
The Activation of JAK/STAT3 Signaling and the Complement System Modulate Inflammation in the Primary Human Dermal Fibroblasts of PXE Patients

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Patient Characteristics
2.3. Cell Culture and Treatment
2.4. Delipidation of FCS
2.5. Isolation of Nucleic Acid
2.6. Gene Expression Analysis
2.7. Immunofluorescence and Western Blot Analysis of Active pSTAT3
2.8. Immunoassays for the Evaluation of IL-6, MCP-1, C3, C3a and C1r Concentration in Cell Culture Supernatant and C3 Concentration in Sera from PXE Patients
2.9. Statistical Analysis
3. Results
3.1. Blocking JAK/STAT3 Signaling Reduced the Protein Level of Active pSTAT3 in PXE Fibroblasts
3.2. Blocking JAK/STAT3 Pathway Partly Reduces Senescence-Associated Secretory Phenotype in PXE Fibroblasts
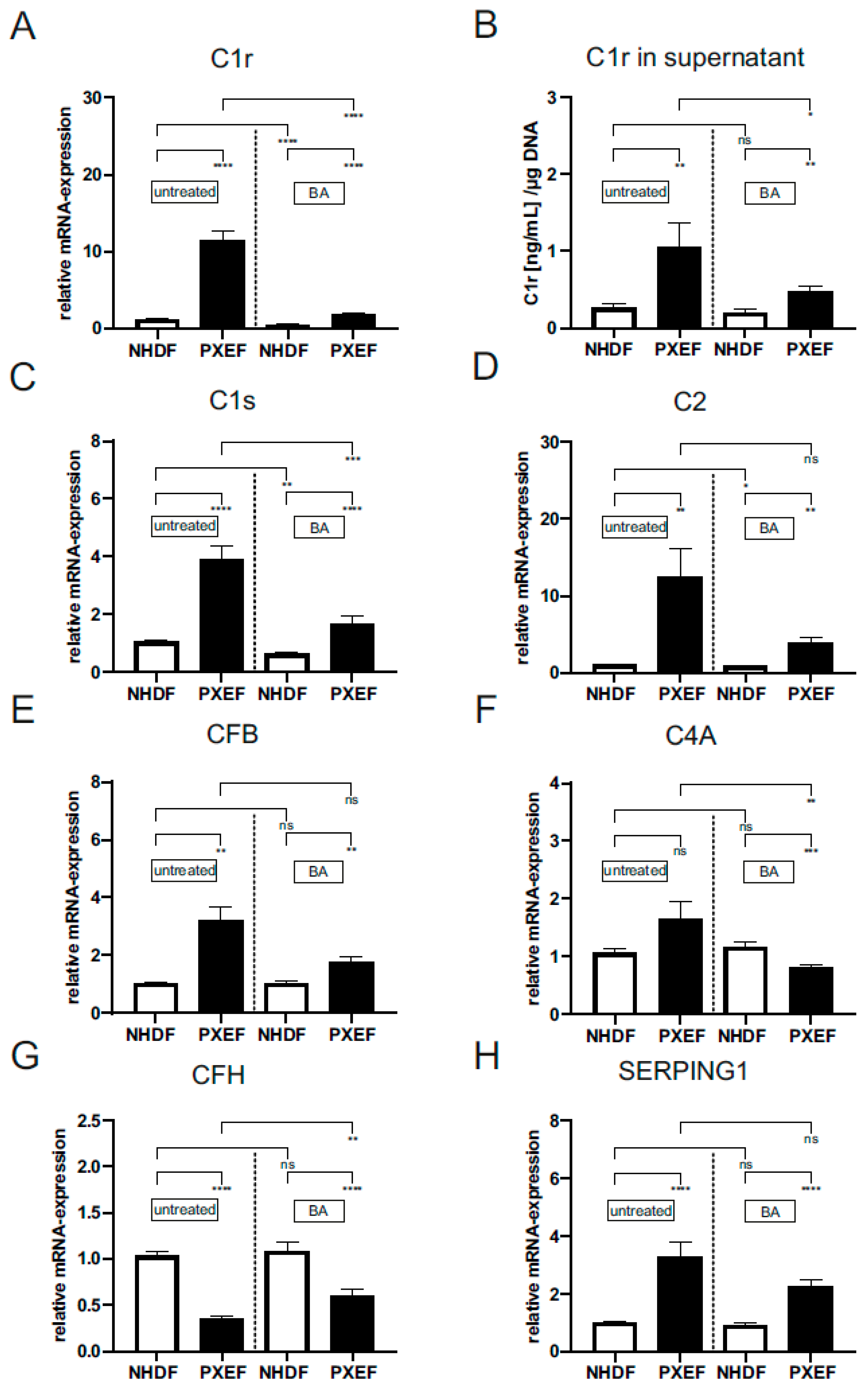
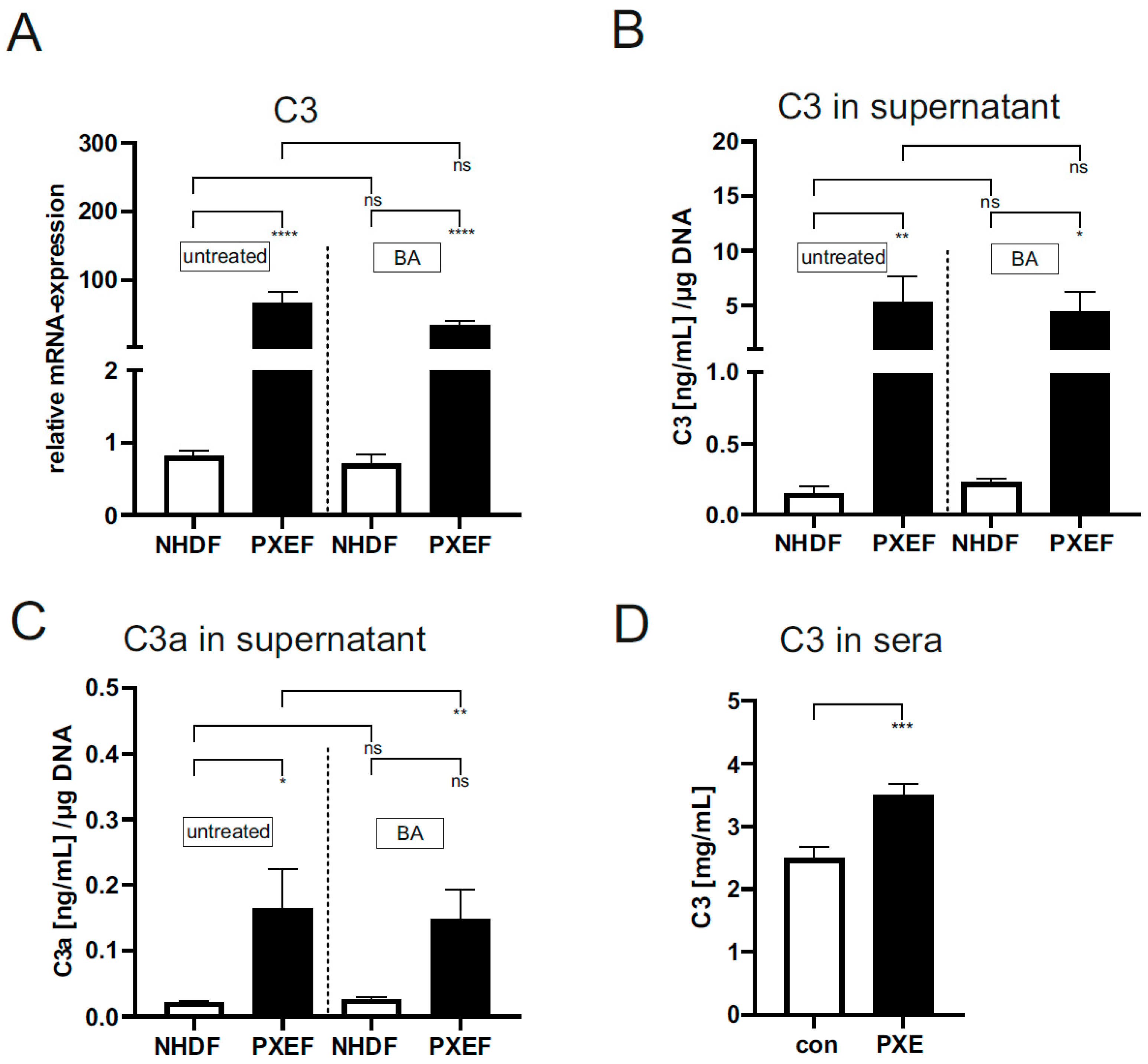
3.3. Increased Gene Expression and Protein Level of Complement Factors in PXE Fibroblasts Are Partially Affected by Blocking JAK/STAT3 Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Contri, M.B.; Boraldi, F.; Taparelli, F.; De Paepe, A.; Ronchetti, I.P. Matrix Proteins with High Affinity for Calcium Ions Are Associated with Mineralization within the Elastic Fibers of Pseudoxanthoma Elasticum Dermis. Am. J. Pathol. 1996, 148, 569–577. [Google Scholar] [PubMed]
