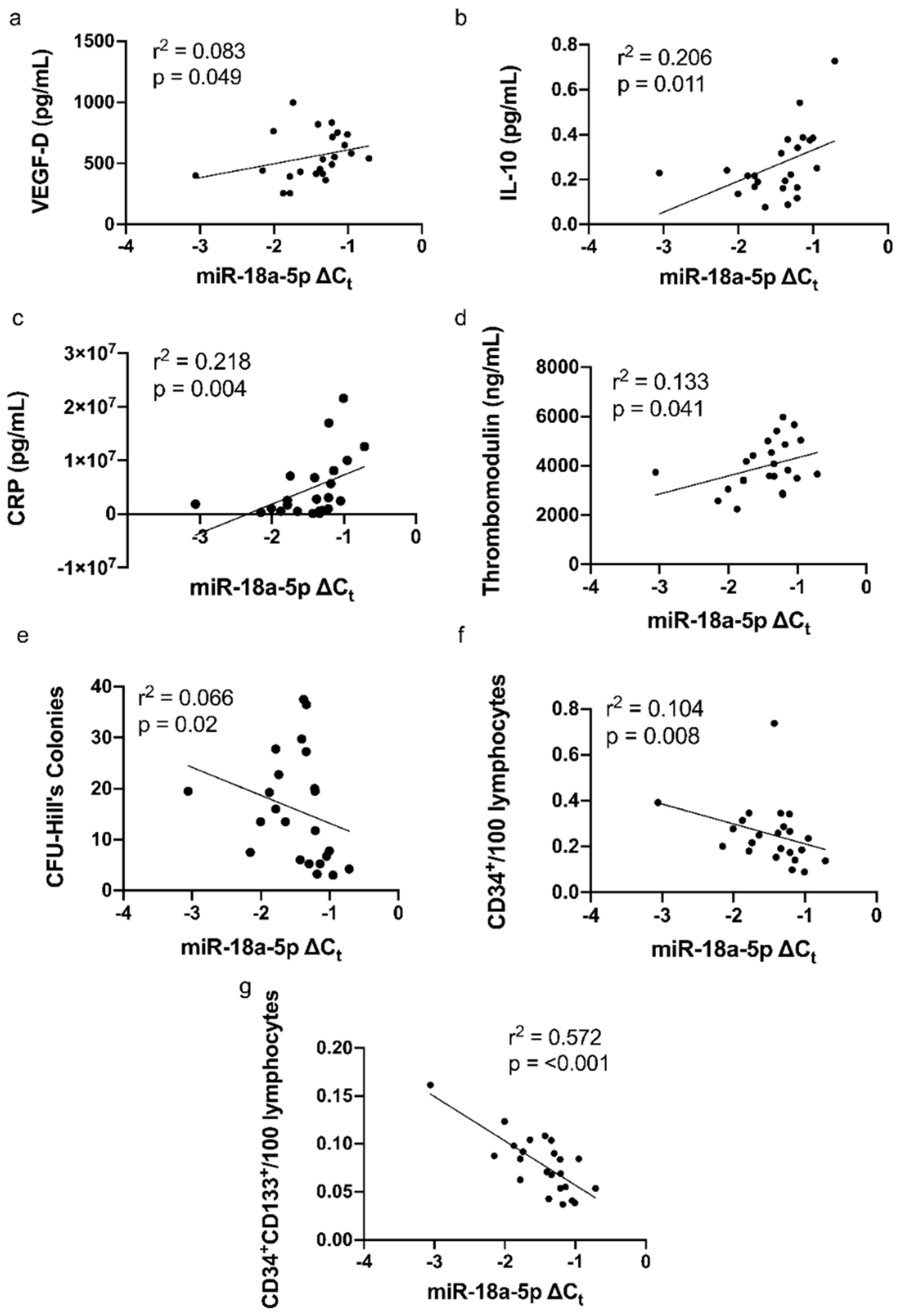
3.4. Correlations of miR-18a-5p with Cytokines, CRP, and Parameters of Vascular Function
3.5. miR-18a-5p Molecular Targets and Functional Pathways
3.6. miR-18a-5p Molecular Targets and Functional Pathways Following Metformin Intervention
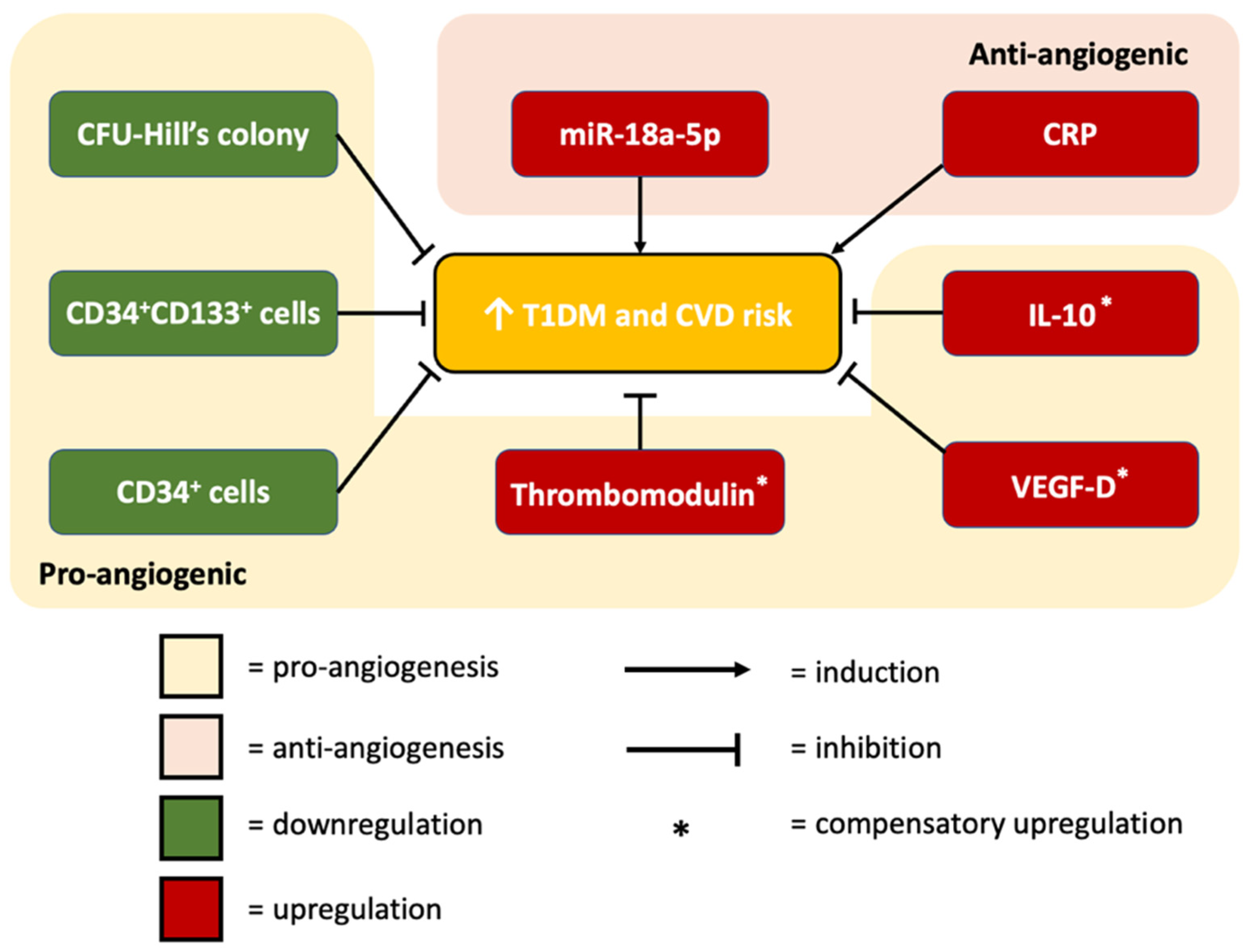
4. Discussion
4.1. miR-18a-5p Expression in T1DM Patients
4.2. The Association between miR-18a-5p and CFU-Hill’s Colonies
4.3. The Association between miR-18a-5p and Vascular Health
4.4. The Association between miR-18a-5p and Inflammatory Markers
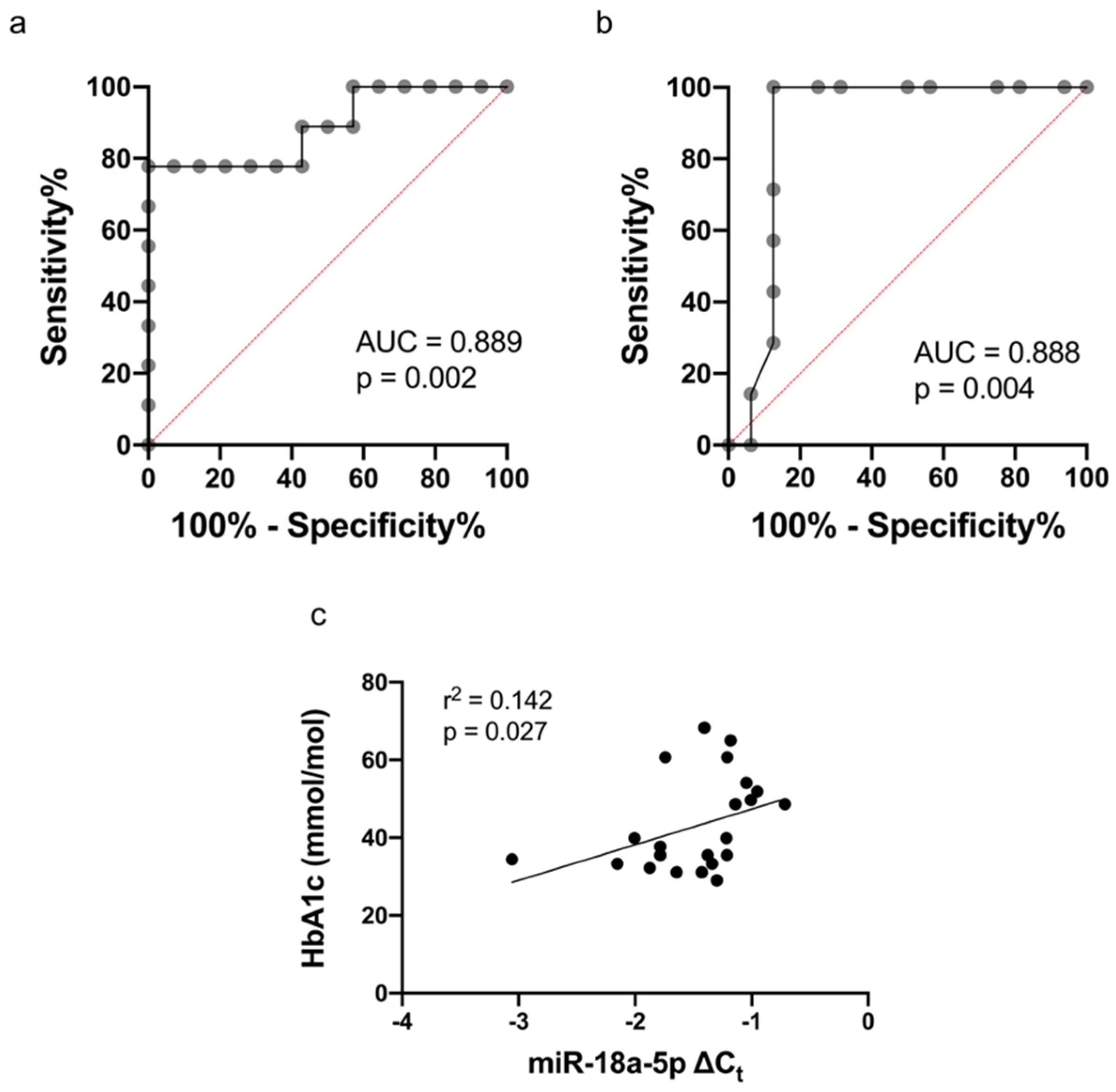
4.5. The Association between miR-18a-5p and HbA1c
4.6. miR-18a-5p Expression Following Metformin Therapy
4.7. Prediction Model: Functional Pathway Analysis of miR-18a-5p in Relation to Cardiovascular Function
4.8. Clinical Applications of Our Research for CVD
4.8.1. miRNA-Based Therapy
4.8.2. IL-10
4.8.3. VEGF-D
4.8.4. Thrombomodulin
4.8.5. IGF1
4.8.6. ESR1
4.8.7. HIF-1α
4.8.8. CCN2
4.8.9. PIAS3
4.8.10. TGFβ1
4.9. Contribution/Causation
4.10. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cardiovascular Diseases (CVDs). 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 20 August 2022).
- World Health Organization. The Challenge of Cardiovascular Disease—Quick Statistics. 2021. Available online: https://www.euro.who.int/en/health-topics/noncommunicable-diseases/cardiovascular-diseases/data-and-statistics (accessed on 10 February 2021).
- Lee, Y.-B.; Han, K.; Kim, B.; Lee, S.-E.; Jun, J.E.; Ahn, J.; Kim, G.; Jin, S.-M.; Kim, J.H. Risk of early mortality and cardiovascular disease in type 1 diabetes: A comparison with type 2 diabetes, a nationwide study. Cardiovasc. Diabetol. 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, S.J.; Levin, D.; Looker, H.C.; Lindsay, R.; Wild, S.H.; Joss, N.; Leese, G.; Leslie, P.; McCrimmon, R.; Metcalfe, W.; et al. Estimated Life Expectancy in a Scottish Cohort with Type 1 Diabetes, 2008–2010. JAMA 2015, 313, 37–44. [Google Scholar] [CrossRef]
- Lind, M.; Svensson, A.-M.; Kosiborod, M.; Gudbjörnsdottir, S.; Pivodic, A.; Wedel, H.; Dahlqvist, S.; Clements, M.; Rosengren, A. Glycemic Control and Excess Mortality in Type 1 Diabetes. N. Engl. J. Med. 2014, 371, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.W.; Rider, R.; Glanville, M.; Narayanan, K.; Razvi, S.; Weaver, J.U. Metformin improves circulating endothelial cells and endothelial progenitor cells in type 1 diabetes: MERIT study. Cardiovasc. Diabetol. 2016, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Asicioglu, E.; Yavuz, D.G.; Koc, M.; Ozben, B.; Yazici, D.; Deyneli, O.; Akalin, S. Circulating endothelial cells are elevated in patients with type 1 diabetes mellitus. Eur. J. Endocrinol. 2010, 162, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Sibal, L.; Aldibbiat, A.; Agarwal, S.C.; Mitchell, G.; Oates, C.; Razvi, S.; Weaver, J.U.; Shaw, J.A.; Home, P.D. Circulating endothelial progenitor cells, endothelial function, carotid intima-media thickness and circulating markers of endothelial dysfunction in people with type 1 diabetes without macrovascular disease or microalbuminuria. Diabetologia 2009, 52, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Snell-Bergeon, J.K.; West, N.A.; Mayer-Davis, E.J.; Liese, A.D.; Marcovina, S.M.; D’Agostino, R.B., Jr.; Hamman, R.F.; Dabelea, D. Inflammatory markers are increased in youth with type 1 diabetes: The SEARCH Case-Control study. J. Clin. Endocrinol. Metab. 