Abstract
Dopamine regulates several functions, such as voluntary movements, spatial memory, motivation, sleep, arousal, feeding, immune function, maternal behaviors, and lactation. Less clear is the role of dopamine in the pathophysiology of type 2 diabetes mellitus (T2D) and chronic complications and conditions frequently associated with it. This review summarizes recent evidence on the role of dopamine in regulating insular metabolism and activity, the pathophysiology of traditional chronic complications associated with T2D, the pathophysiological interconnection between T2D and chronic neurological and psychiatric disorders characterized by impaired dopamine activity/metabolism, and therapeutic implications. Reinforcing dopamine signaling is therapeutic in T2D, especially in patients with dopamine-related disorders, such as Parkinson’s and Huntington’s diseases, addictions, and attention-deficit/hyperactivity disorder. On the other hand, although specific trials are probably needed, certain medications approved for T2D (e.g., metformin, pioglitazone, incretin-based therapy, and gliflozins) may have a therapeutic role in such dopamine-related disorders due to anti-inflammatory and anti-oxidative effects, improvement in insulin signaling, neuroinflammation, mitochondrial dysfunction, autophagy, and apoptosis, restoration of striatal dopamine synthesis, and modulation of dopamine signaling associated with reward and hedonic eating. Last, targeting dopamine metabolism could have the potential for diagnostic and therapeutic purposes in chronic diabetes-related complications, such as diabetic retinopathy.
1. Background
Dopamine has several systemic effects and may have a role in the pathophysiology of chronic diseases. This review aims to summarize the leading evidence regarding the pathophysiological and potentially therapeutic role of dopamine in type 2 diabetes (T2D) and to explore the cross-linking between T2D pathophysiology and the most frequent chronic neurologic and psychiatric disorders associated with impaired dopamine activity/metabolism and T2D in human pathology.
Dopamine (C8H11NO2) is a catecholamine derived by the amino acid tyrosine after a two-step reaction involving (a) the enzyme tyrosine hydroxylase (step 1), which transforms the amino acid tyrosine in L-dihydroxyphenylalanine (L-DOPA), and (b) the enzyme DOPA decarboxylase (step 2), which converts L-DOPA in dopamine [1]. Dopamine is also the precursor of noradrenaline (dopamine β-hydroxylase) and adrenaline (phenyl-ethanolamine N-methyltransferase) [2].
Dopamine is synthesized mainly in the brain and other tissues with neuroectodermal origin, such as the medulla of the adrenal glands and paraganglia, where it serves as the precursor of adrenaline and noradrenaline synthesis. Dopamine is well known as a neurotransmitter, but extracerebral sites of production and dopamine receptors (DRs) are also widely expressed in peripheral tissues.
Four dopaminergic pathways exist in the central nervous system: the mesolimbic, mesocortical, nigrostriatal, and tuberoinfundibular (Figure 1). In the mesolimbic pathway, dopaminergic neurons of the ventral tegmental area (VTA) project to the nucleus accumbens (NAc), anterior cingulate cortex, and amygdala (AMY). The pathway involves pleasure and reward, and its dysfunction is associated with neuropsychiatric disorders such as schizophrenia, depression, and chronic pain [3]. Dopaminergic projections within the mesocortical pathway also originate in the VTA. The firing of VTA dopaminergic neurons travels to some areas in the prefrontal cortex (PC), which regulates critical cognitive functions such as cognition, working memory, and decision making [4]. The nigrostriatal pathway is the foremost dopaminergic system in the brain and is involved in controlling voluntary movement. Dopamine projections start in the substantia nigra and fire to the caudate and putamen in the basal ganglia. This dopaminergic pathway is affected in some neurological disorders involving the extrapyramidal system, such as Parkinson’s disease (PD), and in patients chronically treated with first-generation antipsychotics (D2 receptor antagonists), resulting in irregular muscle contractions manifesting as tremors, spasms, motor restlessness, and tardive dyskinesia. In the tuberoinfundibular pathway, dopamine neurons begin in the hypothalamic arcuate and periventricular nuclei and project to the median eminence of the hypothalamus. After that, dopamine is released into the portal circulation, connecting the median eminence to the pituitary gland, where dopamine inhibits the prolactin release from lactotrophic cells. A mild or moderate increase in serum prolactin is a secondary effect of D2 antagonists [5].
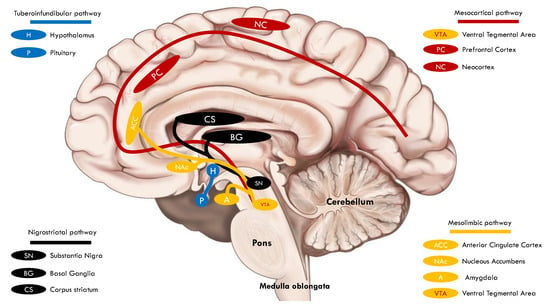
Figure 1.
Simplified depiction of the four dopaminergic pathways in the central nervous system.
2. The Effects of Dopamine on Pancreatic Islets and Insulin and Glucagon Secretion
The potential role of endogenous catecholamines in the pathogenesis of T2D was suggested by landmark studies in the 1970s [6,7,8]. The intravenous administration of L-DOPA increased the pancreatic dopamine concentration, especially within the β-cells, in normal rats [9] and inhibited insulin secretion in several species of golden hamsters [10]. A mechanistic study found that intravenous administration of L-DOPA was accompanied by a subsequent increase in the dopamine-containing grains in β-cells. Accumulation of dopamine-containing grains was found to reduce the release of insulin-containing grains by secretagogues, ultimately indicating that dopamine partially suppressed insulin release from β-cells [11]. Another study confirmed that dopamine suppresses insulin release from β-cells. The dopamine effect was completely inverted after the administration of propranolol (a β-blocker) but was not affected by dopamine antagonists, indicating that the suppression of insulin release by dopamine was mediated by α-adrenergic rather than dopaminergic signaling [12]. In an obese murine model (ob/ob), dopaminergic therapy reduced hyperglycemia and hyperlipidemia and improved islet function by restoring glucose sensitivity in β-cells (assessed by a 1.6-fold increase in the Glucokinase immunoreactivity), stabilizing hyperplasia, enhancing insulin storage, and thus reducing circulating insulin levels [13]. A recent investigation demonstrated that pancreatic islets are a site of dopamine synthesis and that L-DOPA and dopamine reduce glucose-dependent insulin secretion by dropping the frequency of intracellular oscillations of calcium currents. This effect was mediated directly by DR3 stimulation, as demonstrated by experiments using specific dopamine antagonists [14]. In another study, a single administration of the dopamine agonist bromocriptine reduced fasting glucose and insulin levels in patients with T2D. These effects were only mild in healthy controls. They were accompanied by a reduction in prolactin levels in all and growth hormone concentrations only in T2D patients, suggesting that the bromocriptine effect on glucose control could largely depend on an insulin-sensitizing secondary impact, mainly due to a reduction in growth hormone levels [15]. Apart from the effect on insulin secretion, the proliferation rate of β-cells decreases, and the apoptosis increases following dopamine treatment [16]. An inverse correlation between circulating levels of dopamine and c-peptide (a biomarker of insulin secretion from β-cells) was demonstrated in 201 healthy voluntaries [17], in which the insulin suppressive effect of dopamine was mediated by both DR2 and DR3 signaling [18,19]. In a recent study on rodents, dopamine dampened glucose-stimulated insulin secretion after a meal challenge test by counteracting the incretin effect, indicating that dopamine could affect insulin secretion in the post-prandial phase [20]. Glucose intake increases circulating dopamine levels by stimulating the intestinal secretion of dopamine, and this mechanism could work as a brake effect on the incretin actions [21]. As an additional mechanism, dopamine suppresses prolactin secretion. Prolactin stimulates insulin secretion and β-cell proliferation. It plays a role in normal pancreatic development and ameliorates peripheral insulin sensitivity, especially at the level of the adipose tissue [22].
Given the anti-secretive and antiproliferative effects, dopamine may have a role in the pathophysiology of T2D. Monoaminoxidase A and B play a crucial role in the catabolism of catecholamines, including dopamine. Both isoforms are also expressed in β-cells, and a lower level of monoaminoxidase activity is associated with dampened insulin secretion. Therefore, this evidence suggests that blunted dopamine catabolism and, consequently, high intra-islet dopamine concentration may contribute to reducing insulin secretion and raising the number of apoptotic β-cells, both events primarily involved in the pathophysiology of T2D. Interestingly, the transcription of monoaminoxidase A and B genes is under the MAF transcription factor A control [23]. MAF transcription factor A is an essential regulator of β-cell transcriptional activity since it regulates the transcription of genes involved in specific β-cell activities, including insulin biosynthesis and secretion [24]. The activity level and expression of the MAF factor A depend on glucose levels and may be reduced significantly by glucotoxicity due to hyperglycemia and chronic low-grade inflammation observed in prediabetes and diabetes [25]. Experimental models of insulinopenic, such as streptozotocin-induced, diabetes indicated that insulin deficiency increases the activity of circulating dopamine β-hydroxylase (which converts dopamine into noradrenaline), and the administration of insulin significantly reduces the enzymatic activity [26,27]. The phenomenon was associated with an increased dopamine receptor binding (up-regulation) in the striatum [28], which was the probable consequence of reduced dopamine metabolism in the same cerebral area [29,30].
These data suggest that dopamine and insulin may be involved in a potential feedback mechanism in which one negatively regulates the metabolism of the other [31].
Pivotal studies suggested that intravenous dopamine infusion stimulated glucagon release [32] in a dose-dependent manner [33]. Keck et al. found that low-dose dopamine (e.g., 2 mcg/kg/min infused for 6 consecutive hours) did not affect both insulin and glucagon secretion [34], but high-dose dopamine was found to provide relevant hyperglycemia by suppressing insulin and stimulating glucagon secretion in rats and men [32,33]. The effect could be considered an additive mechanism by which dopamine and dopamine agonists could sustain hyperglycemia in healthy and T2D patients. A summary of the mechanisms by which dopamine affects β-cell activity, insulin, and glucagon secretion is shown in Table 1.

Table 1.
Summary of mechanisms of dopamine-mediated modulation of β-cell activity, insulin, and glucagon secretion [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33].
3. Dopamine in the Pathogenesis and Treatment of Traditional Chronic Diabetes-Related Complications
T2D and chronic comorbidities, such as arterial hypertension, overweight/obesity, and dyslipidemia, foster the development of chronic complications over time [35]. Hyperglycemia is the determinant of chronic diabetes-related complications, especially at the microvascular site, and more stringent glucose control is associated with a lower likelihood of the onset and progression of these complications [36]. Moreover, early intensive intervention to achieve optimal control of all risk factors concomitant with T2D is associated with a reduced risk of macrovascular complications [37], and the higher the stability of glucose control over time, the better the attenuation of burdens [38,39]. So far, guidelines recommend comprehensive management of T2D patients to reduce the risk of diabetes-related complications over time by targeting glucose, arterial pressure, body weight, and lipid control, as well as preventing thrombotic events and attenuating thrombotic risks [40]. Evidence is already consolidated to suggest the use of specific classes of medications, such as glucagon-like peptide 1 receptor agonists (GLP-1RAs) and sodium-glucose (co)transporter 2 inhibitors (SGLT2is), to improve hard clinical outcomes, reduce the risk of adverse cardiovascular and renal endpoints, hospital admission due to heart failure and heart failure progression, and diabetes-related mortality [41,42].
The role of dopamine in the pathophysiology of diabetes-related chronic complications is an emerging issue [43]. Comprehending the mechanisms involved in the physiological activities of dopamine and the pathophysiological disruption of dopamine metabolism and dopaminergic pathways in target tissues would have relevant therapeutic implications and advance current treatments (Table 2). Dopaminergic neurons are described in the retina, where dopamine is a neurotransmitter. Here, dopamine diffuses through retinal layers to reach target cells and modulate their activity. Hence, the mechanism of dopamine communication in the retinal tissue is volume-dependent. In other words, dopamine deficiency or impaired metabolism/activity could be associated with retinal disease [44]. Experimental studies suggest that dopamine regulates photoreceptor activity, critical to visual adaptation to daylight [45]. Intraretinal dopamine levels are low in the early phase of retinal damage in diabetes [46], while high intraretinal levels of dopamine are protective against retinal damage and visual field loss [47]. The precise mechanism by which preserving dopamine levels in retinal tissue would prevent retinal damage and visual impairment is unclear. Experimental models found that intravitreal administration of L-DOPA was associated with lower severity of hyperglycemic memory-induced retinal microvascular alterations, including pericyte degeneration, acellular capillary and pericyte ghost generation, and endothelial apoptosis [48]. One mechanistic study in rodents has recently shown that intravitreal administration of L-DOPA reduced intraretinal levels of the vascular endothelial growth factor and insulin-like growth factor 1 receptors via the AKT/ERK pathway after 12 weeks [49]. Nevertheless, the first data available on a few cases did not confirm relevant differences in intraretinal dopamine (metabolites) in patients with diabetes without clinical signs of diabetic retinopathy and those without diabetes [50]. Additional studies are needed to verify whether intraretinal dopamine metabolism in humans differs from what has been seen in experimental models. On the other hand, the results of a pilot trial confirmed that reinforcing the intraretinal dopamine pathway may improve retinal dysfunction in the early stages of diabetic retinopathy [51]. So far, concrete pathophysiological hypotheses suggest a link between neurodegenerative disease and diabetic retinopathy in T2D [52], and evidence supports the role of diagnostic intervention in the early stages of both diseases [53]. From a therapeutic viewpoint, specific trials are currently ongoing to investigate the role of dopamine replacement in early-stage diabetic retinopathy and diabetic macular edema (NCT05132660; NCT02706977; NCT03161652). GLP-1 agonists may accelerate the progression of diabetic retinopathy and can be associated with adverse retinal outcomes while improving glucose control. Although evidence is discordant, data from the literature reported that this effect could be restricted to only some specific analogs and could be related to some background characteristics, such as poor glycemic control, more rapid achievement of glucose targets, higher body weight, and the presence of very high cardiovascular risk [54,55]. The above results align with preclinical evidence suggesting that GLP-1 analogs promote endothelial cell growth and angiogenesis. It could be interesting to assess the role of GLP-1 analogs on the intraretinal dopaminergic pathway. One trial could clarify this issue (NCT02671864).