- Neldner, K.H. Pseudoxanthoma Elasticum. Int. J. Dermatol. 1988, 27, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Plomp, A.S.; van Soest, S.; Wijnholds, J.; de Jong, P.T.V.M.; Bergen, A.A.B. Pseudoxanthoma Elasticum: A Clinical, Histopathological, and Molecular Update. Surv. Ophthalmol. 2003, 48, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Jensen, O.A. Bruch’s Membrane in Pseudoxanthoma Elasticum. Histochemical, Ultrastructural, and x-Ray Microanalytical Study of the Membrane and Angioid Streak Areas. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 1977, 203, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Connor, P.J.; Juergens, J.L.; Perry, H.O.; Hollenhorst, R.W.; Edwards, J.E. Pseudoxanthoma Elasticum and Angioid Streaks. A Review of 106 Cases. Am. J. Med. 1961, 30, 537–543. [Google Scholar] [CrossRef]
- Laube, S.; Moss, C. Pseudoxanthoma Elasticum. Arch. Dis. Child. 2005, 90, 754–756. [Google Scholar] [CrossRef]
- Campens, L.; Vanakker, O.M.; Trachet, B.; Segers, P.; Leroy, B.P.; De Zaeytijd, J.; Voet, D.; De Paepe, A.; De Backer, T.; De Backer, J. Characterization of Cardiovascular Involvement in Pseudoxanthoma Elasticum Families. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2646–2652. [Google Scholar] [CrossRef]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.; Breuning, M.; Dauwerse, H.; Swart, J.; Kool, M.; van Soest, S.; Baas, F.; et al. Mutations in ABCC6 Cause Pseudoxanthoma Elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef]
- Beck, K.; Hayashi, K.; Dang, K.; Hayashi, M.; Boyd, C.D. Analysis of ABCC6 (MRP6) in Normal Human Tissues. Histochem. Cell Biol. 2005, 123, 517–528. [Google Scholar] [CrossRef]
- Pseudoxanthoma Elasticum and ABCC6—ClinVar—NCBI. Available online: https://databases.lovd.nl/shared/genes/ABCC6 (accessed on 19 January 2023).
