
HIF-2α Controls Expression and Intracellular Trafficking of the α2-Subunit of Na,K-ATPase in Hypoxic H9c2 Cardiomyocytes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. HIF Silencing Experiments and the In Vitro Ischemic Heart Model
2.3. RNA Isolation and Quantitative RT-PCR
2.4. Preparation of Nuclear Extracts
2.5. Cell Surface Biotinylation Experiments
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
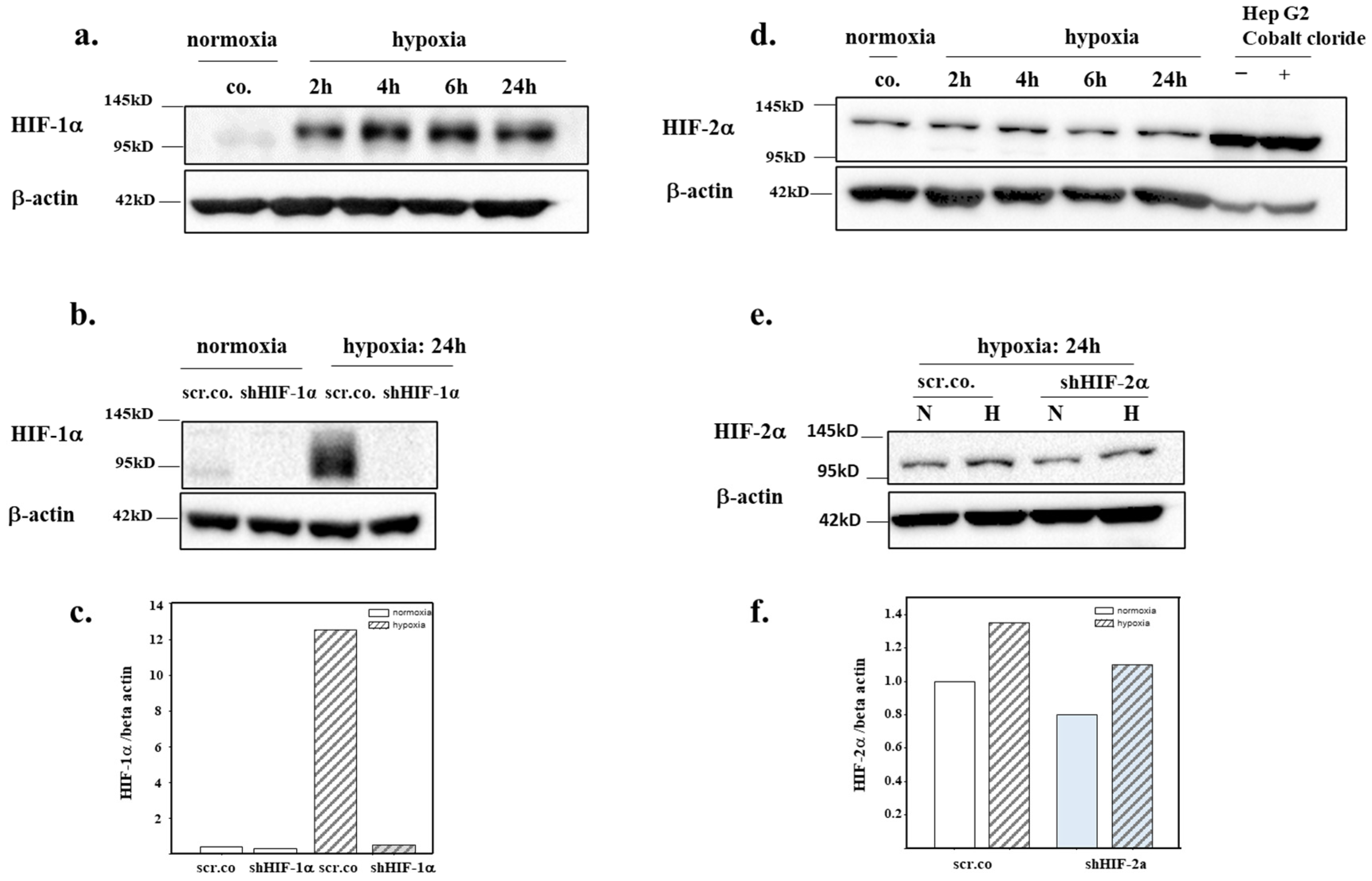
3.1. Silencing Efficiency of HIFs on the mRNA Expression of HIF-1α and HIF-2α
3.2. Silencing Efficiency of HIFs on the Protein Expression of HIF-1α and HIF-2α
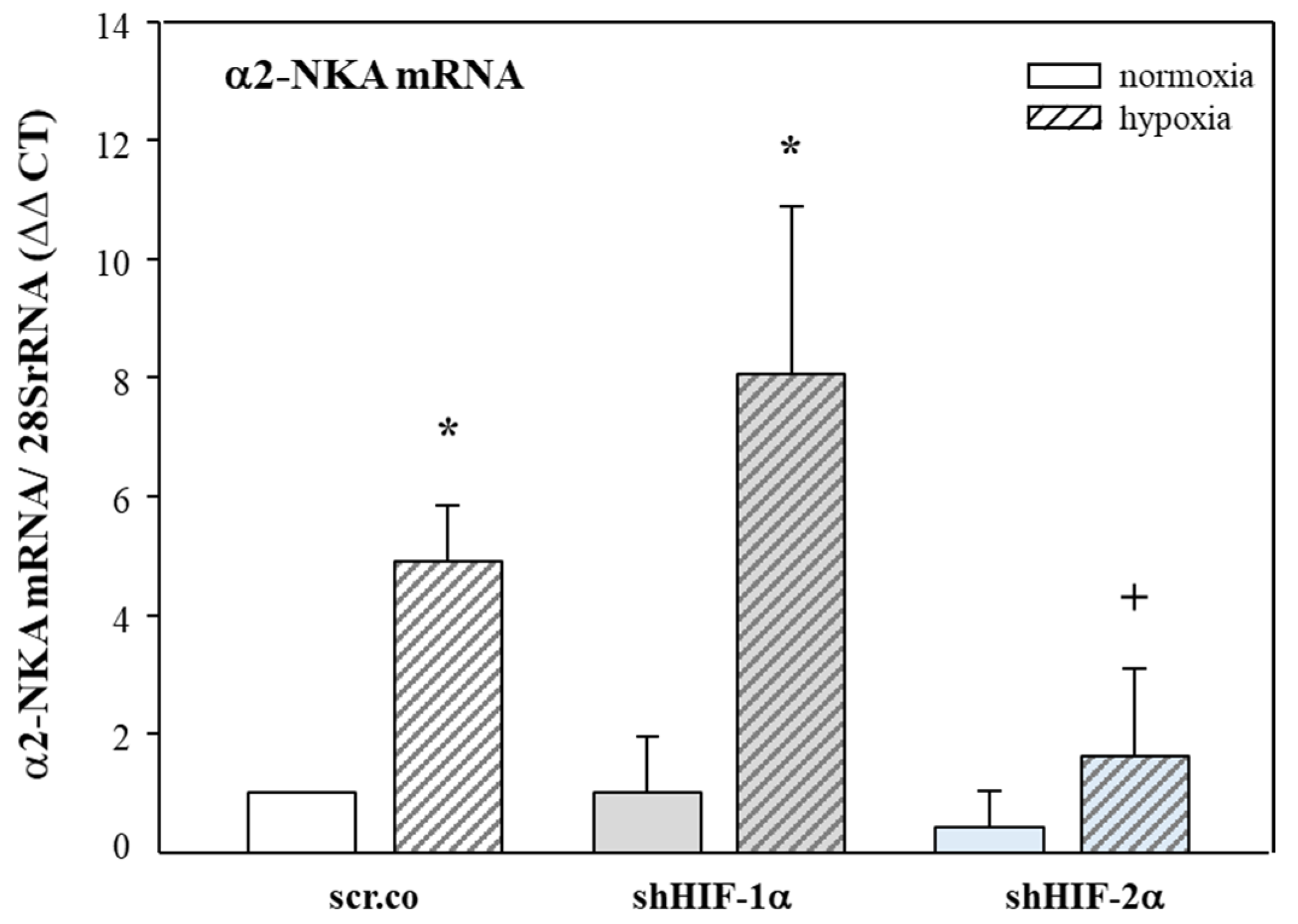
3.3. Effect of Hypoxia and HIF Silencing on the mRNA Expression of α2-NKA
3.4. Effect of Hypoxia and HIF Silencing on α2-NKA Protein Expression and Membrane Insertion
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Baloglu, E. Hypoxic Stress-Dependent Regulation of Na,K-ATPase in Ischemic Heart Disease. Int. J. Mol. Sci. 2023, 24, 7855. [Google Scholar] [CrossRef]
- Geering, K. FXYD proteins: New regulators of Na-K-ATPase. Am. J. Physiol. Renal Physiol. 2006, 290, F241–F250. [Google Scholar] [CrossRef]
- Morth, J.P.; Pedersen, B.P.; Toustrup-Jensen, M.S.; Sorensen, T.L.; Petersen, J.; Andersen, J.P.; Vilsen, B.; Nissen, P. Crystal structure of the sodium-potassium pump. Nature 2007, 450, 1043–1049. [Google Scholar] [CrossRef]
- Geering, K. Functional roles of Na,K-ATPase subunits. Curr. Opin. Nephrol. Hypertens. 2008, 17, 526–532. [Google Scholar] [CrossRef]
- Sweadner, K.J.; Herrera, V.L.; Amato, S.; Moellmann, A.; Gibbons, D.K.; Repke, K.R. Immunologic identification of Na+,K(+)-ATPase isoforms in myocardium. Isoform change in deoxycorticosterone acetate-salt hypertension. Circ. Res. 1994, 74, 669–678. [Google Scholar] [CrossRef]
- Berry, R.G.; Despa, S.; Fuller, W.; Bers, D.M.; Shattock, M.J. Differential distribution and regulation of mouse cardiac Na+/K+-ATPase alpha1 and alpha2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc. Res. 2007, 73, 92–100. [Google Scholar] [CrossRef]