Dopamine and DRs in the nephron tubules are essential in regulating key renal functions, such as electrolytes and water resorption, acid–base balance, and blood pressure regulation. DR1 and DR2 are the most widely expressed receptors mediating dopamine activity in the whole body [56]. In chronic diseases, such as arterial hypertension and diabetes, the expression of dopamine receptors could be significantly impaired in the kidney and the dopamine metabolism altered [57]. Because of these detrimental mechanisms, water exertion and natriuresis can be substantially reduced, thus contributing to water and sodium retention, increased blood pressure, glomerular hyperfiltration, and micro-/macroalbuminuria [58]. Experimental data in rats suggested that high intrarenal levels of dopamine prevent the mentioned effects and protect against glomerular injury and progression of diabetic nephropathy [59]. One pilot study found that administering bromocriptine (a dopamine agonist) compared to placebo reduced blood pressure and the left ventricular mass index without deteriorating the glomerular filtration rate in T2D over 6 months of treatment [60].
Dopamine plays many actions in the human heart, including positive inotropic and chronotropic effects, regulation of coronary flow, and cardiomyocyte metabolism [61]. These effects are mediated directly by dopamine and its interaction with DRs or indirectly by dopamine and noradrenaline action on α-adrenergic receptors [62]. Early evidence suggested the existence of impaired intracardiac dopamine metabolism in patients with diabetes [63]. More recent evidence suggests that early morning dopamine deficiency, frequently described in obese and T2D individuals, is involved in the overactivation of the sympathetic tone and release of corticotropin-stimulating hormone by the hypothalamic paraventricular nucleus. These effects produce substantial variability in daily heart rate, an indicator of cardiac autonomic neuropathy, and are associated with adverse events and dysmetabolic consequences on glucose control [64]. DR2 agonists may improve hemodynamics in T2D patients with heart failure (HF), positively affecting heart-failure-related outcomes [65]. Nevertheless, ergot-derived dopamine agonists are known for their cardiotoxicity due to their co-agonism with serotoninergic receptors [66]. Especially when administered at high doses, ergot-derived dopamine agonists are associated with myocardial valvopathy, thrombosis, arrhythmic events, and HF [66,67]. Antagonizing the serotoninergic effects of these agents may be considered a possible therapeutic strategy in diabetes-related HF [68]. From a therapeutic viewpoint, dopamine agents provide controversial evidence in terms of improvement in hemodynamics, preservation of renal function, and potassium homeostasis while on loop diuretics in advanced and acutely decompensated HF. Combining low-dose dopamine with low-dose loop diuretics effectively improves hemodynamic parameters and preserves glomerular filtration rate deterioration compared to high-dose loop diuretics alone [69]. Nevertheless, the results of two randomized clinical trials did not confirm the efficacy of low-dose dopamine in combination with both low-dose and high-dose diuretics in this clinical setting [70,71]. It is unclear if dopaminergic agents may be therapeutic in less severe clinical stages of HF to prevent adverse outcomes and reduce the risk of hospital admission due to symptomatic HF, but more investigation is ongoing (NCT01901809). It is unclear if positive results provided by SGLT2is on HF-related outcomes could depend, at least in part, on improved intracardiac dopamine metabolism.
Neurologic effects after ischemic stroke largely depend on the location and extension of ischemic areas, time of exposure to ischemic reperfusion injury, and baseline cerebral performance. Generally, ischemic stroke impairs dopamine release, synthesis, and DR activity in the striatum [72]. Dopamine deficiency is associated with cognitive and motor impairment, and evidence suggests that treatments restoring dopamine levels may improve recovery after stroke [73,74]. The mechanisms explaining this potential are that dopamine enhances motivation and improves symptoms of neuropsychiatric disorders related to stroke, complicating the rehabilitative period [75,76]. Nevertheless, no evidence has been provided to confirm the therapeutic rationale as a pharmacological strategy to improve relevant endpoints during post-stroke rehabilitation [77,78]. It is unknown if certain medications, such as thiazolidinediones, GLP-1RAs, and SGLT2is, may affect intracerebral dopamine metabolism as one of the mechanisms by which they benefit the prevention of ischemic stroke.
Table 2.
Summary of evidence highlighting the role of dopamine in the pathogenesis of diabetes-related chronic complications and implication for therapy.
Table 2.
Summary of evidence highlighting the role of dopamine in the pathogenesis of diabetes-related chronic complications and implication for therapy.
| Diabetes-Related Traditional Chronic Complication | Role of Dopamine | Effect | Rationale for Treatment (Dopamine Agonists or Levodopa) |
|---|---|---|---|
| Retinopathy [45,46,47,48,49,50,51,52,53,54,55] | Impaired intraretinal metabolism (deficiency) | Defective photoreceptor adaptation to light | Yes |
| Chronic renal disease [56,57,58,59,60] | Impaired renal metabolism (glomerular filtration-depended reduction) | Dysregulation in water and natrium resorption; promotion of glomerular hyperfiltration; micro- and macroalbuminuria | Scanty evidence or negative results |
| Neuropathy [64,65,66] | Defective axonal transport; impaired metabolism (accumulation due to inadequate conversion to noradrenaline?) | Implication for painful neuropathy | No (dopamine antagonists) |
| Stroke [72,73,74,75,76,77,78] | Impaired cerebral metabolism (deficiency) | Loss of motivation, motor impairment, and pathogenic role in post-stroke neuropsychiatric disorder | Scanty evidence or negative results |
| Cardiovascular diseases [61,62,63,64,65,66,67,68,69,70] | Impaired cardiac metabolism (accumulation due to inadequate conversion to noradrenaline?); striatal deficiency | Increased risk of heart failure, impaired coronary vasodilatation, cardiac autonomic neuropathy | Scanty evidence or negative results |
Table 2 summarizes the leading evidence indicating the role of dopamine in the pathophysiology of traditional diabetes-related chronic complications.
4. The Pathophysiological Link between Type 2 Diabetes and Chronic Disorders Characterized by Impaired Dopamine Activity/Metabolism
Dopamine plays a crucial role in the central nervous system (CNS), as it regulates many activities, including motor control, spatial memory, motivation, sleep, arousal, feeding, immune function, maternal behaviors, and lactation, to cite the main effects [79]. Dopamine signaling disruption is involved in the pathogenesis of several neurological and psychiatric disorders such as Parkinson’s (PD) and Huntington’s (HD) diseases, attention-deficit/hyperactivity disorder (ADHD), and addiction. Here, we discuss the potential and putative pathophysiological interconnection between T2D and these chronic neurological and psychiatric disorders characterized by impaired dopamine activity/metabolism (Table 3).
4.1. Parkinson’s Disease
PD is characterized by a primitive degeneration of a group of neurons in the substantia nigra and largely synapsing with basal ganglia. The distinctive neuropathological hallmarks are the Lewy bodies, α-synuclein aggregates, located in the substantia nigra with overtime spreading to neocortical and cortical regions (late-stage disease) [80]. Dopamine is the neurotransmitter of this intricate system, whose function is essential in planning motorial schemata, beginning and fine regulation of voluntary movements, postural control, and basal muscle tone [81]. Early clinical manifestations of PD include bradykinesia, postural and rest tremors, and muscle rigidity of limbs, neck, and trunk [82]. Nonmotor symptoms are also present. Some may be part of a prodromal syndrome, including depression and anxiety, sleep disorders, and constipation. Late-stage signs and symptoms of PD are related to relevant extrapyramidal imbalance, leading to motorial impairment and postural instability, accompanied by cognitive decline, manifesting in dementia and psychosis [82]. Regarding the frequency of clinical presentation, PD is the second cause of neurodegenerative diseases after Alzheimer’s dementia and affects 1 to 2 per 1000 of the general population [80]. Increasing age, genetic predisposition [83], exposure to pesticides and metals, and history of head trauma are the most common risk factors of PD [84]. T2D was found to increase the risk, accelerate the progression, and increase the severity of PD [85,86]. Diabetes severity, conceived as the number of anti-hyperglycemic agents used to treat hyperglycemia, long diabetes duration, need for insulin, and presence of chronic complications, is associated with an increased risk of developing PD [87]. Moreover, evidence suggests that diabetes affects nigrostriatal dopamine synthesis [88]. Therefore, similar pathways are involved in the pathogenesis of both diseases. The α-synuclein is largely involved in the pathogenesis of PD [89,90]. The protein is encoded by the SNCA gene located in chromosome 4; its native structure and function are still debated, but it is thought that α-synuclein is normally located in presynaptic terminals where it modulates neurotransmitter trafficking by affecting membrane plasticity [91]. Overexpression, posttranscriptional changes, aggregation of α-synuclein, and lysosomal dysfunction may facilitate the intracellular accumulation of the protein that is thought to induce membrane damage as the leading mechanism of neuronal injury [89,92,93]. Neuroinflammation, mitochondrial dysfunction, and impaired autophagy are other mechanisms of neurodegeneration in PD [94], and these mechanisms also characterize the pathophysiology of T2D [95,96]. Alpha-synuclein is essential in regulating insulin secretion at the level of β-cells [97] and glucose homeostasis in skeletal muscles, as demonstrated by experimental models [98]. The relationship between insulin and α-synuclein is interesting, as the latter can be considered a key regulator of insulin synthesis and activity in negative feedback [99]. In other words, the PI3K/Akt/GSK3β signaling could be involved in the pathogenesis of PD and Lewy body dementia as, in rodents, the overactivation of the pathway results in signs of neurodegeneration of the cortex and limbic system while silencing it ameliorates cognitive impairment [100]. Insulin and insulin-like growth factors act exactly by stimulating the phosphatidylinositol 3-kinase/serine-threonine protein kinase/glycogen synthase 3β (PI3K/Akt/GSK3β) pathway. It could be speculated that hyperinsulinemia, commonly found in prediabetes and early-stage T2D, may induce and accelerate neurodegeneration in the human brain [101]. On the other hand, impaired insulin signaling and insulin resistance are associated with the accumulation of α-synuclein (as well as β-amyloid, neurofibrillary tangles, and tau-proteins in Alzheimer’s disease) in PD [102]. Circulating insulin from β-cells can cross the blood–brain barrier and enter the CNS. In addition, insulin can be synthesized into the brain by several types of cells, such as astrocytes and neurons, in different cerebral areas, including the hippocampus, prefrontal cortex, dentate gyrus, thalamus, and olfactory bulb [103]. Insulin receptors are expressed in the same brain areas and VTA and substantia nigra, where insulin signaling regulates reward circuits modulating appetite and food intake [103]. Insulin exerts anti-inflammatory and anti-apoptotic properties, modulates mitochondrial function, improves autophagy and recycling of intracellular matter, and apoptosis in the brain [104]; therefore, as another hypothesis, intracerebral insulin resistance may contribute to neuroinflammation [105] and neurodegeneration [106]. The potential for neurotrophic protection of native GLP-1 is well known [107]. As observed for insulin, native circulating GLP-1 may enter the CNS, but it can also be synthesized in the brain, where several areas express GLP-1 receptors [108]. For example, native GLP-1 modulates appetite and food intake at the hypothalamic level [109,110]. Thanks to this mechanism, native GLP-1 is responsible for a sort of gut–brain talk, which was pivotal for the therapeutic application of GLP-1RAs to reduce appetite and promote weight loss [111]. Mechanistic studies found that GLP-1RAs may protect against neurodegeneration. In two rodent models of nigrostriatal injury, the administration of the GLP-1 agonist extendin-4, one week after the injury, arrested the progression of the nigrostriatal damage and restored the synthesis and release of dopamine [112]. Similarly, extendin-4 was found to promote neurogenesis and improve dopamine metabolism in the substantia nigra in an animal model of PD [113]. Years later, GLP-1RAs demonstrated neuroprotective effects in degenerative, ischemic, and traumatic models of brain injury [114]. Liraglutide provided evidence of reducing chronic neuroinflammation in response to X-ray irradiation [115]. Soon after, the neuroprotective effects of GLP-1RAs were also confirmed by neuroimaging in neurodegenerative diseases, including PD, in which treatment with GLP-1RAs provided a relevant increase in glucose uptake in specific cerebral areas as an indicator of improved metabolic activity [116]. The results of these studies posed the basis for further investigation into human disease [117]. Clinical trials have demonstrated that Exenatide, the most studied GLP-1RAs, improved motor and non-motor symptoms after one year of treatment in patients with PD [118,119,120]. More recently, other GLP-1RAs have shown promising preclinical protective effects on neurodegenerative diseases by providing synaptic protection, improvement in cognition, learning and motor function, amyloid pathology-ameliorating properties, improvement in intracellular calcium currents and endoplasmic stress, anti-inflammatory effects, reduction in oxidative stress, mitochondrial dysfunction and apoptosis, enhancements in the neuronal insulin sensitivity and energy metabolism, improvement in autophagy and mitophagy, and neurogenesis [121,122,123,124,125]. Overall, results for mechanistic and clinical trials indicate a tight relation between PD and T2D. Appropriate pharmacological management of T2D is expected to reinforce the dopamine imbalance in PD, also providing anti-oxidative and anti-neurodegenerative effects with overall improvement in PD-related prognosis.