- Tiemann, J.; Wagner, T.; Vanakker, O.M.; van Gils, M.; Cabrera, J.-L.B.; Ibold, B.; Faust, I.; Knabbe, C.; Hendig, D. Cellular and Molecular Biomarkers Indicate Premature Aging in Pseudoxanthoma Elasticum Patients. Aging Dis. 2020, 11, 536–546. [Google Scholar] [CrossRef]
- Boraldi, F.; Annovi, G.; Bartolomeo, A.; Quaglino, D. Fibroblasts from Patients Affected by Pseudoxanthoma Elasticum Exhibit an Altered PPi Metabolism and Are More Responsive to Pro-Calcifying Stimuli. J. Dermatol. Sci. 2014, 74, 72–80. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Rivera-Torres, J.; Osorio, F.G.; Acín-Pérez, R.; Enriquez, J.A.; López-Otín, C.; Andrés, V. Defective Extracellular Pyrophosphate Metabolism Promotes Vascular Calcification in a Mouse Model of Hutchinson-Gilford Progeria Syndrome That Is Ameliorated on Pyrophosphate Treatment. Circulation 2013, 127, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, Q.; Chou, D.W.; Uitto, J. Atorvastatin Counteracts Aberrant Soft Tissue Mineralization in a Mouse Model of Pseudoxanthoma Elasticum (Abcc6−/−). J. Mol. Med. 2013, 91, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Tiemann, J.; Wagner, T.; Lindenkamp, C.; Plümers, R.; Faust, I.; Knabbe, C.; Hendig, D. Linking ABCC6 Deficiency in Primary Human Dermal Fibroblasts of PXE Patients to P21-Mediated Premature Cellular Senescence and the Development of a Proinflammatory Secretory Phenotype. Int. J. Mol. Sci. 2020, 21, 9665. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.K.; Lee, S.W.; Aaronson, S.A. Role of Progerin-Induced Telomere Dysfunction in HGPS Premature Cellular Senescence. J. Cell Sci. 2010, 123, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 Neutralization Ameliorates Symptoms in Prematurely Aged Mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Kishimoto, T. Interleukin-6: From Basic Science to Medicine--40 Years in Immunology. Annu. Rev. Immunol. 2005, 23, 1–21. [Google Scholar] [CrossRef]