2010, 95, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- West, D.J.; Campbell, M.D.; Gonzalez, J.T.; Walker, M.; Stevenson, E.J.; Ahmed, F.W.; Wijaya, S.; Shaw, J.A.; Weaver, J.U. The inflammation, vascular repair and injury responses to exercise in fit males with and without Type 1 diabetes: An observational study. Cardiovasc. Diabetol. 2015, 14, 71. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C. Human endothelial progenitor cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006692. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Zhou, J.; Gong, R.; Huang, X.; Pansuria, M.; Virtue, A.; Li, X.; Wang, H.; Yang, X.F. Endothelial progenitor cells in atherosclerosis. Front. Biosci. 2012, 17, 2327–2349. [Google Scholar] [CrossRef]
- Hur, J.; Yoon, C.-H.; Kim, H.-S.; Choi, J.-H.; Kang, H.-J.; Hwang, K.-K.; Oh, B.-H.; Lee, M.-M.; Park, Y.-B. Characterization of Two Types of Endothelial Progenitor Cells and Their Different Contributions to Neovasculogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007, 109, 1801–1809. [Google Scholar] [CrossRef]
- Salazar-Martinez, E.; Rodriguez-Valentin, R.; Albavera-Hernandez, C.; Carreon-Rodriguez, A.; Lazcano-Ponce, E. Number of colony-forming unit-Hill colonies among children and teenagers with obesity, dyslipidemia and breastfeeding history. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 534–540. [Google Scholar] [CrossRef] [PubMed]
- MacEneaney, O.J.; Kushner, E.J.; Van Guilder, G.P.; Greiner, J.J.; Stauffer, B.; DeSouza, C.A. Endothelial progenitor cell number and colony-forming capacity in overweight and obese adults. Int. J. Obes. 2008, 33, 219–225. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Coviello, F.; Di Martino, A.; Albanese, G.; Marfella, R.; Sardu, C.; et al. Effects of Metformin in Heart Failure: From Pathophysiological Rationale to Clinical Evidence. Biomolecules 2021, 11, 1834. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2020, 9, 3. [Google Scholar] [CrossRef]
- Paiva, M.; Riksen, N.P.; Davidson, S.M.; Hausenloy, D.J.; Monteiro, P.; Gonçalves, L.; Providência, L.; Rongen, G.A.; Smits, P.; Mocanu, M.M.; et al. Metformin Prevents Myocardial Reperfusion Injury by Activating the Adenosine Receptor. J. Cardiovasc. Pharmacol. 2009, 53, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Chang, J.; Zhang, G.; Fang, Y. MicroRNAs in type 1 diabetes: New research progress and potential directions. Biochem. Cell Biol. 2018, 96, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Assmann, T.S.; Recamonde-Mendoza, M.; De Souza, B.M.; Crispim, D. MicroRNA expression profiles and type 1 diabetes mellitus: Systematic review and bioinformatic analysis. Endocr. Connect. 2017, 6, 773–790. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Kim, G.R.; Cho, S.-N.; Kwon, J.-S.; Ahn, Y.; Kook, H.; Jeong, M.H. miR-18a-5p MicroRNA Increases Vascular Smooth Muscle Cell Differentiation by Downregulating Syndecan4. Korean Circ. J. 2014, 44, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X. miR-18a-5p Promotes Proliferation and Migration of Vascular Smooth Muscle Cells by Activating the AKT/Extracellular Regulated Protein Kinases (ERK) Signaling Pathway. Med. Sci. Monit. 2020, 26, e924625. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-T.; Li, X.-X.; Peng, D.-W.; Zhang, W.-M.; Qu, J.; Lu, F.; D’Amato, R.J.; Chi, Z.-L. MicroRNA-18a-5p Administration Suppresses Retinal Neovascularization by Targeting FGF1 and HIF1A. Front. Pharmacol. 2020, 11, 276. [Google Scholar] [CrossRef]
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Böhm, M.; Nickenig, G. Circulating Endothelial Progenitor Cells and Cardiovascular Outcomes. N. Engl. J. Med. 2005, 353, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D.A.; Caplice, N.M.; Yoder, M.C. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood 2005, 106, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Tamara, A.; Coulson, D.J.; Latief, J.S.; Bakhashab, S.; Weaver, J.U. Upregulated anti-angiogenic miR-424-5p in type 1 diabetes (model of subclinical cardiovascular disease) correlates with endothelial progenitor cells, CXCR1/2 and other parameters of vascular health. Stem Cell Res. Ther. 2021, 12, 249. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, M.; Chen, M.; Zhang, K.; Gu, L.; Li, Q.; Yu, Z.; Li, N.; Meng, Q. miR-18a induces myotubes atrophy by down-regulating IgfI. Int. J. Biochem. Cell Biol. 2017, 90, 145–154. [Google Scholar] [CrossRef]
- Cui, M.; Qu, F.; Wang, L.; Cheng, D.; Liu, X. MiR-18a-5p Facilitates Progression of Hepatocellular Carcinoma by Targeting CPEB3. Technol. Cancer Res. Treat. 2021, 20, 15330338211043976. [Google Scholar] [CrossRef] [PubMed]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Takanashi, M.; Borjigin, N.; Ohno, S.-I.; Fujita, K.; Hoshino, S.; Osaka, Y.; Tsuchida, A.; Kuroda, M.J. MicroRNA-18a modulates STAT3 activity through negative regulation of PIAS3 during gastric adenocarcinogenesis. Br. J. Cancer 2013, 108, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Zhou, Q.; Gu, Y.; Duan, T.; Feng, Y. Luteinizing hormone facilitates angiogenesis in ovarian epithelial tumor cells and metformin inhibits the effect through the mTOR signaling pathway. Oncol. Rep. 2012, 27, 1873–1878. [Google Scholar] [PubMed]
- Rattan, R.; Graham, R.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin Suppresses Ovarian Cancer Growth and Metastasis with Enhancement of Cisplatin Cytotoxicity In Vivo. Neoplasia 2011, 13, 483–491. [Google Scholar] [CrossRef]
- Xiao, H.; Ma, X.; Feng, W.; Fu, Y.; Lu, Z.; Xu, M.; Shen, Q.; Zhu, Y.; Zhang, Y. Metformin attenuates cardiac fibrosis by inhibiting the TGFbeta1-Smad3 signalling pathway. Cardiovasc. Res. 2010, 87, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ye, H.; Xiang, F.; Song, L.J.; Zhou, L.L.; Cai, P.C.; Zhang, J.C.; Yu, F.; Shi, H.Z.; Su, Y.; et al. miR-18a-5p Inhibits Sub-pleural Pulmonary Fibrosis by Targeting TGF-beta Receptor II. Mol. Ther. 2017, 25, 728–738. [Google Scholar] [CrossRef]