- Sweadner, K.J. Isozymes of the Na+/K+-ATPase. Biochim. Biophys. Acta 1989, 988, 185–220. [Google Scholar]
- Rindler, T.N.; Dostanic, I.; Lasko, V.M.; Nieman, M.L.; Neumann, J.C.; Lorenz, J.N.; Lingrel, J.B. Knockout of the Na,K-ATPase alpha(2)-isoform in the cardiovascular system does not alter basal blood pressure but prevents ACTH-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1396–H1404. [Google Scholar] [CrossRef]
- Rindler, T.N.; Lasko, V.M.; Nieman, M.L.; Okada, M.; Lorenz, J.N.; Lingrel, J.B. Knockout of the Na,K-ATPase alpha2-isoform in cardiac myocytes delays pressure overload-induced cardiac dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1147–H1158. [Google Scholar] [CrossRef]
- Dostanic, I.; Lorenz, J.N.; Schultz Jel, J.; Grupp, I.L.; Neumann, J.C.; Wani, M.A.; Lingrel, J.B. The alpha2 isoform of Na,K-ATPase mediates ouabain-induced cardiac inotropy in mice. J. Biol. Chem. 2003, 278, 53026–53034. [Google Scholar] [CrossRef] [PubMed]
- Moseley, A.E.; Cougnon, M.H.; Grupp, I.L.; El Schultz, J.; Lingrel, J.B. Attenuation of cardiac contractility in Na,K-ATPase alpha1 isoform-deficient hearts under reduced calcium conditions. J. Mol. Cell Cardiol. 2004, 37, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Verdonck, F.; Volders, P.G.; Vos, M.A.; Sipido, K.R. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J. Mol. Cell Cardiol. 2003, 35, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Wang, J.; Frank, K.; Muller-Ehmsen, J.; Brixius, K.; McDonough, A.A.; Erdmann, E. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 1999, 99, 2105–2112. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef]
- Pike, M.M.; Luo, C.S.; Clark, M.D.; Kirk, K.A.; Kitakaze, M.; Madden, M.C.; Cragoe, E.J., Jr.; Pohost, G.M. NMR measurements of Na+ and cellular energy in ischemic rat heart: Role of Na(+)-H+ exchange. Am. J. Physiol. 1993, 265, H2017–H2026. [Google Scholar] [CrossRef]