- Gabay, C. Interleukin-6 and Chronic Inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S3. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of Interleukin (IL)-6-Type Cytokine Signalling and Its Regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 Signalling Axis in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Chaves de Souza, J.A.; Nogueira, A.V.B.; Chaves de Souza, P.P.; Kim, Y.J.; Silva Lobo, C.; Pimentel Lopes de Oliveira, G.J.; Cirelli, J.A.; Garlet, G.P.; Rossa, C. SOCS3 Expression Correlates with Severity of Inflammation, Expression of Proinflammatory Cytokines, and Activation of STAT3 and P38 MAPK in LPS-Induced Inflammation in Vivo. Mediat. Inflamm. 2013, 2013, 650812. [Google Scholar] [CrossRef]
- Tulunay, A.; Dozmorov, M.G.; Ture-Ozdemir, F.; Yilmaz, V.; Eksioglu-Demiralp, E.; Alibaz-Oner, F.; Ozen, G.; Wren, J.D.; Saruhan-Direskeneli, G.; Sawalha, A.H.; et al. Activation of the JAK/STAT Pathway in Behcet’s Disease. Genes Immun. 2015, 16, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Doles, J.; Storer, M.; Cozzuto, L.; Roma, G.; Keyes, W.M. Age-Associated Inflammation Inhibits Epidermal Stem Cell Function. Genes Dev. 2012, 26, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Arnold, R.; Henriques, G.; Djabali, K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells 2019, 8, 1276. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK Inhibition Alleviates the Cellular Senescence-Associated Secretory Phenotype and Frailty in Old Age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef]
- Yuan, K.; Ye, J.; Liu, Z.; Ren, Y.; He, W.; Xu, J.; He, Y.; Yuan, Y. Complement C3 Overexpression Activates JAK2/STAT3 Pathway and Correlates with Gastric Cancer Progression. J. Exp. Clin. Cancer Res. 2020, 39, 9. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. Complement in Immune and Inflammatory Disorders: Pathophysiological Mechanisms. J. Immunol. 2013, 190, 3831–3838. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.-C. Complement and Its Role in Innate and Adaptive Immune Responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Mocco, J.; Mack, W.J.; Ducruet, A.F.; Sosunov, S.A.; Sughrue, M.E.; Hassid, B.G.; Nair, M.N.; Laufer, I.; Komotar, R.J.; Claire, M.; et al. Complement Component C3 Mediates Inflammatory Injury Following Focal Cerebral Ischemia. Circ. Res. 2006, 99, 209–217. [Google Scholar] [CrossRef]