4.2. Huntington’s Disease
Huntington’s disease (HD) is a neurodegenerative autosomal dominantly inherited disorder caused by a mutation of the Huntingtin gene located in chromosome 4. The mutation consists of a progressive (cross-generational) trinucleotide (CAG) triplet expansion, resulting in an extended polyglutamine sequence into the Huntingtin protein. When the number of CAG triplet repetitions is more than 35, the mutation makes missense changes in the native Huntingtin structure, compromising its functions, and the disease is most likely to occur [126]. Huntingtin protein is ubiquitously expressed, especially in the CNS, where it regulates the trafficking of vesicles and organelles, transcription, protein handling at the endoplasmic reticulum–Golgi level, and cell survival (anti-apoptotic activity) [127]. Clinical manifestations of HD occur in patients aged 30–50 years and are characterized by motor symptoms consisting of involuntary movements (chorea), memory loss, progressive cognitive decline, and psychiatric disorders [128]. HD evolves through several degrees of muscular and cognitive impairment. Late-stage disease is characterized by severe motor impairment resulting in bradykinesia, akinesia, dysarthria, and dysphagia with relevant deterioration of residual functional capabilities [129]. From a pathophysiological viewpoint, HD is defined by a degeneration of medium-sized spiny neurons of the striatum, with a marked impairment of γ-aminobutyric acid (GABA)-ergic signaling, followed by a progressive retrograde degeneration of cortical pyramidal neurons (which project to the striatum) and neuroanatomical disconnection between the striatum and the substantia nigra [130]. Mechanistic studies found significant neurochemical changes in HD, such as a decreased concentration of the inhibitory neurotransmitter GABA, mostly responsible for the extrapyramidal symptoms of the disease, associated with increased concentrations of dopamine and serotonin in the basal ganglia [131]. Across the stages of the disease, a progressive reduction in cannabinoid, dopamine, and adenosine receptors has been described in the basal ganglia [132]. Studies conducted with animal models demonstrated that the early phase of HD could be characterized by a relevant dopamine deficiency, hence suggesting that nigrostriatal degeneration could be one of the first pathophysiological events of the disease [133]. However, the hyperkinetic manifestation, defining the most common clinical presentation of HD in human pathology, suggests the contrary with regards to dopamine metabolism, so that striatal dopamine excess may have a role in the pathophysiology of HD [134]. The evidence of monoamine-oxidase enzymatic hyperactivity in the basal ganglia could indicate aberrant dopamine metabolism at that level, thus confirming the hypothesis. Excessive dopamine catabolism by monoamine oxidase produces a relevant amount of hydrogen peroxide, which, in turn, contributes to oxidative stress, a well-recognized pattern of damage in HD [135]. Mitochondrial dysfunction, impaired energy metabolism, axonal transport, microglial inflammation, and decreased synthesis of brain-derived neurotrophic factors are the other key factors of HD pathophysiology [136,137]. Beyond the fact that HD is a genetic disorder, it is known that patients with HD exhibit a greater frequency of prediabetes and diabetes than the general population [138,139]. The origin of impaired glucose metabolism in HD is difficult to understand. First, it was demonstrated that age-related decline in β-cell number was more evident and rapid in the HD model than in controls; nevertheless, this phenomenon was not associated with developing insulin deficiency, and most animals survived without developing diabetes lifelong. However, other experiments indicated that Huntingtin mutation was associated with impaired insulin synthesis and vesicular transport, suggesting that hyperglycemia was attributable to insulin deficiency. Conversely, HD models were prone to accumulate weight and expand visceral adipose tissue due to more food intake and hypothalamic dysfunction. As an additional mechanism, the onset of motor symptoms was associated with relevant impairment of cerebral glucose consumption, which could result in less overall glucose utilization and more glucose excretion. The hypothesis was confirmed by the evidence of a significant increase in glycosuria, but diabetes occurred only in a few rats, suggesting that the mechanism was insufficient to explain impaired glucose metabolism. Concurrent and disease-related chronic stress was considered a possible mechanism underlying chronic hyperglycemia, as well as mitochondrial dysfunction and inflammation [140,141,142]. One recent study elucidated the potential mechanism explaining glucose impairment in HD using mouse pancreatic insulinoma cells (line NIT-1) expressing N-terminal mutant Huntingtin containing 160 polyglutamine. More precisely, it was found that insulin receptor substrate 2 (IRS-2) expression decreased, and the remaining was recruited into mutant Huntingtin aggregates [143]. IRS-2 is essential for activating PI3K/AKT/FoxO1 to mediate glucose stimulation into insulin secretion. As an additional mechanism, exogen insulin administration inhibited the formation of mutant Huntingtin aggregates, thus removing the block on the PI3K/AKT/FoxO1 pathway and ameliorating pancreatic insulin reserve [143]. Conversely, T2D is associated with earlier onset and faster progression of HD [144]. The basic mechanism of this relationship involves the impairment of the PI3K/Akt/mTOR pathway described in T2D and HD [145]. The pathway regulates various functions, including cell proliferation, survival, apoptosis, autophagy, protein synthesis, glucose metabolism, angiogenesis, cytoskeletal organization, and vesicular trafficking [146]. Hyperglycemia leads to hyperinsulinemia, insulin resistance, and the formation of advanced glycation end products (AGEs), which interact with a specific receptor (RAGE). The interaction between AGEs and RAGE stimulates different intracellular pathways involved in diabetes-related tissue damage, such as mitochondrial and endoplasmic stress, oxidative stress, intracellular protein aggregation, and inflammation. In addition, AGEs downregulate the PI3K/Akt/mTOR pathway, further impairing cellular functions, as mentioned above, and deterioration of glucose control [147]. This mechanism, which could be defined as a vicious circle, may explain why T2D may deteriorate the prognosis of HD [148,149]. Besides the effect on endogenous dopamine and the above considerations for PD, optimal glucose control would result in better outcomes in HD. Even though no specific clinical trials have been conducted so far, AMP-activated protein kinase AMPK activation (by metformin) has been demonstrated to improve motor and cognitive outcomes in experimental models of HD [150,151,152]. Targeting insulin signaling for restoring insulin sensibility may have a therapeutic rationale in stimulating glial cells to produce neurotrophic factors, attenuating mutant huntingtin precipitates, reducing neuroinflammation and neurotoxicity, and improving autophagy [153,154,155,156,157].
4.3. Attention-Deficit/Hyperactivity Disorder
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder defined by impairing levels of inattention, disorganization, and/or hyperactivity–impulsivity [158]. The estimated prevalence of ADHD is around 6–8%, and it is usually diagnosed in childhood or young adults more in boys than girls [159]. A variable number of gene mutations could be involved in the pathogenesis of ADHD, including those targeting dopaminergic and serotoninergic pathways [160]. It is thought that ADHD, one of the most common inherited mutations, is biologically attributable to impaired dopaminergic signaling rather than impaired dopamine synthesis. Most common polymorphisms of dopamine receptors, such as DR2, have been described, involved in reward to environmental factors, DR4 and DR5 [161]. Real-world data revealed that ADHD is a risk factor for T2D and related comorbidities such as overweight and obesity [162], and it is associated with poor glucose control in young individuals with type 1 diabetes [163]. The results of a meta-analysis confirmed that the adjusted risk of T2D is more than doubled in patients with ADHD than in those without [164]. In addition, ADHD is more frequently diagnosed in individuals born by mothers who developed diabetes during pregnancy, suggesting that maternal hyperglycemia may be an epigenetic risk factor for ADHD [165]. Risks seem to be related to some specific adverse neonatal outcomes such as very and extremely preterm and very and extremely low birth weight as well [166]. The importance of such epidemiological data has been recognized by guidelines recommending implementing medical management of T2D in people living with ADHD [167]. Compulsive eating behaviors and binge eating disorders are frequently associated with ADHD, especially in young patients exposed to a higher risk of weight gain over time [168,169], and complicate glucose management and control of body weight in T2D. Pharmacological management of eating disorders in ADHD is therefore recommended. Most approved medications for ADHD, such as methylphenidate, seem to improve several aspects of behaviors more than placebo or non-pharmacological interventions (e.g., cognitive training and psychotherapy) [170]. Targeting dopaminergic (and noradrenergic) reuptake or stimulating dopamine synthesis represents the rationale of pharmacological intervention in ADHD. Reinforcing dopaminergic pathways in ADHD and obese individuals enhances the reward value of food [171] with positive consequences on food intake, caloric restriction [172], and body weight control [173,174]. Nevertheless, both methylphenidate and amphetamine reduce insulin secretion in a dose-dependent manner. Therefore, it is expected that high-dose treatment may potentially impair glucose control, requiring more appropriate glucose management [175]. So far, no specific clinical trials have been conducted in this cluster of patients, and the management of hyperglycemia is essentially the same in the general population. However, a tight neuroanatomical relation between dopaminergic circuits and GLP-1 sites of action in the brain, such as the amygdala, hypothalamus, hippocampus, and NAc, is known and should not be overlooked [176]. These areas regulate the most relevant cerebral function, including memory, food intake, and motivation. Potentiating dopaminergic pathways, especially DR2 agonism, which is usually impaired in ADHD as well as obesity, by GLP-1RAs would result in caloric restriction and weight loss [177,178]. Besides the controversy on the likelihood that GLP-1RAs act at several brain districts, exactly as observed for native GLP-1, specific trials are needed to clarify if this class of medication has feasible therapeutic opportunities in ADHD patients.
4.4. Addictions
Addiction refers to a psychopathological condition in which one individual is unable to control a specific impulse to carry out reiterating actions or behaviors that hesitate in physical or psychological dependence on something [179]. In pharmacology, addiction is defined as a chronic and relapsing disorder in which individuals are prone to and become dependent on compulsive seeking of abuse substances or drugs for achieving pleasure or feeling better [180]. Addiction is the leading cause of dependence on specific behaviors [181] or chemicals, such as alcohol [182], nicotine [183], certain medications (e.g., opiates) [184], and illicit substances [185]. The underlying mechanisms of addiction are complex and involve several brain areas of the reward circuit and dopaminergic (mesolimbic/mesocortical) pathways [186]. While nigro-striatal dopaminergic pathways are involved in regulating feeding [187] and impaired dopamine activity is associated with reduced food intake at this level, mesolimbic dopaminergic pathways seem to be involved in other aspects of food intake such as motivation, reward, and hedonic food [188]. Food and eating addiction [189], carbohydrates, and fat addiction are considered neurobehavioral, psychopathological, and maladaptive dysfunctions associated with exaggerated caloric intake, obesity, and possibly eating disorders, including binge eating [190]. Recent research confirms that the same mechanisms underlying dependence on drugs and substances of abuse are involved in food addiction [191], with dopaminergic pathways and reward circuitry playing a crucial role [191]. Importantly, food intake is also associated with gratifying stimuli from the limbic system in a way that can be neurobiologically translated into an increase in mesolimbic and mesocortical dopamine levels and activation of reward circuits, as observed in alcohol dependence [192]. Although food addiction is not currently classified as an eating disorder or an independent condition, it is more frequently observed among overweight and obese patients [193]. Moreover, being far from describing a well-established pathophysiological link between food addiction and hormonal parameters, some interesting glucometabolic, inflammatory, and neurohormonal biomarkers could relate to this condition [194]. Overeating is associated with specific neurochemical and neurobiological changes in the CNS, mostly attributable to an imbalance between homeostatic, cognitive, and hedonic homeostasis. Neuroimaging studies found that obese compared to lean people had reduced mesolimbic and mesocortical expression of DR2 and reduced neuronal metabolic activity in these areas, leading to an impaired reward system [195]. Conversely, obese individuals have a greater baseline metabolic activity in the somatosensitive cortex representing the mouth, lips, and tongue, in other words, the leading cerebral region involved in the conscious processing of food palatability [196]. Low and very low calorie diets, selectively restrictive diets, diets plus behavioral interventions, or physical exercise are the most effective non-pharmacological treatments to induce weight loss and improve body composition in patients with obesity [197,198]. Despite optimal results in the short term, only a high adherence to diet recommendations and calorie restrictions may provide long-term benefits to prevent weight gain or weight regain after dietary-induced weight loss [199]. No specific diet protocols have been shown to ensure weight maintenance over time (usually no more than 6 months) [200,201], so weight regain is frequently observed in patients who discontinued calorie restriction or those not receiving adequate food education or behavioral intervention [202]. The causes of this common but difficult-to-manage phenomenon include several mechanisms that can be considered as an adaptation to weight loss following calorie restriction. The drastic decline of leptin levels and shortage of circulating free fatty acids, both associated with diet-induced adipose tissue shrinkage, are the peripheral mechanisms of weight regain after diets. Inflammation, metabolic adaptation to calorie restriction, metabolic shift toward carbohydrate utilization upon their reintroduction (i.e., after ketogenic diets), and neuroendocrine adaptation to weight loss are other common mechanisms of weight regain [203,204,205]. Most importantly, diet and a healthy lifestyle cannot reverse brain attraction to food intake, and this effect may be considered as another important mechanism of weight regain after diet discontinuation [206]. The VTA is crucial in perceiving rewarding environmental stimuli and activating specific behaviors to obtain future rewards. Function crosstalk in the mesolimbic system between the VTA, NAc, hippocampus, and AMY is necessary to carry out the functions mentioned above, and dopamine is the key neurotransmitter. Food intake directly stimulates VTA activation by an intricate series of neurotransmitters and neuromodulators that include opioids, GABA, glutamate, and acetylcholine [207]. After the activation, neurons in the VTA fire to dopaminergic neurons in the NAc, resulting in dopamine synthesis and the release and activation of reward circuits in response to food intake [207]. Most importantly, orexigenic hormones affect dopaminergic activity at that level, being involved in food reward and hedonic eating [208]. Ghrelin, a stomach-derived polypeptide, is secreted in response to calorie restriction. Ghrelin receptors have been described in the VTA, where the hormone exerts a bimodal effect on dopamine synthesis and release. In the presence of normal food intake or consumption of palatable food, ghrelin was found to stimulate the mentioned dopaminergic pathway, hence motivating reward and gratification after meal ingestion [209,210]. This effect is thought to be mediated by mu and kappa opioid receptors in the VTA [211] and neuropeptide Y [212]. On the other side, ghrelin stimulation without food intake attenuates dopamine release. Other orexigenic hormones may modulate dopamine release in the VTA, as observed for ghrelin, while anorexigenic hormones may act in the opposite way [213]. Interesting experiments in rodents have found that GLP-1RAs may also affect dopaminergic pathways involved in addictions. Intracerebral administration of exendin 4 in the NAc, but not in the VTA, significantly attenuated alcohol-induced locomotor stimulation and memory of alcohol reward, as well as decreased alcohol intake [214]. Similarly, peripheral and central administration of exendin 4 attenuated cocaine-induced locomotion and abuse by blunting dopamine release in the VTA and NAc in response to cocaine administration [215]. Despite some controversial results, GLP-1RAs may reduce dopamine release by the mesolimbic/mesocortical pathways in response to food intake [216,217]. In other words, besides a direct effect on appetite, GLP-1RAs may reduce meal-induced gratification after food intake, foster lower caloric intake, and promote weight loss. Targeting dopamine metabolism also positively affects energy balance and weight loss, as observed with specific dopaminergic/noradrenergic reuptake inhibitors [218,219,220]. Overall, GLP-1RAs have the potential to modulate dopamine metabolism in addictions by contrasting positive rewards related to chronic and repetitive exposure to novice stimuli, such as pathological food intake or exposure to substances of abuse.