- Semb, S.O.; Lunde, P.K.; Holt, E.; Tonnessen, T.; Christensen, G.; Sejersted, O.M. Reduced myocardial Na+, K(+)-pump capacity in congestive heart failure following myocardial infarction in rats. J. Mol. Cell Cardiol. 1998, 30, 1311–1328. [Google Scholar] [CrossRef]
- Bossuyt, J.; Ai, X.; Moorman, J.R.; Pogwizd, S.M.; Bers, D.M. Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ. Res. 2005, 97, 558–565. [Google Scholar] [CrossRef]
- Ishino, K.; Botker, H.E.; Clausen, T.; Hetzer, R.; Sehested, J. Myocardial adenine nucleotides, glycogen, and Na, K-ATPase in patients with idiopathic dilated cardiomyopathy requiring mechanical circulatory support. Am. J. Cardiol. 1999, 83, 396–399. [Google Scholar] [CrossRef]
- Swift, F.; Birkeland, J.A.; Tovsrud, N.; Enger, U.H.; Aronsen, J.M.; Louch, W.E.; Sjaastad, I.; Sejersted, O.M. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc. Res. 2008, 78, 71–78. [Google Scholar] [CrossRef]
- Jager, H.; Wozniak, G.; Akinturk, I.H.; Hehrlein, F.W.; Scheiner-Bobis, G. Expression of sodium pump isoforms and other sodium or calcium ion transporters in the heart of hypertensive patients. Biochim. Biophys. Acta 2001, 1513, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.D.; Schmidt, T.A.; Marsh, J.D.; Kjeldsen, K. Na,K-ATPase expression in normal and failing human left ventricle. Basic. Res. Cardiol. 1992, 87, 87–94. [Google Scholar] [PubMed]
- Magyar, C.E.; Wang, J.; Azuma, K.K.; McDonough, A.A. Reciprocal regulation of cardiac Na-K-ATPase and Na/Ca exchanger: Hypertension, thyroid hormone, development. Am. J. Physiol. 1995, 269, C675–C682. [Google Scholar] [CrossRef] [PubMed]
- Zahler, R.; Gilmore-Hebert, M.; Baldwin, J.C.; Franco, K.; Benz, E.J., Jr. Expression of alpha isoforms of the Na,K-ATPase in human heart. Biochim. Biophys. Acta 1993, 1149, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef]
- Lucero Garcia Rojas, E.Y.; Villanueva, C.; Bond, R.A. Hypoxia Inducible Factors as Central Players in the Pathogenesis and Pathophysiology of Cardiovascular Diseases. Front. Cardiovasc. Med. 2021, 8, 709509. [Google Scholar]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N. Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef]