- Lubbers, R.; Sutherland, J.S.; Goletti, D.; de Paus, R.A.; Dijkstra, D.J.; van Moorsel, C.H.M.; Veltkamp, M.; Vestjens, S.M.T.; Bos, W.J.W.; Petrone, L.; et al. Expression and Production of the SERPING1-Encoded Endogenous Complement Regulator C1-Inhibitor in Multiple Cohorts of Tuberculosis Patients. Mol. Immunol. 2020, 120, 187–195. [Google Scholar] [CrossRef]
- Rodríguez de Córdoba, S.; Esparza-Gordillo, J.; Goicoechea de Jorge, E.; Lopez-Trascasa, M.; Sánchez-Corral, P. The Human Complement Factor H: Functional Roles, Genetic Variations and Disease Associations. Mol. Immunol. 2004, 41, 355–367. [Google Scholar] [CrossRef]
- Zheng, R.; Zhang, Y.; Zhang, K.; Yuan, Y.; Jia, S.; Liu, J. The Complement System, Aging, and Aging-Related Diseases. Int. J. Mol. Sci. 2022, 23, 8689. [Google Scholar] [CrossRef] [PubMed]
- Plomp, A.S.; Toonstra, J.; Bergen, A.A.B.; van Dijk, M.R.; de Jong, P.T.V.M. Proposal for Updating the Pseudoxanthoma Elasticum Classification System and a Review of the Clinical Findings. Am. J. Med. Genet. A 2010, 152A, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Hendig, D.; Langmann, T.; Kocken, S.; Zarbock, R.; Szliska, C.; Schmitz, G.; Kleesiek, K.; Götting, C. Gene Expression Profiling of ABC Transporters in Dermal Fibroblasts of Pseudoxanthoma Elasticum Patients Identifies New Candidates Involved in PXE Pathogenesis. Lab. Investig. 2008, 88, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Legrand, A.; Cornez, L.; Samkari, W.; Mazzella, J.-M.; Venisse, A.; Boccio, V.; Auribault, K.; Keren, B.; Benistan, K.; Germain, D.P.; et al. Mutation Spectrum in the ABCC6 Gene and Genotype-Phenotype Correlations in a French Cohort with Pseudoxanthoma Elasticum. Genet. Med. 2017, 19, 909–917. [Google Scholar] [CrossRef]
- Gibson, K.M.; Hoffmann, G.; Schwall, A.; Broock, R.L.; Aramaki, S.; Sweetman, L.; Nyhan, W.L.; Brandt, I.K.; Wappner, R.S.; Lehnert, W. 