Table 3.
Pathophysiological mechanisms linking type 2 diabetes and chronic disorder characterized by impaired dopamine activity/metabolism.
Table 3.
Pathophysiological mechanisms linking type 2 diabetes and chronic disorder characterized by impaired dopamine activity/metabolism.
| Diseases and Conditions | Pathophysiological Mechanisms |
|---|---|
| Diabetes and Parkinson’s disease [85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125] |
|
| Diabetes and Huntington’s disease [135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157] |
|
| Diabetes and ADHD [161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178] |
|
| Diabetes and addictions [191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220] |
|
Table 3 summarizes the mechanisms linking the pathophysiology of T2D and the most relevant diseases in which dopamine activity/metabolism is impaired. Insulin resistance, mitochondrial dysfunction, neuroinflammation, impaired autophagy, and impaired neurogenesis are the most common mechanisms linking T2D and neurogenerative diseases (such as Parkinson’s and Huntington’s diseases). In ADHD and addictions, behavioral disorders inducing eating disorders are crucial elements that expose affected patients to T2D and related complications. Improving dopaminergic pathways results in improved clinical outcomes.
5. Therapeutic Implications
Here, we discuss the pharmacological role of dopamine in T2D and of anti-hyperglycemic medications in T2D and concomitant diseases in which dopamine activity/metabolism is impaired to overview existing and lacking evidence indicating that specific drugs may improve the pathophysiology and clinical manifestation of the above-mentioned disturbances. Most evidence refers to PD due to its more epidemiological relevance than other dopamine-related conditions. However, more information about the potential effects of approved treatments for T2D and chronic neurologic and psychiatric disorders characterized by impaired dopamine activity/metabolism on specific disease-related outcomes has been summarized in Table 4.
The intrapancreatic uptake of levodopa increases after glucose exposure, and hyperglycemia may sustain the machinery of dopamine synthesis in β-cells. Animal models showed that dopamine directly suppresses insulin secretion by curbing the duration of the action potential in response to β-cell glucose entry. The leading effect is mediated by the D2 receptor signaling [221]. In an experimental model, D2 overexpression abolished the glucose-stimulated Ca2+ influx and insulin secretion in β-cells. This toxic effect was partially reverted after the treatment with a D1-D2 heterodimer agonist (D1 and D2 dimerized on β-cell surface), suggesting that the D1 receptor may protect β-cells from the harmful effects of dopamine by modulating D2 signaling [222]. Given the role of D2 receptors in mediating the intrapancreatic effects of dopamine [223], specific treatment could be necessary to prevent or improve metabolic disorders associated with neurological and psychiatric conditions and their related treatment [224,225].
Bromocriptine, a DR2 agonist, is an FDA-approved oral medication for T2D, available in the US as tablets of 0.8 mg. The maximal approved daily dose is 4.8 mg, corresponding to 8 tables per day [226]. The tables are administered at fasting as food intake significantly affects the gastric adsorption of bromocriptine. Bromocriptine peaks 60 min after the table assumption, with a half-life of around 6 h, and undergoes relevant first-pass hepatic extraction and clearance so that only less than 10% of the assumed dose has a therapeutical effect [227]. Bromocriptine seems to replace the reduced dopaminergic tone in the hypothalamus, observed in insulin-resistant, obese, and diabetic patients, and reduce the serotoninergic tone, which is responsible for increased appetite and preference for carbohydrate-rich and hypercaloric foods [226]. Thanks to this mechanism, bromocriptine reduces the appetite and improves peripheral insulin sensitivity. In addition, bromocriptine reduces insulin and glucagon secretion by acting directly on DR2 on β- and α-cells, respectively [228]. Bromocriptine produces a 0.6–0.7% reduction in HbA1c levels in T2D when assumed alone (monotherapy) or in variable combinations with other anti-hyperglycemic agents also providing cardiovascular benefits [229]. Dopamine agonists, including bromocriptine, have been approved for PD [230]. Dopamine agonists effectively improve motor symptoms, delay levodopa replacement, and reduce fluctuation in dopamine levels over time [231]. The benefit of dopamine agonists could depend, at least in part, on low oxidative stress due to replacing dopamine deficiency instead of stimulating the neurotransmitter synthesis by L-DOPA supplementation [232,233]. However, the long-term efficacy of bromocriptine is lower than that of L-DOPA, and treatment with dopamine agonists is usually weighted down by adverse events such as mitral valve damage, impulse control disorders, and compulsive behaviors [234,235]. Other typical adverse events are headache, dizziness, hypersomnia, dyskinesia, psychosis, hypotension, tachycardia, nausea, and nasal congestion. Overall, dopamine agonists may be preferred as early-stage treatment of PD and could be considered a treatment option to improve glucose control in patients with both conditions (PD and T2D).
An experimental study indicated that the expression of molecules involved in DR2 signaling is increased in islets from high-fat-diet obese mice. The whole grain-derived γ-oryzanol, a mixture of vegetal sterols, improved glucose control, promoted weight loss, and reduced appetite and caloric intake by acting at the hypothalamic level [236]. Oryzanol is found in rice and bran oils, tomatoes, and green peas. Given its lipophilic nature, γ-oryzanol crosses the blood–brain barrier and enters the CNS, where it provides anti-oxidative and anti-inflammatory properties and reduces the endoplasmic reticulum stress (an indicator of relevant cell damage) [237]. With a similar mechanism, γ-oryzanol may protect against β-cell damage [238]. Moreover, γ-oryzanol enhances glucose-dependent insulin secretion by activating the protein kinase A pathway. This mechanism is thought to suppress the dopamine signaling, thus providing a beneficial effect on insulin secretion and β-cell survival [239]. Preclinical studies confirmed that γ-oryzanol may reduce neuromotor deficits, dopamine depletion, and oxidative stress in models of PD [240,241]. Overall, γ-oryzanol may positively affect T2D by stimulating insulin secretion and neurodegenerative disorders due to anti-oxidative and anti-inflammatory effects. However, it may hamper dopamine signaling. The magnitude of this effect is unclear in specific conditions such as PD and HD and should be elucidated.
Metformin is a synthetic biguanide approved as first-line treatment in T2D due to several effects, including mild suppression of gluconeogenesis in the liver, reduction in glucose absorption in the gut, enhancement of glucose utilization by mitochondria in peripheral tissues, and induction of weight loss [242]. Most of the therapeutic effects of metformin are mediated by the activation of AMPK, which is impaired in T2D and chronic neurodegenerative diseases, including PD [243]. Metformin-mediated AMPK activation can result in potentially favorable effects in PD as it regulates cellular energy metabolism, autophagy, mitochondrial performance, redox homeostasis, and anti-inflammation [244,245,246]. The branched-chain amino acid transferase (BCAT-1) is a critical regulator enzyme involved in the metabolism of leucine, isoleucine, and valine to transmute them into terminal products of the mitochondrial oxidative chain such as acetoacetate and succinyl-CoA [247]. Early-stage PD is characterized by mitochondrial hyperactivity, excessive oxidative stress, and production of oxygen-reactive species, and BCAT-1 has a role in mediating mitochondrial-hyperactivity-induced neurotoxicity. As metformin attenuates mitochondrial hyperactivity, it could have a therapeutic role in early-stage PD [248]. The activation of AMPK in the brain decreases the expression of β-secretase 1 protein, which plays a crucial role in producing cleavage products such as β-amyloid and α-synuclein [249]. Moreover, metformin may increase the level of acetylcholine at the presynaptic site by suppressing the synthesis and activity of the acetylcholinesterase and, therefore, may play a crucial role in maintaining memory function [250]. Besides the positive results of preclinical mechanistic studies, a few data have been published in humans with controversial data on the efficacy and effectiveness of metformin in PD. One real-life study found that low-dose but not high-dose exposure to metformin (considering 2 g of metformin as the defined daily dose) reduced the odds of PD in T2D [251]. Hence, one could speculate that low-dose metformin may be sufficient to provide positive effects, as described above, while exposure to high-dose metformin may result in potential toxicity. The actual mechanism of this “metformin-related” neurotoxicity could be attributable to some secondary (and dose-dependent) effects of chronic treatment. One of these is the reduction in circulating levels of Vitamin B12, as metformin was found to impair its absorption at the intestinal level in a dose-dependent manner [252]. Vitamin B12 deficiency has been found to affect cognition and may play a role in cognitive impairment and motorial deterioration [253]. Being far from confirming this hypothesis, baseline assessment of Vitamin B12 and periodic monitoring of its circulating levels during the follow-up could be reasonable in patients with T2D on metformin, especially those with or at risk of cognitive impairment or neurodegenerative disorders, including PD. Other common side effects of metformin include gastrointestinal discomfort, nausea, diarrhea, and abdominal pain. Adverse effects of metformin are usually mild and self-limiting, and specific advice usually reduces their incidence and severity. Metformin should be prescribed at low doses (e.g., 500 mg once daily) with gradual titration over time (usually a few days or weeks). Moreover, the post-meal assumption of metformin usually attenuates gastrointestinal discomfort. In case of persisting or recurring adverse events, switching to long-release formulations is recommended. Overall, more trials are needed to understand better and elucidate the therapeutic role of metformin in patients with PD (e.g., NCT05781711).
Acarbose, an α-glucosidase inhibitor, attenuates the intestinal absorption of carbohydrates. Thanks to this mechanism, acarbose improves glucose control, especially by reducing post-prandial glucose amplitude and insulin spikes [254]. In addition, acarbose may have a role in suppressing the synthesis of proinflammatory cytokines by increasing a key regulator microRNA (10a-5p) in the ileum that is overexpressed in T2D and PD [255]. These effects are probably insufficient to explain the potential rationale of acarbose in treating T2D patients with PD. To our knowledge, no specific studies have been published, nor are there ongoing trials to assess the efficacy/effectiveness of acarbose in this cluster of patients.
Apart from the anti-hyperglycemic properties, peroxisome-proliferator-activated receptor (PPAR) agonists have shown promising neuroprotective effects. In T2D, the foremost PPAR-γ agonist, pioglitazone, has relevant anti-inflammatory and anti-atherosclerotic properties [256]. Several mechanisms explain the anti-atherosclerotic effects of PPAR-γ agonists, inducing a potent insulin-sensitizing action that improves glucose uptake and metabolism by peripheral tissues (skeletal muscle and adipose tissue); a potent modulating effect on lipid metabolism by reducing circulating levels of free fatty acids and low-density lipoprotein and increasing high-density lipoprotein; and relevant modulation of the cytokine signature of adipose tissue by simulating adipocyte to release Adiponectin and Leptin and suppressing the release of resistin [257]. In addition, pioglitazone has been demonstrated to reduce neuroinflammation after cerebral injuries (i.e., ischemia, brain trauma) [258]. Preclinical trials showed that pioglitazone administered subcutaneously during the reperfusion phase in a rat model of experimental cerebral ischemia provided a relevant reduction in cerebral infarction by around 30% [259]. Thanks to these effects, pioglitazone has gained much interest in preventing or treating cerebrovascular diseases in T2D [260]. Clinical data showed that pioglitazone was associated with a reduced risk of cerebrovascular events by 38% and 24% in two secondary prevention trials, respectively [261,262]. Other trials confirmed the cardio and cerebrovascular protection of pioglitazone in T2D patients [263]. The rationale of pioglitazone as a background treatment in chronic cerebrovascular diseases has also been considered for other conditions, including degenerative and traumatic cerebral diseases [264,265] and cognitive impairment [266]. Despite the solid rationale [267,268,269,270], the level of evidence is limited, and the results of clinical trials are still inconclusive or debated [271,272]. In one meta-analysis of clinical studies [273], pioglitazone reduced the risk of PD in patients with T2D [274,275]. Moreover, a Finnish nationwide case-control study found that, among the anti-hyperglycemic medications, only pioglitazone decreased the risk of incident PD in T2D [276]. Whether or not the results of these studies may be translated into real benefit in PD irrespective of T2D and how pioglitazone may modify the progression of PD in the early stage of the disease are unclear [277].
Dipeptidyl peptidase IV inhibitors (DPP-IVis) are a class of oral gliptins approved for T2D. These agents compete with endogenous incretins at the catalytic site of the enzyme and delay the incretin degradation [278]. Therefore, DPP-IVis improve glucose control by extending the circulating half-life of endogenous incretins, especially in the post-prandial phase, with satisfactory efficacy and durability [279,280]. Mechanistic preclinical studies using animal models of PD found that DPP-IVis had the rationale for targeting some specific mechanisms involved in the pathophysiology of motor degeneration as observed for Vildagliptin (suppressed the nuclear factor-kB and normalized the expression of the RAGE) [281,282], Sitagliptin (anti-inflammatory and anti-apoptotic properties; enhanced the expression of brain-derived neurotrophic factor) [283,284], Linagliptin (increased the levels of superoxide dismutase, catalase, and glutathione; elevated the levels of striatal dopamine; reduced the levels of proinflammatory cytokines) [285,286], and, with some controversial results, Saxagliptin [287,288]. Translating preclinic data into clinical potential is challenging. No specific clinical trials have been conducted, but data from real-life studies reported a relevant reduction in PD risk in patients with T2D on any DPP-IVis [289,290]. A relevant revitalizing effect of DPP-IVis against aging-related nigrostriatal deterioration has also been reported [291]. These positive effects may result from DPP-IVis capability to enhance dopamine synthesis in the basal ganglia [292]. However, controversial results were also published, and a recent meta-analysis did not confirm the therapeutical potential of DPP-IVis [293], over other agents, such as pioglitazone [276].