- Cowburn, A.S.; Takeda, N.; Boutin, A.T.; Kim, J.W.; Sterling, J.C.; Nakasaki, M.; Southwood, M.; Goldrath, A.W.; Jamora, C.; Nizet, V.; et al. HIF isoforms in the skin differentially regulate systemic arterial pressure. Proc. Natl. Acad. Sci. USA 2013, 110, 17570–17575. [Google Scholar] [CrossRef]
- Jurgensen, J.S.; Rosenberger, C.; Wiesener, M.S.; Warnecke, C.; Horstrup, J.H.; Grafe, M.; Philipp, S.; Griethe, W.; Maxwell, P.H.; Frei, U.; et al. Persistent induction of HIF-1alpha and -2alpha in cardiomyocytes and stromal cells of ischemic myocardium. FASEB J. 2004, 18, 1415–1417. [Google Scholar] [CrossRef]
- Sui, X.; Wei, H.; Wang, D. Novel mechanism of cardiac protection by valsartan: Synergetic roles of TGF-beta1 and HIF-1alpha in Ang II-mediated fibrosis after myocardial infarction. J. Cell Mol. Med. 2015, 19, 1773–1782. [Google Scholar] [CrossRef]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Takeda, N.; Isagawa, T.; Semba, H.; Nishimura, S.; Morioka, M.S.; Nakagama, Y.; Sato, T.; Soma, K.; Koyama, K.; et al. Macrophage hypoxia signaling regulates cardiac fibrosis via Oncostatin M. Nat. Commun. 2019, 10, 2824. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [PubMed]
- Baloglu, E.; Nonnenmacher, G.; Seleninova, A.; Berg, L.; Velineni, K.; Ermis-Kaya, E.; Mairbaurl, H. The role of hypoxia-induced modulation of alveolar epithelial Na(+)- transport in hypoxemia at high altitude. Pulm. Circ. 2020, 10, 50–58. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]
- Kennedy, D.; Omran, E.; Periyasamy, S.M.; Nadoor, J.; Priyadarshi, A.; Willey, J.C.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J. Am. Soc. Nephrol. 2003, 14, 90–97. [Google Scholar] [CrossRef]
- Sato, T.; Takeda, N. The roles of HIF-1alpha signaling in cardiovascular diseases. J. Cardiol. 2023, 81, 202–208. [Google Scholar] [CrossRef]
- Liu, M.; Galli, G.; Wang, Y.; Fan, Q.; Wang, Z.; Wang, X.; Xiao, W. Novel Therapeutic Targets for Hypoxia-Related Cardiovascular Diseases: The Role of HIF-1. Front. Physiol. 2020, 11, 774. [Google Scholar] [CrossRef]