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Activity in Cultured Fibroblasts from Patients with Mevalonate Kinase Deficiency: Differential Response to Lipid Supplied by Fetal Bovine Serum in Tissue Culture Medium. J. Lipid Res. 1990, 31, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Kuzaj, P.; Kuhn, J.; Dabisch-Ruthe, M.; Faust, I.; Götting, C.; Knabbe, C.; Hendig, D. ABCC6- a New Player in Cellular Cholesterol and Lipoprotein Metabolism? Lipids Health Dis. 2014, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C. Clinical Efficacy of Launched JAK Inhibitors in Rheumatoid Arthritis. Rheumatology 2019, 58, i17–i26. [Google Scholar] [CrossRef]
- Williams, J.J.L.; Munro, K.M.A.; Palmer, T.M. Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression. Cells 2014, 3, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 Feed-Forward Loop Drives Tumorigenesis and Metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Recchia Luciani, G.; Soli, M.; Dall’Asta, V.; Rotoli, B.M. The JAK1/2 Inhibitor Baricitinib Mitigates the Spike-Induced Inflammatory Response of Immune and Endothelial Cells In Vitro. Biomedicines 2022, 10, 2324. [Google Scholar] [CrossRef]
- Dhawan, P.; Richmond, A. Role of CXCL1 in Tumorigenesis of Melanoma. J. Leukoc. Biol. 2002, 72, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Vries, M.H.M.; Wagenaar, A.; Verbruggen, S.E.L.; Molin, D.G.M.; Dijkgraaf, I.; Hackeng, T.H.; Post, M.J. Erratum to: CXCL1 Promotes Arteriogenesis through Enhanced Monocyte Recruitment into the Peri-Collateral Space. Angiogenesis 2015, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating Monocytes Promote Brain Inflammation and Exacerbate Neuronal Damage after Status Epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef]
- Zegeye, M.M.; Lindkvist, M.; Fälker, K.; Kumawat, A.K.; Paramel, G.; Grenegård, M.; Sirsjö, A.; Ljungberg, L.U. Activation of the JAK/STAT3 and PI3K/AKT Pathways Are Crucial for IL-6 Trans-Signaling-Mediated pro-Inflammatory Response in Human Vascular Endothelial Cells. Cell Commun. Signal. 2018, 16, 55. [Google Scholar] [CrossRef]
- Choi, J.M.; Rotimi, O.O.; O’Carroll, S.J.; Nicholson, L.F.B. IL-6 Stimulates a Concentration-Dependent Increase in MCP-1 in Immortalised Human Brain Endothelial Cells. F1000Research 2016, 5, 270. [Google Scholar] [CrossRef]