GLP-1RAs are promising agents. As mentioned above, Exenatide provided reliable evidence of improving motor and non-motor symptoms in PD, but its role in reducing the pathophysiology and background brain injury in humans currently needs to be clarified [294]. Promising results have also been elucidated by a preclinical study with Dulaglutide, in which GLP-1RAs were demonstrated to reduce neuroinflammation and promote neurodegeneration [295]. However, more investigation is essential to clarify several issues, including the potential for preventing the onset of degenerative diseases, including PD, in predisposing clinical conditions, such as T2D, efficacy and safety, and durability of treatment in the early and late-stage disease. With these purposes, ongoing trials are investigating the role of GLP-1RA analogs in PD; these include Liraglutide (NCT02953665), Semaglutide (NCT03659682), Exenatide again (NCT04232969, 48-week study; NCT04154072-NCT04305002, early-stage PD), and Lixisenatide (NCT03439943). It should be considered that GLP-1RAs induce relevant weight loss and reduce appetite, thus limiting their use in patients with low-weight-related concerns, undernourishment, or reduced motivation for food intake. GLP-1RA-associated adverse events are usually mild-to-moderate and self-limiting over a few weeks of treatment. Additionally, concomitant drugs (e.g., metformin) may increase the risk and severity of gastrointestinal adverse events. Adequate titration of GLP-1RAs is essential to minimize the risk of adverse events and to manage better side effects once they have occurred. A lower effective dose should be prescribed in sensitive patients, and in the case of persisting or relapsing adverse events, the switch to other GLP-1RAs or other classes of drugs is recommended. The potential for GLP-1RA-related retinal injury should also be considered.
More recently, the glucose-dependent insulinotropic peptide (GIP) has gained endorsement as a promising therapy in T2D. Dual agonists GIP/GLP-1RA, rather than single GIP agonists, have been shown to improve glucose control and induce relevant weight loss significantly better than previously observed with GLP-1RAs alone [296,297,298]. Moreover, promising results have been obtained by experimental models in mice where dual agonists compared to single GLP-1RAs attenuated more significantly relevant signs of neuroinflammation and neurodegeneration (such as α-synuclein) and boosted regenerative stimuli [299,300,301]. It is expected that dual GIP/GLP-1RA agonists may have a therapeutic role in such neurodegenerative disorders [302,303], and specific clinical trials are urgently needed.
SGLT2is impairs glucose resorption at the level of the proximal tubule in the kidney [304]. Thanks to this mechanism, SGLT2is induce a relevant reduction in renal glucose resorption threshold with a relevant increase in the rate of daily renal glucose excretion (glycosuria) [305], exerting an insulin-independent anti-hyperglycemic effect [306,307]. SGLT2is and GLP-1RAs [308] provided evidence to improve relevant extra-glycemic outcomes in T2D, especially at the cardiovascular and renal levels [309]. SGLT2is is demonstrated to affect glucose metabolism positively, provide more efficient energy utilization at the cellular site, and reduce oxidative stress and inflammation, which could be considered relevant targets for treating neurodegenerative diseases [310]. Dapagliflozin (1 mg/kg/day, for 3 weeks) improved motor coordination, diminished the expression of α-synuclein and related pathological alterations, augmented the level of expression of tyrosine hydroxylase and, consequently, dopamine in the basal ganglia, and reduced oxidative stress and neuroinflammation (by decreasing the activity of the NF-κB pathway and TNF-α levels) of a PD rodent model [311]. Similar findings were obtained with Empagliflozin [312], which also reduced endoplasmic reticulum stress and improved autophagy in rodents [313,314]. More recently, a computational study found that SGLT2is (Canagliflozin and Empagliflozin) displayed binding affinity and stability to the distal ubiquitin-binding domain, serving as possible inhibitors of the ubiquitin-specific protease 30 or USP30 that plays a crucial role in the pathogenesis of mitochondrial dysfunction in PD [315]. Real-world data show that SGLT2is can work to prevent neurodegenerative diseases in T2D [316], but available data did not include specific studies assessing the potential benefit of this class of medications on neurological outcomes [317].
Table 4.
Summary of potential effects of approved treatments for type 2 diabetes and chronic disorders characterized by impaired dopamine activity/metabolism on specific disease-related outcomes. The present list includes the most common approved medication summarized by specific therapeutic areas and conditions for which the drug has been approved.
Table 4.
Summary of potential effects of approved treatments for type 2 diabetes and chronic disorders characterized by impaired dopamine activity/metabolism on specific disease-related outcomes. The present list includes the most common approved medication summarized by specific therapeutic areas and conditions for which the drug has been approved.
| List of Medications | Therapeutic Area | Approved for | Mechanism of Action | Beneficial Effects * | Detrimental Effects * |
|---|---|---|---|---|---|
| Metformin | Diabetology | Diabetes mellitus | AMPK activator | Enhancement of cellular energy metabolism, improvement of autophagy and mitochondrial performance and redox homeostasis, anti-inflammatory effect, reduction in β-secretase 1 expression (AMPK activation) [244,245,246,247,248,249,250] | Dose-dependent adverse effects (abdominal pain or discomfort, nausea, diarrhea), impaired intestinal adsorption of Vitamin B12 [251,252,253] |
| Acarbose | Diabetology | Diabetes mellitus | Intestine α-glucosidase inhibitor | Reduction in synthesis of biomarkers associated with adverse outcomes (proinflammatory cytokines, microRNA 10a-5p) [254,255] | Adverse intestinal effects |
| Pioglitazone | Diabetology | Diabetes mellitus | PPAR-γ agonism | Anti-inflammatory and anti-atherosclerotic properties, insulin-sensitizing effect, attenuation of neuroinflammation [264,265,266,267,268,269,270,271,272,273,274] | Weight gain, water and sodium retention, intensive monitoring, or contraindication in case of heart failure, renal insufficiency, and macular edema |
| Gliptins | Diabetology | Diabetes mellitus | DPP-IV inhibitors | Suppression of NFkB, reduction in the expression of RAGE, anti-inflammatory and anti-apoptotic properties, enhancement of brain-derived neurotrophic factors, reinforcement of anti-oxidative systems, increase in striatal dopamine synthesis [281,282,283,284,285,286,287,288,289,290,291,292] | - |
| GLP-1RAs | Diabetology | Diabetes mellitus | GLP-1 agonism | Contrasting nigrostriatal injury and promoting neurogenesis, improvement of neuroinflammation and neuronal metabolic activity [118,119,120,121,122,123,124,125] | Relevant weight loss and reduction in appetite, potential for retinal injury |
| Gliflozins | Diabetology | Diabetes mellitus | SGLT2 inhibitors | Improvement of energy utilization, reduction in oxidative stress and neuroinflammation, improvement of endoplasmic stress and autophagy, potential for exogen USP30 inhibitor [310,311,312,313,314,315,316,317] | Potential risk of genitourinary infections, hypotension, dehydration, and rapid decline of renal function, especially in older patients |
| Secretagogues (Sulphonylureas, Glinides) | Diabetology | Diabetes mellitus | K inward channel inhibitors | Relevant improvement in short-term glucose control [318] | High risk of hypoglycemia, short durability, lack of evidence of extra-glycemic benefits, increased risk of dementia [319] |
| Cabergoline, Bromocriptine, Apomorphine, Pramipexole, Rotigotine | Endocrinology/ Neurology | Diabetes mellitus Parkinson’s disease | Dopamine agonism | Improved glucose control; improved motor symptoms; reduced oxidative stress; possible CV benefits [226,227,228,229,230,231,232,233,234,235] | Mitral valve damage; impulse control disorders; short-term efficacy |
| Entacapone, Tolcapone, Opicapone | Neurology | Parkinson’s disease | COMT inhibitors | Not well established | - |
| Rivastigmine | Neurology | Parkinson’s disease | Acetylcholinesterase inhibitor | Not well established | - |
| Amantadine | Neurology | Parkinson’s disease | Dopamine enhancer | Suppression of glucagon synthesis and stimulation of insulin release in response to oral glucose load [320] | Hypoglycemia (?) |
| Istradefylline | Neurology | Parkinson’s disease | Adenosine (A2A) receptor antagonists | Potential for relevant impairment of intestinal glucose absorption (amelioration of non-fasting glycemia) [321] | - |
| Pimavanserin | Neurology | Parkinson’s disease | Serotonin (5-HT2A) receptor inverse agonism | Mitigation of appetite, delaying gastric emptying, weight loss [322] | - |
| Safinamide | Neurology | Parkinson’s disease | MAO-B reversible inhibitor | - | Potential affection of insulin secretion, apoptosis of β-cells (hyperglycemia and risk of new-onset T2D) [323] |
| Deutetrabenazine, Tetrabenazine | Neurology | Huntington’s disease | VMAT2 reversible inhibitors | Neutral effect on glucose and metabolic parameters [324] | Slight weight gain [325] |
| Methylphenidate, Lisdexamfetamine, Atomoxetine | Psychiatry | ADHD (adults) | Noradrenaline and dopamine reuptake inhibitors | Antidepressant effect, significant improvement of eating disorders, weight loss, improved glucose control [326] | Gastrointestinal discomfort, weight loss, or inability to gain weight |
| Buprenorphine | Neurology | Addictions | Opiate (mu) receptor partial agonism | Restriction of sugar consumption, caloric intake, and weight loss [327] | Constipation, nausea, and vomiting |
| Lofexidine | Psychiatry | Addictions | Central adrenergic (α2) receptor agonism | - | Enhancement of glucagon secretion, reduction in insulin secretion, lipolysis, gluconeogenesis, hyperglycemia [328] |
| Naltrexone | Psychiatry | Addictions | Opiate (mu) receptor antagonism | Relevant attenuation of impulsive eating and purging behaviors, weight loss, improvement of glucose control in patients with diabetes [329] | Constipation, nausea |
| Buprenorphine/Naloxone | Psychiatry | Addictions | Combining partial agonism and antagonism on opiate (mu) receptor | Restriction of sugar consumption, caloric intake, and weight loss [327] | Constipation, nausea, and vomiting |
* Beneficial and detrimental effects are intended as mutual effects of concomitant medications on disease-related outcomes (e.g., in patients with T2D and PD, beneficial or harmful neurologic effects have been described for antihyperglycemic drugs, and vice versa). Abbreviations: AMPK = AMP-activated protein kinase; RNA = ribonucleic acid; PPAR = peroxisome-proliferator-activated receptor; DPP = dipeptidyl peptidase; GLP-1 = glucagon-like peptide-1; SGLT2 = sodium-glucose (co)transporter 2; USP30 = ubiquitin-specific protease 30; COMT = catechol-O-methyltransferase; MAO = monoamine oxidase; VAMT = vesicular monoamine transporter; ADHD = attention-deficit/hyperactivity disorder.
6. Conclusions and Future Perspectives
Dopamine signaling is involved in the fine regulation of several physiologic mechanisms that control cerebral functions, cognition, eating behaviors and reward, maintenance of glucose balance, and retinal, renal, and cardiovascular homeostasis. Impaired dopamine synthesis, metabolism, or activity is associated with neurological and psychiatric diseases, impaired glucose metabolism and T2D, and diabetes-related chronic complications. Evidence suggests that reinforcing dopamine signaling has a therapeutic role in T2D. The therapeutic implication should be better investigated in patients with dopamine-related disorders, such as PD, HD, addictions, and ADHD, as they are exposed to an increased risk of T2D, indicating the existence of a cross-link among these conditions. Understanding the potential interaction between pharmacological interventions in such disorders may ameliorate the management of patients, but specific trials are needed to confirm the therapeutic potential of certain medications. Last, emerging evidence suggests that dopamine imbalance is involved in developing chronic diabetes-related complications, and targeting dopamine metabolism would have the rationale for diagnostic and therapeutic purposes, as found in diabetic retinopathy.
Author Contributions
G.L. conceived the review; G.L. and A.D.T. drafted the manuscript; E.G., V.T., V.A.G., M.I., and O.D. read the text and provided feedback. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
L-DihydrOxyPhenylAlanine = L-DOPA; Dopamine Receptor = DR; Ventral Tegmental Area = VTA; Nucleus Accumbens = NAc; Prefrontal Cortex = PC; Amygdala = AMY; Parkinson’s Disease = PD; Type 2 Diabetes (mellitus) = T2D; Heart Failure = HF; Glucagon-Like Peptide-1 Receptor Agonists = GLP-1RAs; Sodium-Glucose (co)Transporter 2 Inhibitors (SGLT2is); Central Nervous System = CNS; Insulin Receptor Substrate 2 = IRS-2; Advanced Glycation End Products = AGEs; Advanced Glycation End Product Receptor = RAGE.
References
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Dopamine (accessed on 1 August 2023).
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Boudier-Revéret, M.; Choo, Y.J.; Chang, M.C. Association between Chronic Pain and Alterations in the Mesolimbic Dopaminergic System. Brain Sci. 2020, 10, 701. [Google Scholar] [CrossRef]
- Yadav, S.K.; Prakash, J.; Chouhan, S.; Westfall, S.; Verma, M.; Singh, T.D.; Singh, S.P. Comparison of the neuroprotective potential of Mucuna pruriens seed extract with estrogen in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mice model. Neurochem. Int. 2014, 65, 1–13. [Google Scholar] [CrossRef]
- Guzmán, F. The Four Dopamine Pathways Relevant to Antipsychotics Pharmacology. Available online: http://psychopharmacologyinstitute.com/antipsychotics-videos/dopamine-pathways-antipsychotics-pharmacology/ (accessed on 15 November 2016).