- de Candia, P.; Spinetti, G.; Specchia, C.; Sangalli, E.; La Sala, L.; Uccellatore, A.; Lupini, S.; Genovese, S.; Matarese, G.; Ceriello, A. A unique plasma microRNA profile defines type 2 diabetes progression. PLoS ONE 2017, 12, e0188980. [Google Scholar] [CrossRef]
- Garcia-Contreras, M.; Shah, S.H.; Tamayo, A.; Robbins, P.D.; Golberg, R.B.; Mendez, A.J.; Ricordi, C. Plasma-derived exosome characterization reveals a distinct microRNA signature in long duration Type 1 diabetes. Sci. Rep. 2017, 7, 5998. [Google Scholar] [CrossRef] [PubMed]
- Assmann, T.S.; Recamonde-Mendoza, M.; Punales, M.; Tschiedel, B.; Canani, L.H.; Crispim, D. MicroRNA expression profile in plasma from type 1 diabetic patients: Case-control study and bioinformatic analysis. Diabetes Res. Clin. Pract. 2018, 141, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Erener, S.; Marwaha, A.; Tan, R.; Panagiotopoulos, C.; Kieffer, T.J. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight 2017, 2, e89656. [Google Scholar] [CrossRef] [Green Version]
- Tavano, F.; Fontana, A.; Mazza, T.; Gioffreda, D.; Biagini, T.; Palumbo, O.; Carella, M.; Andriulli, A. Early-Onset Diabetes as Risk Factor for Pancreatic Cancer: miRNA Expression Profiling in Plasma Uncovers a Role for miR-20b-5p, miR-29a, and miR-18a-5p in Diabetes of Recent Diagnosis. Front. Oncol. 2020, 10, 1567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, R.; Su, H.; Li, K.; Chen, C.; Xie, R. miR-18a increases insulin sensitivity by inhibiting PTEN. Aging 2020, 13, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Briguori, C.; Testa, U.; Riccioni, R.; Colombo, A.; Petrucci, E.; Condorelli, G.; Mariani, G.; D’Andrea, D.; De Micco, F.; Rivera, N.V.; et al. Correlations between progression of coronary artery disease and circulating endothelial progenitor cells. FASEB J. 2010, 24, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- De Ferranti, S.D.; De Boer, I.H.; Fonseca, V.; Fox, C.S.; Golden, S.H.; Lavie, C.J.; Magge, S.N.; Marx, N.; McGuire, D.K.; Orchard, T.J.; et al. Type 1 diabetes mellitus and cardiovascular disease: A scientific statement from the American Heart Association and American Diabetes Association. Diabetes Care 2014, 37, 2843–2863. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.L.; Coulson, D.J.; Yeoh, M.L.Y.; Tamara, A.; Latief, J.S.; Bakhashab, S.; Weaver, J.U. The Role of miR-342 in Vascular Health. Study in Subclinical Cardiovascular Disease in Mononuclear Cells, Plasma, Inflammatory Cytokines and PANX2. Int. J. Mol. Sci. 2020, 21, 7217. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Kuo, C.-H.; Shi, G.-Y.; Wu, H.-L. The role of thrombomodulin lectin-like domain in inflammation. J. Biomed. Sci. 2012, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Thachil, J.; Asakura, H.; Levy, J.H.; Iba, T. Thrombomodulin in disseminated intravascular coagulation and other critical conditions—A multi-faceted anticoagulant protein with therapeutic potential. Crit. Care 2019, 23, 280. [Google Scholar] [CrossRef]