- Gheduzzi, D.; Sammarco, R.; Quaglino, D.; Bercovitch, L.; Terry, S.; Taylor, W.; Ronchetti, I.P. Extracutaneous Ultrastructural Alterations in Pseudoxanthoma Elasticum. Ultrastruct. Pathol. 2003, 27, 375–384. [Google Scholar] [CrossRef]
- Hosen, M.J.; Lamoen, A.; De Paepe, A.; Vanakker, O.M. Histopathology of Pseudoxanthoma Elasticum and Related Disorders: Histological Hallmarks and Diagnostic Clues. Scientifica 2012, 2012, 598262. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Hussell, T.; Gilliland, S.M.; Holden, D.W.; Paton, J.C.; Ehrenstein, M.R.; Walport, M.J.; Botto, M. The Classical Pathway Is the Dominant Complement Pathway Required for Innate Immunity to Streptococcus Pneumoniae Infection in Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 16969–16974. [Google Scholar] [CrossRef]
- Xavier, S.; Sahu, R.K.; Bontha, S.V.; Mass, V.; Taylor, R.P.; Megyesi, J.; Thielens, N.M.; Portilla, D. Complement C1r Serine Protease Contributes to Kidney Fibrosis. Am. J. Physiol. Renal Physiol. 2019, 317, F1293–F1304. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Viiklepp, K.; Nissinen, L.; Farshchian, M.; Kallajoki, M.; Kivisaari, A.; Meri, S.; Peltonen, J.; Peltonen, S.; Kähäri, V.-M. Tumour-Cell-Derived Complement Components C1r and C1s Promote Growth of Cutaneous Squamous Cell Carcinoma. Br. J. Dermatol. 2020, 182, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Götting, C.; Hendig, D.; Zarbock, R.; Szliska, C.; Kleesiek, K. Complement Factor H Variant p.Y402H in Pseudoxanthoma Elasticum Patients. Genet. Test. 2008, 12, 431–436. [Google Scholar] [CrossRef]
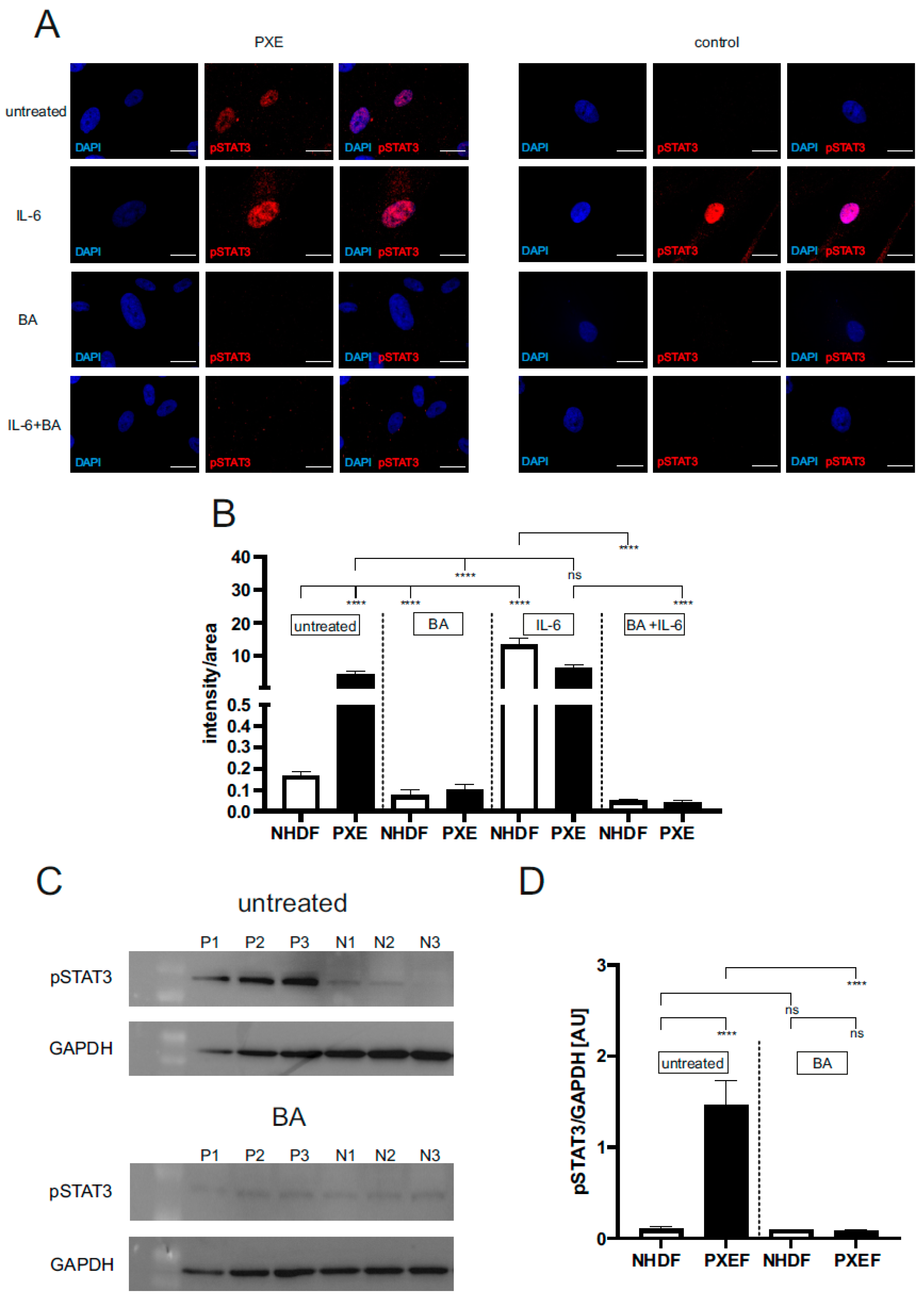





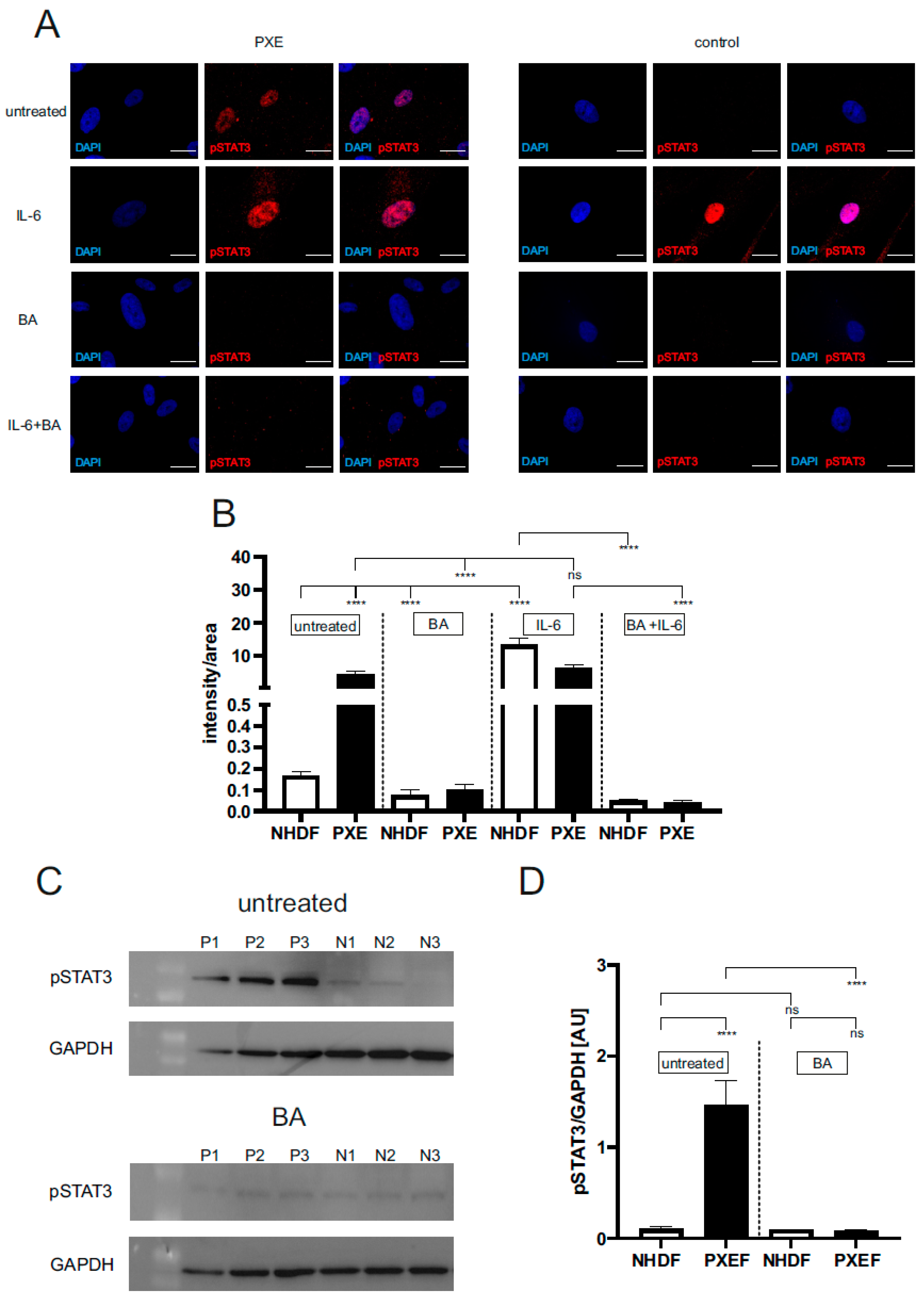{kind=link}
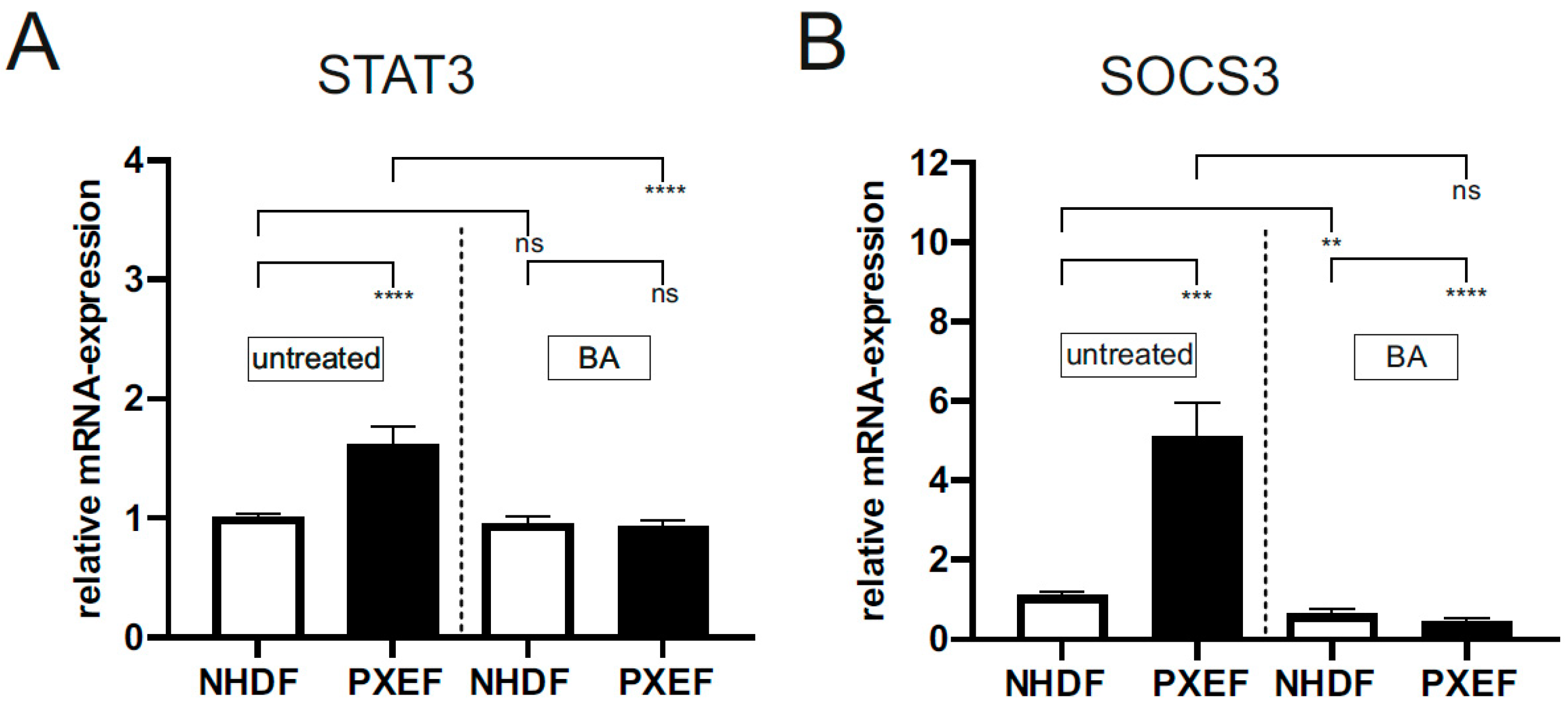{kind=link}
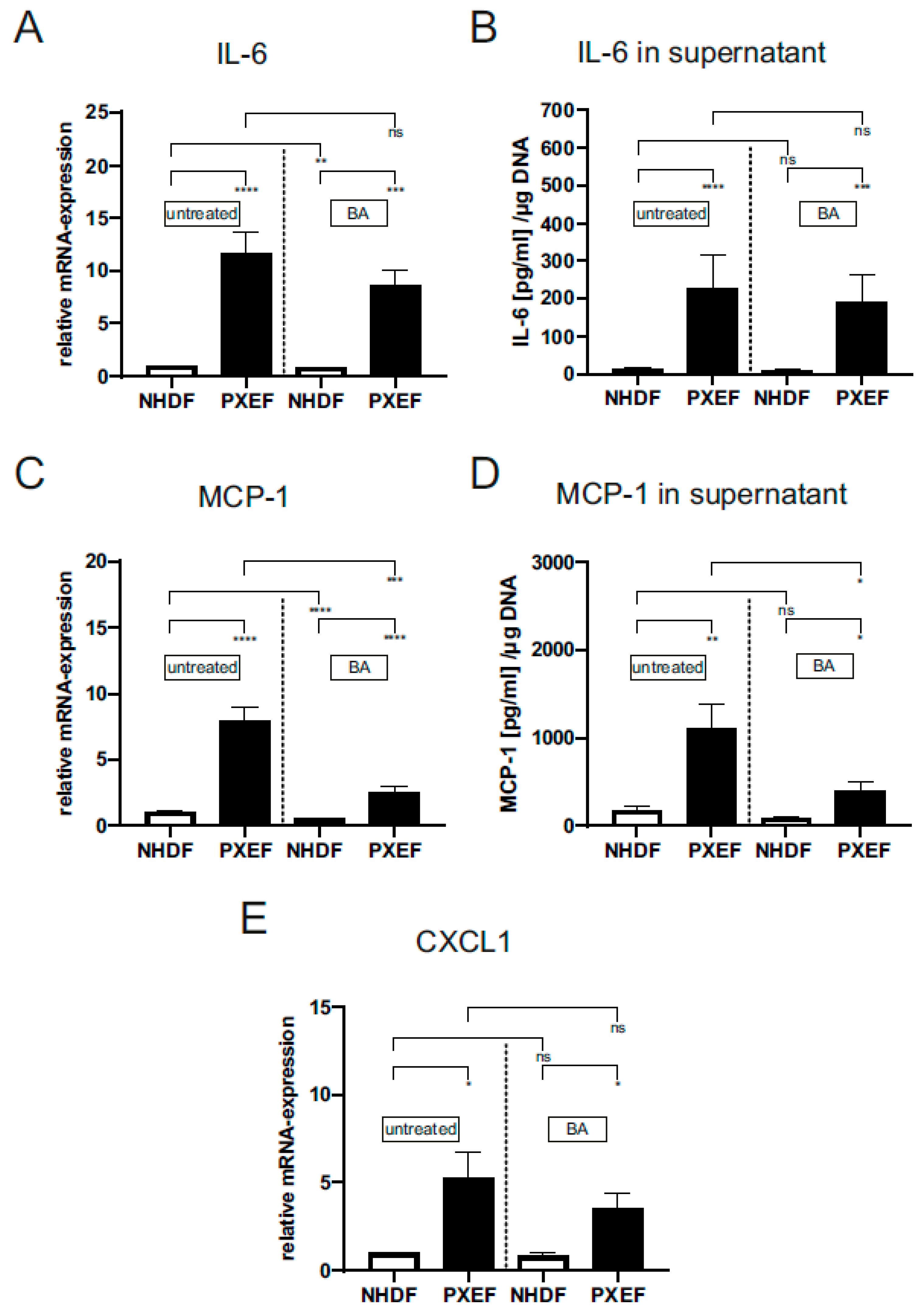{kind=link}
{kind=link}
{kind=link}
| Sample ID | Gender | Age 1 | Biopsy Source | ABCC6 Genotype 2 | Genotype Status | Phenodex Score 3 | |
|---|---|---|---|---|---|---|---|
| PXE Patients | |||||||
| P3M a | Male | 57 | Neck | c.3421C > T (p.Arg1141*) | c.3883_6G > A (SSM) | cht | S3, V2, C0 |
| P128M a | Male | 51 | Neck | c.3769_ 3370insC (p.Leu1259fs*18) | c.3769_3770insC (p.Leu1259fs*18) | hm | S2, E2, G0, C1 |