- Quickel, K.E., Jr.; Feldman, J.M.; Lebovitz, H.E. Enhancement of insulin secretion in adult onset diabetics by methysergide maleate: Evidence for an endogenous biogenic monoamine mechanism as a factor in the impaired insulin secretion in diabetes mellitus. J. Clin. Endocrinol. Metab. 1971, 33, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, I. Insulin secretion. Its regulation by monoamines and acid amyloglucosidase. Acta Physiol. Scand. Suppl. 1971, 372, 1–47. [Google Scholar]
- Lebovitz, H.E.; Feldman, J.M. Pancreatic biogenic amines and insulin secretion in health and disease. Fed. Proc. 1973, 32, 1797–1802. [Google Scholar] [PubMed]
- Gagliardino, J.J.; Iturriza, F.C.; Hernandez, R.E.; Zieher, L.M. Effect of catecholamines precursors on insulin secretion. Endocrinology 1970, 87, 823–825. [Google Scholar] [CrossRef] [PubMed]
- Quickel, K.E., Jr.; Feldman, J.M.; Lebovitz, H.E. Inhibition of insulin secretion by serotonin and dopamine: Species variation. Endocrinology 1971, 89, 1295–1302. [Google Scholar] [CrossRef]
- Ericson, L.E.; Håkanson, R.; Lundquist, I. Accumulation of dopamine in mouse pancreatic B-cells following injection of L-DOPA. Localization to secretory granules and inhibition of insulin secretion. Diabetologia 1977, 13, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furman, B.L.; Gerich, J.E. Dopaminergic suppression of pancreatic somatostatin secretion. Acta Endocrinol. 1982, 101, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Jetton, T.L.; Liang, Y.; Cincotta, A.H. Systemic treatment with sympatholytic dopamine agonists improves aberrant β-cell hyperplasia and GLUT2, glucokinase, and insulin immunoreactive levels in ob/ob mice. Metabolism 2001, 50, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Ustione, A.; Piston, D.W. Dopamine synthesis and D3 receptor activation in pancreatic β-cells regulates insulin secretion and intracellular [Ca(2+)] oscillations. Mol. Endocrinol. 2012, 26, 1928–1940. [Google Scholar] [CrossRef]
- Barnett, A.H.; Chapman, C.; Gailer, K.; Hayter, C.J. Effect of bromocriptine on maturity onset diabetes. Postgrad. Med. J. 1980, 56, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Garcia Barrado, M.J.; Iglesias Osma, M.C.; Blanco, E.J.; Carretero Hernández, M.; Sánchez Robledo, V.; Catalano Iniesta, L.; Carrero, S.; Carretero, J. Dopamine modulates insulin release and is involved in the survival of rat pancreatic beta cells. PLoS ONE 2015, 10, e0123197. [Google Scholar] [CrossRef] [PubMed]
- Tomaschitz, A.; Ritz, E.; Kienreich, K.; Pieske, B.; März, W.; Boehm, B.O.; Drechsler, C.; Meinitzer, A.; Pilz, S. Circulating dopamine and C-peptide levels in fasting nondiabetic hypertensive patients: The Graz Endocrine Causes of Hypertension study. Diabetes Care 2012, 35, 1771–1773. [Google Scholar] [CrossRef]
- Simpson, N.; Maffei, A.; Freeby, M.; Burroughs, S.; Freyberg, Z.; Javitch, J.; Leibel, R.L.; Harris, P.E. Dopamine-mediated autocrine inhibitory circuit regulating human insulin secretion in vitro. Mol. Endocrinol. 2012, 26, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Kopf, D.; Gilles, M.; Paslakis, G.; Medlin, F.; Lederbogen, F.; Lehnert, H.; Deuschle, M. Insulin secretion and sensitivity after single-dose amisulpride, olanzapine or placebo in young male subjects: Double blind, cross-over glucose clamp study. Pharmacopsychiatry 2012, 45, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Maffei, A.; Segal, A.M.; Alvarez-Perez, J.C.; Garcia-Ocaña, A.; Harris, P.E. Anti-incretin, Anti-proliferative Action of Dopamine on β-Cells. Mol. Endocrinol. 2015, 29, 542–557. [Google Scholar] [CrossRef]
- Tavares, G.; Rosendo-Silva, D.; Simões, F.; Eickhoff, H.; Marques, D.; Sacramento, J.F.; Capucho, A.M.; Seiça, R.; Conde, S.V.; Matafome, P. Circulating Dopamine Is Regulated by Dietary Glucose and Controls Glucagon-like 1 Peptide Action in White Adipose Tissue. Int. J. Mol. Sci. 2023, 24, 2464. [Google Scholar] [CrossRef]
- Chien, H.Y.; Chen, S.M.; Li, W.C. Dopamine receptor agonists mechanism of actions on glucose lowering and their connections with prolactin actions. Front. Clin. Diabetes Healthc. 2023, 4, 935872. [Google Scholar] [CrossRef] [PubMed]
- Ganic, E.; Johansson, J.K.; Bennet, H.; Fex, M.; Artner, I. Islet-specific monoamine oxidase A and B expression depends on MafA transcriptional activity and is compromised in type 2 diabetes. Biochem. Biophys. Res. Commun. 2015, 468, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/gene/389692 (accessed on 31 August 2023).
- Oetjen, E.; Blume, R.; Cierny, I.; Schlag, C.; Kutschenko, A.; Krätzner, R.; Stein, R.; Knepel, W. Inhibition of MafA transcriptional activity and human insulin gene transcription by interleukin-1beta and mitogen-activated protein kinase kinase kinase in pancreatic islet beta cells. Diabetologia 2007, 50, 1678–1687. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schmidt, R.E.; Geller, D.M.; Johnson, E.M., Jr. Characterization of increased plasma dopamine-β-hydroxylase activity in rats with experimental diabetes. Diabetes 1981, 30, 416–423. [Google Scholar] [CrossRef]
- Hurst, J.H.; Nisula, B.C.; Stolk, J.M. Circulating dopamine-β-hydroxylase in the rat: Importance of altered disposal pathways in experimental diabetes. J. Pharmacol. Exp. Ther. 1982, 220, 108–114. [Google Scholar] [PubMed]
- Lozovsky, D.; Saller, C.F.; Kopin, I.J. Dopamine receptor binding is increased in diabetic rats. Science 1981, 214, 1031–1033. [Google Scholar] [CrossRef]
- Kwok, R.P.; Juorio, A.V. Concentration of striatal tyramine and dopamine metabolism in diabetic rats and effect of insulin administration. Neuroendocrinology 1986, 43, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.P.; Walls, E.K.; Juorio, A.V. The concentration of dopamine, 5-hydroxytryptamine, and some of their acid metabolites in the brain of genetically diabetic rats. Neurochem. Res. 1985, 10, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.; Bernardes, M.; Harris, P.E.; Maffei, A. Gastrointestinal dopamine as an anti-incretin and its possible role in bypass surgery as therapy for type 2 diabetes with associated obesity. Minerva Endocrinol. 2016, 41, 43–56. [Google Scholar]
- Leblanc, H.; Lachelin, G.C.; Abu-Fadil, S.; Yen, S.S. The effect of dopamine infusion on insulin and glucagon secretion in man. J. Clin. Endocrinol. Metab. 1977, 44, 196–198. [Google Scholar] [CrossRef]
- Pernet, A.; Hammond, V.A.; Blesa-Malpica, G.; Burrin, J.; Orskov, H.; Alberti, K.G.; Johnston, D.G. The metabolic effects of dopamine in man. Eur. J. Clin. Pharmacol. 1984, 26, 23–28. [Google Scholar] [CrossRef]
- Keck, F.S.; Foldenauer, A.; Wolf, C.F.; Zeller, G.; Meyerhoff, C.; Dolderer, M.; Loos, U.; Pfeiffer, E.F. Differential effects of dopamine on glucoregulatory hormones in rats. Diabetes Res. Clin. Pract. 1990, 8, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of diabetes and diabetes-related complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef]
- Gaster, B.; Hirsch, I.B. The effects of improved glycemic control on complications in type 2 diabetes. Arch. Intern. Med. 1998, 158, 134–140. [Google Scholar] [CrossRef]
- Nørgaard, C.H.; Mosslemi, M.; Lee, C.J.; Torp-Pedersen, C.; Wong, N.D. The Importance and Role of Multiple Risk Factor Control in Type 2 Diabetes. Curr. Cardiol. Rep. 2019, 21, 35. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Colette, C.; Schlienger, J.L.; Bauduceau, B.; ROwens, D. Glucocentric risk factors for macrovascular complications in diabetes: Glucose ‘legacy’ and ‘variability’-what we see, know and try to comprehend. Diabetes Metab. 2019, 45, 401–408. [Google Scholar] [CrossRef]
- Ceriello, A.; Prattichizzo, F. Variability of risk factors and diabetes complications. Cardiovasc. Diabetol. 2021, 20, 101. [Google Scholar] [CrossRef]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. Addendum. 3. Prevention or Delay of Type 2 Diabetes and Associated Comorbidities: Standards of Care in Diabetes–2023. Diabetes Care 2023, 46 (Suppl. S1), S41–S48, Erratum in Diabetes Care 2023, 46, 1716–1717. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Drexel, H.; Jornayvaz, F.R.; Pataky, Z.; Seferović, P.M.; Wanner, C. Cardiovascular outcomes trials: A paradigm shift in the current management of type 2 diabetes. Cardiovasc. Diabetol. 2022, 21, 144. [Google Scholar] [CrossRef] [PubMed]
- Dardano, A.; Miccoli, R.; Bianchi, C.; Daniele, G.; Del Prato, S. Invited review. Series: Implications of the recent CVOTs in type 2 diabetes: Which patients for GLP-1RA or SGLT-2 inhibitor? Diabetes Res. Clin. Pract. 2020, 162, 108112. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Siwik, D.; Gajewska, M.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J.; Postuła, M.; Eyileten, C. Early Biomarkers of Neurodegenerative and Neurovascular Disorders in Diabetes. J. Clin. Med. 2020, 9, 2807. [Google Scholar] [CrossRef] [PubMed]
- Popova, E. Role of Dopamine in Retinal Function. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. Available online: https://www.ncbi.nlm.nih.gov/books/NBK561740/ (accessed on 28 May 2020).
- Jain, V.; Liang, P.J.M.; Raja, S.; Mikhael, M.; Cameron, M.A. Light activation of the dopaminergic system occurs after eye-opening in the mouse retina. Front. Ophthalmol. 2023, 3, 1184627. [Google Scholar] [CrossRef]
- Wubben, T.J. Dopamine and Early Retinal Dysfunction in Diabetes: Insights From a Phase 1 Study. Diabetes 2020, 69, 1339–1340. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.S.; Khayat, C.T.; Feola, A.J.; Win, A.S.; Grubman, A.R.; Chesler, K.C.; He, L.; Dixon, J.A.; Kern, T.S.; Iuvone, P.M.; et al. Diabetic rats with high levels of endogenous dopamine do not show retinal vascular pathology. Front. Neurosci. 2023, 17, 1125784. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Jeon, H.Y.; Lee, A.J.; Kim, M.; Ha, K.S. Dopamine ameliorates hyperglycemic memory-induced microvascular dysfunction in diabetic retinopathy. FASEB J. 2022, 36, e22643. [Google Scholar] [CrossRef] [PubMed]
- Upreti, S.; Sen, S.; Nag, T.C.; Ghosh, M.P. Insulin like growth factor-1 works synergistically with dopamine to attenuate diabetic retinopathy by downregulating vascular endothelial growth factor. Biomed. Pharmacother. 2022, 149, 112868. [Google Scholar] [CrossRef]
- Hendrick, A.; Smith, J.; Stelton, C.; Barb, S.; Yan, J.; Cribbs, B.; Jain, N.; Yeh, S.; Hubbard, G.B.; He, L.; et al. Dopamine metabolite levels in the vitreous of diabetic and non-diabetic humans. Exp. Eye Res. 2020, 195, 108040. [Google Scholar] [CrossRef]
- Motz, C.T.; Chesler, K.C.; Allen, R.S.; Bales, K.L.; Mees, L.M.; Feola, A.J.; Maa, A.Y.; Olson, D.E.; Thule, P.M.; Iuvone, P.M.; et al. Novel Detection and Restorative Levodopa Treatment for Preclinical Diabetic Retinopathy. Diabetes 2020, 69, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, Y.; Zhao, H.; Xu, J.; Yang, X. Association Between Pathophysiological Mechanisms of Diabetic Retinopathy and Parkinson’s Disease. Cell. Mol. Neurobiol. 2022, 42, 665–675. [Google Scholar] [CrossRef]
- Chen, P.; Li, J.; Li, Z.; Yu, D.; Ma, N.; Xia, Z.; Meng, X.; Liu, X. 18F-FP-CIT dopamine transporter PET findings in the striatum and retina of type 1 diabetic rats. Ann. Nucl. Med. 2023, 37, 219–226. [Google Scholar] [CrossRef]
- Bethel, M.A.; Diaz, R.; Castellana, N.; Bhattacharya, I.; Gerstein, H.C.; Lakshmanan, M.C. HbA1c Change and Diabetic Retinopathy During GLP-1 Receptor Agonist Cardiovascular Outcome Trials: A Meta-analysis and Meta-regression. Diabetes Care 2021, 44, 290–296. [Google Scholar] [CrossRef]
- Kapoor, I.; Sarvepalli, S.M.; D’Alessio, D.; Grewal, D.S.; Hadziahmetovic, M. GLP-1 receptor agonists and diabetic retinopathy: A meta-analysis of randomized clinical trials. Surv. Ophthalmol. 2023, 68, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Hernández, A.; Figuero-Pérez, L.; Cruz-Hernandez, J.J.; González Sarmiento, R.; Usategui-Martin, R.; Miramontes-González, J.P. Dopamine Receptors and the Kidney: An Overview of Health- and Pharmacological-Targeted Implications. Biomolecules 2021, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Ohashi, N.; Ishigaki, S.; Isobe, S.; Tsuji, N.; Fujikura, T.; Tsuji, T.; Kato, A.; Miyajima, H.; Yasuda, H. The Relationship between the Intrarenal Dopamine System and Intrarenal Renin-angiotensin System Depending on the Renal Function. Intern. Med. 2018, 57, 3241–3247. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Tomoda, F.; Koike, T.; Yamazaki, H.; Ohara, M.; Liu, H.; Kagitani, S.; Inoue, H. Imbalance of renal production between 5-hydroxytryptamine and dopamine in patients with essential hypertension complicated by microalbuminuria. Am. J. Hypertens. 2013, 26, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Yao, B.; Yang, S.; Yang, H.; Wang, S.; Fan, X.; Yin, H.; Fogo, A.B.; Moeckel, G.W.; Harris, R.C. Intrarenal dopamine inhibits progression of diabetic nephropathy. Diabetes 2012, 61, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Rodríguez, O.; Herrera-Abarca, J.E.; Ceballos-Reyes, G.; Avila-Diaz, M.; Prado-Uribe, C.; Belio-Caro, F.; Salinas-González, A.; Vega-Gomez, H.; Alvarez-Aguilar, C.; Lindholm, B.; et al. Cardiovascular and renal effects of bromocriptine in diabetic patients with stage 4 chronic kidney disease. Biomed. Res. Int. 2013, 2013, 104059. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Hofmann, B.; Dhein, S.; Gergs, U. Role of Dopamine in the Heart in Health and Disease. Int. J. Mol. Sci. 2023, 24, 5042. [Google Scholar] [CrossRef] [PubMed]