- Ronkainen, V.P.; Skoumal, R.; Tavi, P. Hypoxia and HIF-1 suppress SERCA2a expression in embryonic cardiac myocytes through two interdependent hypoxia response elements. J. Mol. Cell Cardiol. 2011, 50, 1008–1016. [Google Scholar] [CrossRef]
- Williams, A.L.; Walton, C.B.; Pinell, B.; Khadka, V.S.; Dunn, B.; Lee, K.; Anagaran, M.C.T.; Avelar, A.; Shohet, R.V. Ischemic heart injury leads to HIF1-dependent differential splicing of CaMK2gamma. Sci. Rep. 2021, 11, 13116. [Google Scholar] [CrossRef]
- Skoumal, R.; Szokodi, I.; Aro, J.; Foldes, G.; Gooz, M.; Seres, L.; Sarman, B.; Lako-Futo, Z.; Papp, L.; Vuolteenaho, O.; et al. Involvement of endogenous ouabain-like compound in the cardiac hypertrophic process in vivo. Life Sci. 2007, 80, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Gao, X.; Guo, X.; Wang, N.; Wang, X. Research Progress in Pharmacological Activities and Applications of Cardiotonic Steroids. Front. Pharmacol. 2022, 13, 902459. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Catalogue Number |
|---|---|
| rat 28SrRNA | Rn_Rnr1_1_SG QuantiTect Primer Assay QT00199374 |
| rat HIF-1α | Rn_Hif1a_1_SG QuantiTect Primer Assay QT00182532 |
| rat HIF-2α | Rn_Epas1_1_SG QuantiTect Primer Assay QT00192059 |
| rat α2-NKA | Rn_Atp1a2_1_SG QuantiTect Primer Assay QT00175924 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baloglu, E. HIF-2α Controls Expression and Intracellular Trafficking of the α2-Subunit of Na,K-ATPase in Hypoxic H9c2 Cardiomyocytes. Biomedicines 2023, 11, 2879. https://doi.org/10.3390/biomedicines11112879
Baloglu E. HIF-2α Controls Expression and Intracellular Trafficking of the α2-Subunit of Na,K-ATPase in Hypoxic H9c2 Cardiomyocytes. Biomedicines. 2023; 11(11):2879. https://doi.org/10.3390/biomedicines11112879
Chicago/Turabian StyleBaloglu, Emel. 2023. "HIF-2α Controls Expression and Intracellular Trafficking of the α2-Subunit of Na,K-ATPase in Hypoxic H9c2 Cardiomyocytes" Biomedicines 11, no. 11: 2879. https://doi.org/10.3390/biomedicines11112879