| P255F a | Female | 48 | Arm | c.3421C > T (p.Arg1141*) | c.2787 + 1C > T (SSM) | cht | S3, E2, G0, C0 |
| P265F a | Female | 62 | Neck | c.1132C > T (p.Gln378*fs) | c.3421C > T (p.Arg1141*) | cht | S3, E3, G0, V1, C1 |
| Healthy Controls | |||||||
| M57A b (AG13145) | Male | 57 | Arm | - | - | wt | None |
| M52A b (AG11482) | Male | 52 | Arm | - | - | wt | None |
| F48A b (AG14284) | Female | 48 | Arm | - | - | wt | None |
| F63A b (AG12786) | Female | 63 | Arm | - | - | wt | None |
| Step | Time [s] | Temperature [°C] | Cycle |
|---|---|---|---|
| Preincubation | 300 | 95 | 1 |
| Denaturation | 10 | 95 | |
| Annealing | 15 | Ta * | 40 |
| Elongation | 20 | 72 | |
| Detection | - | 72 | |
| Melting curve | 5 | 95 | |
| 60 | 65 | 1 | |
| Cooling | 60 | 40 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindenkamp, C.; Plümers, R.; Osterhage, M.R.; Vanakker, O.M.; Van Wynsberghe, J.; Knabbe, C.; Hendig, D. The Activation of JAK/STAT3 Signaling and the Complement System Modulate Inflammation in the Primary Human Dermal Fibroblasts of PXE Patients. Biomedicines 2023, 11, 2673. https://doi.org/10.3390/biomedicines11102673
Lindenkamp C, Plümers R, Osterhage MR, Vanakker OM, Van Wynsberghe J, Knabbe C, Hendig D. The Activation of JAK/STAT3 Signaling and the Complement System Modulate Inflammation in the Primary Human Dermal Fibroblasts of PXE Patients. Biomedicines. 2023; 11(10):2673. https://doi.org/10.3390/biomedicines11102673
Chicago/Turabian StyleLindenkamp, Christopher, Ricarda Plümers, Michel R. Osterhage, Olivier M. Vanakker, Judith Van Wynsberghe, Cornelius Knabbe, and Doris Hendig. 2023. "The Activation of JAK/STAT3 Signaling and the Complement System Modulate Inflammation in the Primary Human Dermal Fibroblasts of PXE Patients" Biomedicines 11, no. 10: 2673. https://doi.org/10.3390/biomedicines11102673
APA StyleLindenkamp, C., Plümers, R., Osterhage, M. R., Vanakker, O. M., Van Wynsberghe, J., Knabbe, C., & Hendig, D. (2023). The Activation of JAK/STAT3 Signaling and the Complement System Modulate Inflammation in the Primary Human Dermal Fibroblasts of PXE Patients. Biomedicines, 11(10), 2673. https://doi.org/10.3390/biomedicines11102673