- Lokhandwala, M.F.; Barrett, R.J. Cardiovascular dopamine receptors: Physiological, pharmacological and therapeutic implications. J. Auton. Pharmacol. 1982, 2, 189–215. [Google Scholar] [CrossRef]
- Neubauer, B.; Christensen, N.J. Norepinephrine, epinephrine, and dopamine contents of the cardiovascular system in long-term diabetics. Diabetes 1976, 25, 6–10. [Google Scholar] [CrossRef]
- Vinik, A.I.; Casellini, C.; Parson, H.K.; Colberg, S.R.; Nevoret, M.L. Cardiac Autonomic Neuropathy in Diabetes: A Predictor of Cardiometabolic Events. Front. Neurosci. 2018, 12, 591. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Vaduganathan, M.; Butler, J. Glucose-Lowering Therapies and Heart Failure in Type 2 Diabetes Mellitus: Mechanistic Links, Clinical Data, and Future Directions. Circulation 2018, 137, 1060–1073. [Google Scholar] [CrossRef]
- Frishman, W.H.; Grewall, P. Serotonin and the heart. Ann. Med. 2000, 32, 195–209. [Google Scholar] [CrossRef]
- Mokhles, M.M.; Trifirò, G.; Dieleman, J.P.; Haag, M.D.; van Soest, E.M.; Verhamme, K.M.; Mazzaglia, G.; Herings, R.; de Luise, C.; Ross, D.; et al. The risk of new onset heart failure associated with dopamine agonist use in Parkinson’s disease. Pharmacol. Res. 2012, 65, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Fouad Shalaby, M.A.; Abd El Latif, H.A.; El Yamani, M.; Galal, M.A.; Kamal, S.; Sindi, I.; Masaood, R. Therapeutic activity of sarpogrelate and dopamine D2 receptor agonists on cardiovascular and renal systems in rats with alloxan-induced diabetes. BMC Pharmacol. Toxicol. 2021, 22, 64. [Google Scholar] [CrossRef]
- Giamouzis, G.; Butler, J.; Starling, R.C.; Karayannis, G.; Nastas, J.; Parisis, C.; Rovithis, D.; Economou, D.; Savvatis, K.; Kirlidis, T.; et al. Impact of dopamine infusion on renal function in hospitalized heart failure patients: Results of the Dopamine in Acute Decompensated Heart Failure (DAD-HF) Trial. J. Card. Fail. 2010, 16, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.K.; Butler, J.; Karayannis, G.; Starling, R.C.; Filippatos, G.; Wolski, K.; Parissis, J.; Parisis, C.; Rovithis, D.; Koutrakis, K.; et al. Efficacy and safety of high dose versus low dose furosemide with or without dopamine infusion: The Dopamine in Acute Decompensated Heart Failure II (DAD-HF II) trial. Int. J. Cardiol. 2014, 172, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Vaishnav, J.; Kalathiya, R.; Hu, J.R.; Miller, J.; Shah, N.; Hill, T.; Sharp, M.; Tsao, A.; Alexander, K.M.; et al. Randomized Evaluation of Heart Failure With Preserved Ejection Fraction Patients With Acute Heart Failure and Dopamine: The ROPA-DOP Trial. JACC Heart Fail. 2018, 6, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.; Tiberi, M. The Intersection of Central Dopamine System and Stroke: Potential Avenues Aiming at Enhancement of Motor Recovery. Front. Synaptic Neurosci. 2018, 10, 18. [Google Scholar] [CrossRef]
- Vitrac, C.; Nallet-Khosrofian, L.; Iijima, M.; Rioult-Pedotti, M.S.; Luft, A. Endogenous dopamine transmission is crucial for motor skill recovery after stroke. IBRO Neurosci. Rep. 2022, 13, 15–21. [Google Scholar] [CrossRef]
- Stinear, C.M. Dopamine for motor recovery after stroke: Where to from here? Lancet Neurol. 2019, 18, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Sami, M.B.; Faruqui, R. The effectiveness of dopamine agonists for treatment of neuropsychiatric symptoms post brain injury and stroke. Acta Neuropsychiatr. 2015, 27, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.; Martínez-Vega, M.; Del Pozo, A.; Muneta-Arrate, I.; Gómez-Soria, A.; Muguruza, C.; de Hoz-Rivera, M.; Romero, A.; Silva, L.; Callado, L.F.; et al. The Role of the Dopamine System in Post-Stroke Mood Disorders in Newborn Rats. Int. J. Mol. Sci. 2023, 24, 3229. [Google Scholar] [CrossRef]
- Ford, G.A.; Bhakta, B.B.; Cozens, A.; Cundill, B.; Hartley, S.; Holloway, I.; Meads, D.; Pearn, J.; Ruddock, S.; Sackley, C.M.; et al. Dopamine Augmented Rehabilitation in Stroke (DARS): A multicentre double-blind, randomised controlled trial of co-careldopa compared with placebo, in addition to routine NHS occupational and physical therapy, delivered early after stroke on functional recovery. Effic. Mech. Eval. 2019, 6, 1–172. [Google Scholar]
- Scheidtmann, K.; Fries, W.; Müller, F.; Koenig, E. Effect of levodopa in combination with physiotherapy on functional motor recovery after stroke: A prospective, randomised, double-blind study. Lancet 2001, 358, 787–790. [Google Scholar] [CrossRef]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell. Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- O’Reilly, S.; Loncin, M.; Cooksey, B. Dopamine and basal ganglia disorders. Neurology 1965, 15, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Váradi, C. Clinical Features of Parkinson’s Disease: The Evolution of Critical Symptoms. Biology 2020, 9, 103. [Google Scholar] [CrossRef]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Chohan, H.; Senkevich, K.; Patel, R.K.; Bestwick, J.P.; Jacobs, B.M.; Bandres Ciga, S.; Gan-Or, Z.; Noyce, A.J. Type 2 Diabetes as a Determinant of Parkinson’s Disease Risk and Progression. Mov. Disord. 2021, 36, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Komici, K.; Femminella, G.D.; Bencivenga, L.; Rengo, G.; Pagano, G. Diabetes Mellitus and Parkinson’s Disease: A Systematic Review and Meta-Analyses. J. Park. Dis. 2021, 11, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Kim, B.; Lee, S.H.; Kim, M.K. A nationwide cohort study on diabetes severity and risk of Parkinson disease. NPJ Park. Dis. 2023, 9, 11. [Google Scholar] [CrossRef]
- Gallego, M.; Setién, R.; Izquierdo, M.J.; Casis, O.; Casis, E. Diabetes-induced biochemical changes in central and peripheral catecholaminergic systems. Physiol. Res. 2003, 52, 735–741. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gu, X.; Wang, X. α-Synuclein in Parkinson’s disease and advances in detection. Clin. Chim. Acta 2022, 529, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K. Initiation and progression of α-synuclein pathology in Parkinson’s disease. Cell. Mol. Life Sci. 2022, 79, 210. [Google Scholar] [CrossRef]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef] [PubMed]
- Asslih, S.; Damri, O.; Agam, G. Neuroinflammation as a Common Denominator of Complex Diseases (Cancer, Diabetes Type 2, and Neuropsychiatric Disorders). Int. J. Mol. Sci. 2021, 22, 6138. [Google Scholar] [CrossRef]
- Sarparanta, J.; García-Macia, M.; Singh, R. Autophagy and Mitochondria in Obesity and Type 2 Diabetes. Curr. Diabetes Rev. 2017, 13, 352–369. [Google Scholar] [CrossRef]
- Geng, X.; Lou, H.; Wang, J.; Li, L.; Swanson, A.L.; Sun, M.; Beers-Stolz, D.; Watkins, S.; Perez, R.G.; Drain, P. α-Synuclein binds the K(ATP) channel at insulin-secretory granules and inhibits insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E276–E286. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Ahrens, R.; Wu, L.; Langman, T.; Tandon, A.; Fraser, P.E. α-Synuclein Regulates Peripheral Insulin Secretion and Glucose Transport. Front. Aging Neurosci. 2021, 13, 665348. [Google Scholar] [CrossRef]
- Vidal-Martinez, G.; Yang, B.; Vargas-Medrano, J.; Perez, R.G. Could α-Synuclein Modulation of Insulin and Dopamine Identify a Novel Link Between Parkinson’s Disease and Diabetes as Well as Potential Therapies? Front. Mol. Neurosci. 2018, 11, 465. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Liu, L.; Xie, A. The Role of Insulin/IGF-1/PI3K/Akt/GSK3β Signaling in Parkinson’s Disease Dementia. Front. Neurosci. 2018, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Dong, M.; de la Monte, S.M. Brain insulin-like growth factor and neurotrophin resistance in Parkinson’s disease and dementia with Lewy bodies: Potential role of manganese neurotoxicity. J. Alzheimers Dis. 2009, 16, 585–599. [Google Scholar] [CrossRef]
- Bhattamisra, S.K.; Shin, L.Y.; Saad, H.I.B.M.; Rao, V.; Candasamy, M.; Pandey, M.; Choudhury, H. Interlink Between Insulin Resistance and Neurodegeneration with an Update on Current Therapeutic Approaches. CNS Neurol. Disord. Drug Targets 2020, 19, 174–183. [Google Scholar] [CrossRef] [PubMed]
- De Iuliis, A.; Montinaro, E.; Fatati, G.; Plebani, M.; Colosimo, C. Diabetes mellitus and Parkinson’s disease: Dangerous liaisons between insulin and dopamine. Neural Regen. Res. 2022, 17, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Kim, S.J. Insulin on activation of autophagy with integrins and syndecans against MPP+-induced α-synuclein neurotoxicity. Neurosci. Lett. 2016, 633, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Vissel, B. Therapeutic implications of how TNF links apolipoprotein E, phosphorylated tau, α-synuclein, amyloid-β and insulin resistance in neurodegenerative diseases. Br. J. Pharmacol. 2018, 175, 3859–3875. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pozo, V.A.; Tamayo-Trujillo, R.; Cadena-Ullauri, S.; Frias-Toral, E.; Guevara-Ramírez, P.; Paz-Cruz, E.; Chapela, S.; Montalván, M.; Morales-López, T.; Simancas-Racines, D.; et al. The Molecular Mechanisms of the Relationship between Insulin Resistance and Parkinson’s Disease Pathogenesis. Nutrients 2023, 15, 3585. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Mattson, M.P.; Perry, T.; Chan, S.L.; Giordano, T.; Sambamurti, K.; Rogers, J.T.; Ovadia, H.; Lahiri, D.K. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann. N. Y. Acad. Sci. 2004, 1035, 290–315. [Google Scholar] [CrossRef] [PubMed]
- Cabou, C.; Burcelin, R. GLP-1, the gut-brain, and brain-periphery axes. Rev. Diabet. Stud. 2011, 8, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Katsurada, K.; Yada, T. Neural effects of gut- and brain-derived glucagon-like peptide-1 and its receptor agonist. J. Diabetes Investig. 2016, 7 (Suppl. S1), 64–69. [Google Scholar] [CrossRef]
- Chen, X.Y.; Chen, L.; Yang, W.; Xie, A.M. GLP-1 Suppresses Feeding Behaviors and Modulates Neuronal Electrophysiological Properties in Multiple Brain Regions. Front. Mol. Neurosci. 2021, 14, 793004. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Glucagon-like peptide-1 receptors in the brain: Controlling food intake and body weight. J. Clin. Investig. 2014, 124, 4223–4226. [Google Scholar] [CrossRef] [PubMed]
- Harkavyi, A.; Abuirmeileh, A.; Lever, R.; Kingsbury, A.E.; Biggs, C.S.; Whitton, P.S. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J. Neuroinflammation 2008, 5, 19. [Google Scholar] [CrossRef]
- Bertilsson, G.; Patrone, C.; Zachrisson, O.; Andersson, A.; Dannaeus, K.; Heidrich, J.; Kortesmaa, J.; Mercer, A.; Nielsen, E.; Rönnholm, H.; et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 326–338. [Google Scholar] [CrossRef]
- Rachmany, L.; Tweedie, D.; Li, Y.; Rubovitch, V.; Holloway, H.W.; Miller, J.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age 2013, 35, 1621–1636. [Google Scholar] [CrossRef] [PubMed]
- Parthsarathy, V.; Hölscher, C. The type 2 diabetes drug liraglutide reduces chronic inflammation induced by irradiation in the mouse brain. Eur. J. Pharmacol. 2013, 700, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Edison, P. Evaluation of neuroprotective effect of glucagon-like peptide 1 analogs using neuroimaging. Alzheimers Dement. 2014, 10 (Suppl. S1), S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Brundin, P. Is exenatide the next big thing in Parkinson’s disease? J. Park. Dis. 2014, 4, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Kahan, J.; Ell, P.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J. Park. Dis. 2014, 4, 337–344. [Google Scholar] [CrossRef]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Hölscher, C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 2018, 136, 251–259. [Google Scholar] [CrossRef]
- Reich, N.; Hölscher, C. The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: An in-depth review. Front. Neurosci. 2022, 16, 970925. [Google Scholar] [CrossRef]
- Cheng, D.; Yang, S.; Zhao, X.; Wang, G. The Role of Glucagon-Like Peptide-1 Receptor Agonists (GLP-1 RA) in Diabetes-Related Neurodegenerative Diseases. Drug Des. Dev. Ther. 2022, 16, 665–684. [Google Scholar] [CrossRef]