- Kikuchi, T.; Lin, L.; Horigome, H. Soluble thrombomodulin and cardiovascular disease risk factors in Japanese children. Blood Coagul. Fibrinolysis 2021, 32, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Salomaa, V.; Matei, C.; Aleksic, N.; Sansores-Garcia, L.; Folsom, A.R.; Juneja, H.; Chambless, L.E.; Wu, K.K. Soluble thrombomodulin as a predictor of incident coronary heart disease and symptomless carotid artery atherosclerosis in the Atherosclerosis Risk in Communities (ARIC) Study: A case-cohort study. Lancet 1999, 353, 1729–1734. [Google Scholar] [CrossRef]
- Hsu, Y.Y.; Shi, G.Y.; Wang, K.C.; Ma, C.Y.; Cheng, T.L.; Wu, H.L. Thrombomodulin promotes focal adhesion kinase activation and contributes to angiogenesis by binding to fibronectin. Oncotarget 2016, 7, 68122–68139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirici, N.; Soligo, D.; Caneva, L.; Servida, F.; Bossolasco, P.; Deliliers, G.L. Differentiation and expansion of endothelial cells from human bone marrow CD133(+) cells. Br. J. Haematol. 2001, 115, 186–194. [Google Scholar] [CrossRef]
- Raghavachari, N.; Liu, P.; Barb, J.J.; Yang, Y.; Wang, R.; Nguyen, Q.T.; Munson, P.J. Integrated analysis of miRNA and mRNA during differentiation of human CD34+ cells delineates the regulatory roles of microRNA in hematopoiesis. Exp. Hematol. 2014, 42, 14–27.e2. [Google Scholar] [CrossRef] [PubMed]
- Dessapt, C.; Karalliedde, J.; Hernandez-Fuentes, M.; Martin, P.P.; Maltese, G.; Dattani, N.; Atkar, R.; Viberti, G.; Gnudi, L. Circulating Vascular Progenitor Cells in Patients with Type 1 Diabetes and Microalbuminuria. Diabetes Care 2010, 33, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Rigato, M.; Cappellari, R.; Bonora, B.M.; Avogaro, A. Long-term Prediction of Cardiovascular Outcomes by Circulating CD34+ and CD34 + CD133+ Stem Cells in Patients with Type 2 Diabetes. Diabetes Care 2017, 40, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Miyamoto, Y.; Kikuchi-Taura, A.; Soma, T.; Taguchi, A.; Kishimoto, I. Decreased levels of circulating CD 34 + cells are associated with coronary heart disease in Japanese patients with type 2 diabetes. J. Diabetes Investig. 2015, 6, 473–478. [Google Scholar] [CrossRef]
- Stacker, S.; Achen, M. Emerging Roles for VEGF-D in Human Disease. Biomolecules 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, J.; Smith, J.G.; Johnson, L.S.B.; Söderholm, M.; Borné, Y.; Melander, O.; Orho-Melander, M.; Nilsson, J.; Engström, G. Increased vascular endothelial growth factor D is associated with atrial fibrillation and ischaemic stroke. Heart 2019, 105, 553–558. [Google Scholar] [CrossRef]
- Borne, Y.; Gransbo, K.; Nilsson, J.; Melander, O.; Orho-Melander, M.; Smith, J.G.; Engstrom, G. Vascular Endothelial Growth Factor D, Pulmonary Congestion, and Incidence of Heart Failure. J. Am. Coll. Cardiol. 2018, 71, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Lange, J.; Kranz, A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation 2000, 102, 185–190. [Google Scholar] [CrossRef]
- Luan, Y.-Y.; Yao, Y.-M. The Clinical Significance and Potential Role of C-Reactive Protein in Chronic Inflammatory and Neurodegenerative Diseases. Front. Immunol. 2018, 9, 1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paffen, E.; DeMaat, M.P. C-reactive protein in atherosclerosis: A causal factor? Cardiovasc. Res. 2006, 71, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Pasceri, V.; Willerson, J.T.; Yeh, E.T.H. Direct Proinflammatory Effect of C-Reactive Protein on Human Endothelial Cells. Circulation 2000, 102, 2165–2168. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Xu, D.Y.; Jialal, I. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: Implications for the metabolic syndrome and atherothrombosis. Circulation 2003, 107, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.; Seybold, K.; Petzi, S.; Wasmeier, G.; Raaz, D.; Yilmaz, A.; Anger, T.; Daniel, W.G.; Garlichs, C.D. Interleukin-10 improves left ventricular function in rats with heart failure subsequent to myocardial infarction. Eur. J. Heart Fail. 2008, 10, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.K.; Surendran, A.; Malik, A.; Jassal, D.S.; Ravandi, A.; Singal, P.K. IL-10 attenuates OxPCs-mediated lipid metabolic responses in ischemia reperfusion injury. Sci. Rep. 2020, 10, 12120. [Google Scholar] [CrossRef]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Gay, S.; Speich, R.; Huber, L.C. MicroRNA-18a enhances the interleukin-6-mediated production of the acute-phase proteins fibrinogen and haptoglobin in human hepatocytes. J. Biol. Chem. 2011, 286, 40142–40150. [Google Scholar] [CrossRef]
- Steensberg, A.; Fischer, C.P.; Keller, C.; Moller, K.; Pedersen, B.K. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E433–E437. [Google Scholar] [CrossRef]
- Szodoray, P.; Timár, O.; Vereš, K.; Der, H.; Szomjak, E.; Lakos, G.; Aleksza, M.; Nakken, B.; Szegedi, G.; Soltész, P. Th1/Th2 Imbalance, Measured by Circulating and Intracytoplasmic Inflammatory Cytokines—Immunological Alterations in Acute Coronary Syndrome and Stable Coronary Artery Disease. Scand. J. Immunol. 2006, 64, 336–344. [Google Scholar] [CrossRef]
- Huang, Y.; Li, T.; Sane, D.C.; Li, L. IRAK1 Serves as a Novel Regulator Essential for Lipopolysaccharide-induced Interleukin-10 Gene Expression. J. Biol. Chem. 2004, 279, 51697–51703. [Google Scholar] [CrossRef]
- Wang, S.-S.; Li, Y.-Q.; Liang, Y.-Z.; Dong, J.; He, Y.; Zhang, L.; Yan, Y.-X. Expression of miR-18a and miR-34c in circulating monocytes associated with vulnerability to type 2 diabetes mellitus and insulin resistance. J. Cell. Mol. Med. 2017, 21, 3372–3380. [Google Scholar] [CrossRef] [Green Version]
- Rodbard, H.W.; Jellinger, P.S.; Davidson, J.A.; Einhorn, D.; Garber, A.J.; Grunberger, G.; Handelsman, Y.; Horton, E.S.; Lebovitz, H.; Levy, P.; et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: An algorithm for glycemic control. Endocr. Pract. 2009, 15, 540–559. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Health and Care Excellence. Type 1 Diabetes in Adults: Diagnosis and Management. 2021. Available online: http://www.nice.org.uk/ (accessed on 25 November 2021).
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.C.G.J.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): A double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 597–609. [Google Scholar] [CrossRef]
- Giuliani, A.; Londin, E.; Ferracin, M.; Mensà, E.; Prattichizzo, F.; Ramini, D.; Marcheselli, F.; Recchioni, R.; Rippo, M.R.; Bonafè, M.; et al. Long-term exposure of human endothelial cells to metformin modulates miRNAs and isomiRs. Sci. Rep. 2020, 10, 21782. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, J.; Xu, Z.; Feng, Y.; Zhang, M.; Liu, J.; Chen, R.; Shen, J.; Wu, J.; Lu, Z.; et al. Metformin is a novel suppressor for transforming growth factor (TGF)-beta1. Sci. Rep. 2016, 6, 28597. [Google Scholar] [CrossRef]
- Yi, Q.Y.; Deng, G.; Chen, N.; Bai, Z.S.; Yuan, J.S.; Wu, G.H.; Wang, Y.W.; Wu, S.J. Metformin inhibits development of diabetic retinopathy through inducing alternative splicing of VEGF-A. Am. J. Transl. Res. 2016, 8, 3947–3954. [Google Scholar] [PubMed]
- Lin, B.; Feng, D.; Xu, J. Cardioprotective effects of microRNA-18a on acute myocardial infarction by promoting cardiomyocyte autophagy and suppressing cellular senescence via brain derived neurotrophic factor. Cell Biosci. 2019, 9, 38. [Google Scholar] [CrossRef]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Mäkinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhao, W.; Chen, Y.; Liu, L.; Ahokas, R.A.; Sun, Y. Differential expression of vascular endothelial growth factor isoforms and receptor subtypes in the infarcted heart. Int. J. Cardiol. 2013, 167, 2638–2645. [Google Scholar] [CrossRef]
- Hartikainen, J.; Hassinen, I.; Hedman, A.; Kivela, A.; Saraste, A.; Knuuti, J.; Husso, M.; Mussalo, H.; Hedman, M.; Rissanen, T.T.; et al. Adenoviral intramyocardial VEGF-DDeltaNDeltaC gene transfer increases myocardial perfusion reserve in refractory angina patients: A phase I/IIa study with 1-year follow-up. Eur. Heart J. 2017, 38, 2547–2555. [Google Scholar] [CrossRef]
- Wei, H.J.; Li, Y.H.; Shi, G.Y.; Liu, S.L.; Chang, P.C.; Kuo, C.H.; Wu, H.L. Thrombomodulin domains attenuate atherosclerosis by inhibiting thrombin-induced endothelial cell activation. Cardiovasc. Res. 2011, 92, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, H.; Maruyama, I.; Shimazaki, S.; Yamamoto, Y.; Aikawa, N.; Ohno, R.; Hirayama, A.; Matsuda, T.; Asakura, H.; Nakashima, M.; et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: Results of a phase III, randomized, double-blind clinical trial. J. Thromb. Haemost. 2007, 5, 31–41. [Google Scholar] [CrossRef]
- Higashi, Y.; Sukhanov, S.; Shai, S.-Y.; Danchuk, S.; Tang, R.; Snarski, P.; Li, Z.; Lobelle-Rich, P.; Wang, M.; Wang, D.; et al. Insulin-Like Growth Factor-1 Receptor Deficiency in Macrophages Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Apolipoprotein E–Deficient Mice. Circulation 2016, 133, 2263–2278. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, Q.; Shao, X.; Zhang, T.; Xue, C.; Shi, S.; Zhao, D.; Lin, Y. IGF-1 promotes angiogenesis in endothelial cells/adipose-derived stem cells co-culture system with activation of PI3K/Akt signal pathway. Cell Prolif. 2017, 50, e12390. [Google Scholar] [CrossRef] [PubMed]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Eurell, T.E.; Cooke, P.S.; Lubahn, D.B.; Gross, D.R. Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1640–H1647. [Google Scholar] [CrossRef] [PubMed]
- Fatima, L.A.; Campello, R.S.; Santos, R.D.S.; Freitas, H.S.; Frank, A.P.; Machado, U.F.; Clegg, D.J. Estrogen receptor 1 (ESR1) regulates VEGFA in adipose tissue. Sci. Rep. 2017, 7, 16716. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shen, H.; Shao, L.; Teng, X.; Chen, Y.; Liu, X.; Yang, Z.; Shen, Z. HIF-1α overexpression in mesenchymal stem cell-derived exosomes mediates cardioprotection in myocardial infarction by enhanced angiogenesis. Stem Cell Res. Ther. 2020, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, C.; Bin, J.; Wang, Y.; Chen, J.; Xiu, J.; Pei, J.; Lai, Y.; Chen, D.; Fan, C.; et al. Mutant hypoxia inducible factor-1alpha improves angiogenesis and tissue perfusion in ischemic rabbit skeletal muscle. Microvasc. Res. 2011, 81, 26–33. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Gravning, J.; Martinov, V.N.; von Lueder, T.G.; Edvardsen, T.; Czibik, G.; Moe, I.T.; Vinge, L.E.; Oie, E.; Valen, G.; et al. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1291–H1302. [Google Scholar] [CrossRef] [PubMed]
- Panek, A.N.; Posch, M.G.; Alenina, N.; Ghadge, S.K.; Erdmann, B.; Popova, E.; Perrot, A.; Geier, C.; Dietz, R.; Morano, I.; et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS ONE 2009, 4, e6743. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Xu, L.; Lin, Y.; Yang, X.; Bai, L.; Chen, Y.; Zhao, S.; Fan, J.; Cheng, X.; et al. Protein Inhibitor of Activated STAT3 Suppresses Oxidized LDL-induced Cell Responses during Atherosclerosis in Apolipoprotein E-deficient Mice. Sci. Rep. 2016, 6, 36790. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Jia, J.; Yu, Z.; Duanmu, Z.; He, H.; Chen, S.; Qu, C. Inhibition of JAK2/STAT3/SOCS3 signaling attenuates atherosclerosis in rabbit. BMC Cardiovasc. Disord. 2020, 20, 133. [Google Scholar] [CrossRef] [PubMed]
- Villar, A.V.; Cobo, M.; Llano, M.; Montalvo, C.; Gonzalez-Vilchez, F.; Martin-Duran, R.; Hurle, M.A.; Nistal, J.F. Plasma levels of transforming growth factor-beta1 reflect left ventricular remodeling in aortic stenosis. PLoS ONE 2009, 4, e8476. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.E.J.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Investig. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- Ferreira, R.R.; Abreu, R.D.S.; Vilar-Pereira, G.; Degrave, W.; Meuser-Batista, M.; Ferreira, N.V.C.; da Cruz Moreira, O.; da Silva Gomes, N.L.; Mello de Souza, E.; Ramos, I.P.; et al. TGF-beta inhibitor therapy decreases fibrosis and stimulates cardiac improvement in a pre-clinical study of chronic Chagas’ heart disease. PLoS Negl. Trop. Dis. 2019, 13, e0007602. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Kostoulas, N.; Vergoulis, T.; Georgakilas, G.; Reczko, M.; Maragkakis, M.; Paraskevopoulou, M.D.; Prionidis, K.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA miRPath v.2.0: Investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012, 40, W498–W504. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Fluorochrome | Volume |
|---|---|---|
| CD34 | PerCP-Cy5.5 | 20 μL |
| CD133 | APC | 5 μL |
| VEGFR2 (KDR) | PE | 5 μL |
| CD144 | FITC | 10 μL |
| CD45 | V500 | 5 μL |
| Target Gene | Representative Transcript | Gene Name | Transcript Position | Predicted Consequential Pairing of Target Region Transcript (Top) and miRNA (Bottom) | Site Type |
|---|---|---|---|---|---|
| IGF1 | ENST00000337514.6 | Insulin-like growth factor 1 | 181–187 3′ UTR | (transcript) 5′ CUUUAGGAGUGAUUUGCACCUUG (miRNA) 3′ GAUAGACGUGAUCUACGUGGAAU | 7mer-8 |
| ESR1 | ENST00000440973.1 | Estrogen Receptor 1 | 1938–1945 3′ UTR | (transcript) 5′ UAGUUUGUUUAAGAAGCACCUUA (miRNA) 3′ GAUAGACGUGAUCUACGUGGAAU | 8mer |
| HIF-1α | ENST00000323441.6 | Hypoxia Inducible Factor 1 Subunit Alpha | 409–415 3′ UTR | (transcript) 5′ AUCAUUUUAAAAAAUGCACCUUU (miRNA) 3′ GAUAGACGUGAUCUACGUGGAAU | 7mer-m8 |
| CCN2 (CTGF) | ENST00000367976.3 | Cellular Communication Network Factor 2 (Connective Tissue Growth Factor; CTGF) | 1046–1052 3′ UTR | (transcript) 5′ AAAAGUUACAUGUUUGCACCUUU (miRNA) 3′ GAUAGACGUGAUCUACGUGGAAU | 7mer-m8 |
| PIAS3 | ENST00000393045.2 | Protein Inhibitor of Activated STAT 3 | 774–780 3′ UTR | (transcript) 5′ GGCCUGGCUCAUUCUGCACCUUG (miRNA) 3′ GAUAGACGUGAUCUACGUGGAAU | 7mer-m8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phowira, J.; Ahmed, F.W.; Bakhashab, S.; Weaver, J.U. Upregulated miR-18a-5p in Colony Forming Unit-Hill’s in Subclinical Cardiovascular Disease and Metformin Therapy; MERIT Study. Biomedicines 2022, 10, 2136. https://doi.org/10.3390/biomedicines10092136
Phowira J, Ahmed FW, Bakhashab S, Weaver JU. Upregulated miR-18a-5p in Colony Forming Unit-Hill’s in Subclinical Cardiovascular Disease and Metformin Therapy; MERIT Study. Biomedicines. 2022; 10(9):2136. https://doi.org/10.3390/biomedicines10092136
Chicago/Turabian StylePhowira, Jason, Fahad W. Ahmed, Sherin Bakhashab, and Jolanta U. Weaver. 2022. "Upregulated miR-18a-5p in Colony Forming Unit-Hill’s in Subclinical Cardiovascular Disease and Metformin Therapy; MERIT Study" Biomedicines 10, no. 9: 2136. https://doi.org/10.3390/biomedicines10092136
APA StylePhowira, J., Ahmed, F. W., Bakhashab, S., & Weaver, J. U. (2022). Upregulated miR-18a-5p in Colony Forming Unit-Hill’s in Subclinical Cardiovascular Disease and Metformin Therapy; MERIT Study. Biomedicines, 10(9), 2136. https://doi.org/10.3390/biomedicines10092136