- Chen, S.D.; Chuang, Y.C.; Lin, T.K.; Yang, J.L. Alternative role of glucagon-like Peptide-1 receptor agonists in neurodegenerative diseases. Eur. J. Pharmacol. 2023, 938, 175439. [Google Scholar] [CrossRef] [PubMed]
- Nowell, J.; Blunt, E.; Gupta, D.; Edison, P. Antidiabetic agents as a novel treatment for Alzheimer’s and Parkinson’s disease. Ageing Res. Rev. 2023, 89, 101979. [Google Scholar] [CrossRef]
- Andhale, R.; Shrivastava, D. Huntington’s Disease: A Clinical Review. Cureus 2022, 14, e28484. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Reiner, A.; Anderson, K.D.; Dure, L.S., 4th; Handelin, B.; Balfour, R.; Whetsell, W.O., Jr.; Penney, J.B.; Young, A.B. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann. Neurol. 1992, 31, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Bird, E.D. Chemical pathology of Huntington’s disease. Annu. Rev. Pharmacol. Toxicol. 1980, 20, 533–551. [Google Scholar] [CrossRef]
- Glass, M.; Dragunow, M.; Faull, R.L. The pattern of neurodegeneration in Huntington’s disease: A comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience 2000, 97, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Bibb, J.A.; Yan, Z.; Svenningsson, P.; Snyder, G.L.; Pieribone, V.A.; Horiuchi, A.; Nairn, A.C.; Messer, A.; Greengard, P. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. USA 2000, 97, 6809–6814. [Google Scholar] [CrossRef]
- Jakel, R.J.; Maragos, W.F. Neuronal cell death in Huntington’s disease: A potential role for dopamine. Trends Neurosci. 2000, 23, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kalonia, H.; Kumar, A. Huntington’s disease: Pathogenesis to animal models. Pharmacol. Rep. 2010, 62, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Illarioshkin, S.N.; Klyushnikov, S.A.; Vigont, V.A.; Seliverstov, Y.A.; Kaznacheyeva, E.V. Molecular Pathogenesis in Huntington’s Disease. Biochemistry 2018, 83, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.J.; Renoir, T.; Gray, L.J.; Hannan, A.J. Huntington’s Disease: Pathogenic Mechanisms and Therapeutic Targets. Adv. Neurobiol. 2017, 15, 93–128. [Google Scholar] [CrossRef]
- Farrer, L.A. Diabetes mellitus in Huntington disease. Clin. Genet. 1985, 27, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Ro, J.S.; Jung, H.; Kim, M.; Jeon, B.; Lee, J.Y. Increased 10-Year Prevalence of Huntington’s Disease in South Korea: An Analysis of Medical Expenditure Through the National Healthcare System. J. Clin. Neurol. 2023, 19, 147–155. [Google Scholar] [CrossRef]
- Montojo, M.T.; Aganzo, M.; González, N. Huntington’s Disease and Diabetes: Chronological Sequence of its Association. J. Huntingt. Dis. 2017, 6, 179–188. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.S.; Lerskiatiphanich, T.; Woodruff, T.M.; McCombe, P.A.; Lee, J.D. Potential mechanisms to modify impaired glucose metabolism in neurodegenerative disorders. J. Cereb. Blood Flow Metab. 2023, 43, 26–43. [Google Scholar] [CrossRef]
- Podlacha, M.; Pierzynowska, K.; Gaffke, L.; Jerzemowska, G.; Piotrowska, E.; Węgrzyn, G. Behavioral- and blood-based biomarkers for Huntington’s disease: Studies on the R6/1 mouse model with prospects for early diagnosis and monitoring of the disease. Brain Behav. Immun. Health 2022, 23, 100482. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, Y.; Zhang, Y.; Wang, W.; Ye, C. Mutant Huntingtin Impairs Pancreatic β-cells by Recruiting IRS-2 and Disturbing the PI3K/AKT/FoxO1 Signaling Pathway in Huntington’s Disease. J. Mol. Neurosci. 2021, 71, 2646–2658. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, A.C.; Gonzalez-Alegre, P.; Schultz, J.L. Diabetes Mellitus Is Associated With an Earlier Age of Onset of Huntington’s Disease. Mov. Disord. 2021, 36, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. The Metabolic Basis for Nervous System Dysfunction in Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease. Curr. Neurovascular Res. 2023, 20, 314. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys. Acta 2010, 1804, 433–439. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Han, Y.; Chen, Y.; Liu, L.; Lang, J.; Yang, C.; Luo, H.; Ning, J. Advanced glycation end-products suppress autophagy by AMPK/mTOR signaling pathway to promote vascular calcification. Mol. Cell. Biochem. 2020, 471, 91–100. [Google Scholar] [CrossRef]
- Ramasubbu, K.; Devi Rajeswari, V. Impairment of insulin signaling pathway PI3K/Akt/mTOR and insulin resistance induced AGEs on diabetes mellitus and neurodegenerative diseases: A perspective review. Mol. Cell. Biochem. 2023, 478, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Brás, I.C.; König, A.; Outeiro, T.F. Glycation in Huntington’s Disease: A Possible Modifier and Target for Intervention. J. Huntingt. Dis. 2019, 8, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millán, J.M.; Weiss, A.; Déglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, T.C.; Ho, H.L.; Kuo, C.Y.; Li, H.H.; Korolenko, T.A.; Chen, W.J.; Lai, T.J.; Ho, Y.J.; Lin, C.L. GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling. Int. J. Mol. Sci. 2018, 19, 2505. [Google Scholar] [CrossRef]
- Shawki, S.M.; Saad, M.A.; Rahmo, R.M.; Wadie, W.; El-Abhar, H.S. Liraglutide Improves Cognitive and Neuronal Function in 3-NP Rat Model of Huntington’s Disease. Front. Pharmacol. 2021, 12, 731483. [Google Scholar] [CrossRef]
- Martin, B.; Golden, E.; Carlson, O.D.; Pistell, P.; Zhou, J.; Kim, W.; Frank, B.P.; Thomas, S.; Chadwick, W.A.; Greig, N.H.; et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes 2009, 58, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.H.; Fathy, N.; Kortam, M.A.; Rabie, M.A.; Mohamed, A.F.; Kamel, A.S. Correction to: Vildagliptin Attenuates Huntington’s Disease Through Activation of GLP-1 Receptor/PI3K/Akt/BDNF Pathway in 3-Nitropropionic Acid Rat Model. Neurotherapeutics 2022, 17, 252–268, Erratum in Neurotherapeutics 2022, 19, 686. [Google Scholar] [CrossRef] [PubMed]
- El-Sahar, A.E.; Rastanawi, A.A.; El-Yamany, M.F.; Saad, M.A. Dapagliflozin improves behavioral dysfunction of Huntington’s disease in rats via inhibiting apoptosis-related glycolysis. Life Sci. 2020, 257, 118076. [Google Scholar] [CrossRef] [PubMed]
- Koutsoklenis, A.; Honkasilta, J. ADHD in the DSM-5-TR: What has changed and what has not. Front. Psychiatry 2023, 13, 1064141. [Google Scholar] [CrossRef] [PubMed]
- Salari, N.; Ghasemi, H.; Abdoli, N.; Rahmani, A.; Shiri, M.H.; Hashemian, A.H.; Akbari, H.; Mohammadi, M. The global prevalence of ADHD in children and adolescents: A systematic review and meta-analysis. Ital. J. Pediatr. 2023, 49, 48. [Google Scholar] [CrossRef] [PubMed]
- Faraone, S.V.; Larsson, H. Genetics of attention deficit hyperactivity disorder. Mol. Psychiatry 2019, 24, 562–575. [Google Scholar] [CrossRef]
- Blum, K.; Chen, A.L.; Braverman, E.R.; Comings, D.E.; Chen, T.J.; Arcuri, V.; Blum, S.H.; Downs, B.W.; Waite, R.L.; Notaro, A.; et al. Attention-deficit-hyperactivity disorder and reward deficiency syndrome. Neuropsychiatr. Dis. Treat. 2008, 4, 893–918. [Google Scholar] [CrossRef] [PubMed]
- Cortese, S.; Moreira-Maia, C.R.; St Fleur, D.; Morcillo-Peñalver, C.; Rohde, L.A.; Faraone, S.V. Association Between ADHD and Obesity: A Systematic Review and Meta-Analysis. Am. J. Psychiatry 2016, 173, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Mazor-Aronovitch, K.; Pinhas-Hamiel, O.; Pivko-Levy, D.; Modan-Moses, D.; Levek, N.; Miller, S.; Yackobovitch-Gavan, M.; Gruber, N.; Ben-Ami, M.; Stern, E.; et al. Dual diagnosis of type 1 diabetes mellitus and attention deficit hyperactivity disorder. Pediatr. Diabetes 2021, 22, 649–655. [Google Scholar] [CrossRef]
- Garcia-Argibay, M.; Li, L.; Du Rietz, E.; Zhang, L.; Yao, H.; Jendle, J.; Ramos-Quiroga, J.A.; Ribasés, M.; Chang, Z.; Brikell, I.; et al. The association between type 2 diabetes and attention- deficit/hyperactivity disorder: A systematic review, meta-analysis, and population-based sibling study. Neurosci. Biobehav. Rev. 2023, 147, 105076. [Google Scholar] [CrossRef]
- Kittel-Schneider, S.; Arteaga-Henriquez, G.; Vasquez, A.A.; Asherson, P.; Banaschewski, T.; Brikell, I.; Buitelaar, J.; Cormand, B.; Faraone, S.V.; Freitag, C.M.; et al. Non-mental diseases associated with ADHD across the lifespan: Fidgety Philipp and Pippi Longstocking at risk of multimorbidity? Neurosci. Biobehav. Rev. 2022, 132, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.P.; Bolat, G.U.; Bolat, H.; Matijasevich, A.; Santos, I.S.; Silveira, R.C.; Procianoy, R.S.; Rohde, L.A.; Moreira-Maia, C.R. Attention-Deficit/Hyperactivity Disorder and Very Preterm/Very Low Birth Weight: A Meta-analysis. Pediatrics 2018, 141, e20171645. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Mechanick, J.I.; Brett, E.M.; Garber, A.J.; Hurley, D.L.; Jastreboff, A.M.; Nadolsky, K.; Pessah-Pollack, R.; Plodkowski, R.; Reviewers of the AACE/ACE Obesity Clinical Practice Guidelines. American association of clinical endocrinologists and american college of endocrinology comprehensive clinical practice guidelines for Medical care of patients with obesity. Endocr. Pract. 2016, 22 (Suppl. S3), 1–203. [Google Scholar] [CrossRef]
- Villa, F.M.; Crippa, A.; Rosi, E.; Nobile, M.; Brambilla, P.; Delvecchio, G. ADHD and eating disorders in childhood and adolescence: An updated minireview. J. Affect. Disord. 2023, 321, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Nazar, B.P.; Bernardes, C.; Peachey, G.; Sergeant, J.; Mattos, P.; Treasure, J. The risk of eating disorders comorbid with attention-deficit/hyperactivity disorder: A systematic review and meta-analysis. Int. J. Eat. Disord. 2016, 49, 1045–1057. [Google Scholar] [CrossRef]
- Catalá-López, F.; Hutton, B.; Núñez-Beltrán, A.; Page, M.J.; Ridao, M.; Macías Saint-Gerons, D.; Catalá, M.A.; Tabarés-Seisdedos, R.; Moher, D. The pharmacological and non-pharmacological treatment of attention deficit hyperactivity disorder in children and adolescents: A systematic review with network meta-analyses of randomised trials. PLoS ONE 2017, 12, e0180355. [Google Scholar] [CrossRef]
- Wang, G.J.; Volkow, N.D.; Fowler, J.S. The role of dopamine in motivation for food in humans: Implications for obesity. Expert. Opin. Ther. Targets 2002, 6, 601–609. [Google Scholar] [CrossRef]
- Danilovich, N.; Mastrandrea, L.D.; Cataldi, L.; Quattrin, T. Methylphenidate decreases fat and carbohydrate intake in obese teenagers. Obesity 2014, 22, 781–785. [Google Scholar] [CrossRef]
- Goldfield, G.S.; Lorello, C.; Doucet, E. Methylphenidate reduces energy intake and dietary fat intake in adults: A mechanism of reduced reinforcing value of food? Am. J. Clin. Nutr. 2007, 86, 308–315. [Google Scholar] [CrossRef]
- Liu, L.L.; Li, B.M.; Yang, J.; Wang, Y.W. Does dopaminergic reward system contribute to explaining comorbidity obesity and ADHD? Med. Hypotheses 2008, 70, 1118–1120. [Google Scholar] [CrossRef]
- Charach, G.; Karniel, E.; Grosskopf, I.; Rabinovich, A.; Charach, L. Methylphenidate has mild hyperglycemic and hypokalemia effects and increases leukocyte and neutrophil counts. Medicine 2020, 99, e20931. [Google Scholar] [CrossRef] [PubMed]
- Skibicka, K.P. The central GLP-1: Implications for food and drug reward. Front. Neurosci. 2013, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- van Bloemendaal, L.; IJzerman, R.G.; Ten Kulve, J.S.; Barkhof, F.; Konrad, R.J.; Drent, M.L.; Veltman, D.J.; Diamant, M. GLP-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes 2014, 63, 4186–4196. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, R.H.; Anefors, C.; Bergquist, F.; Nissbrandt, H.; Skibicka, K.P. Dopamine signaling in the amygdala, increased by food ingestion and GLP-1, regulates feeding behavior. Physiol. Behav. 2014, 136, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Alavi, S.S.; Ferdosi, M.; Jannatifard, F.; Eslami, M.; Alaghemandan, H.; Setare, M. Behavioral Addiction versus Substance Addiction: Correspondence of Psychiatric and Psychological Views. Int. J. Prev. Med. 2012, 3, 290–294. [Google Scholar] [PubMed]
- Chen, C.Y.; Storr, C.L.; Anthony, J.C. Early-onset drug use and risk for drug dependence problems. Addict. Behav. 2009, 34, 319–322. [Google Scholar] [CrossRef]
- Haug, S.; Castro, R.P.; Kwon, M.; Filler, A.; Kowatsch, T.; Schaub, M.P. Smartphone use and smartphone addiction among young people in Switzerland. J. Behav. Addict. 2015, 4, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Dick, D.M.; King, A.; Ray, L.A.; Sher, K.J.; Vena, A.; Vendruscolo, L.F.; Acion, L. Mechanisms of Alcohol Addiction: Bridging Human and Animal Studies. Alcohol Alcohol. 2020, 55, 603–607. [Google Scholar] [CrossRef]
- Caron, L.; Karkazis, K.; Raffin, T.A.; Swan, G.; Koenig, B.A. Nicotine addiction through a neurogenomic prism: Ethics, public health, and smoking. Nicotine Tob. Res. 2005, 7, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Salsitz, E.A. Chronic Pain, Chronic Opioid Addiction: A Complex Nexus. J. Med. Toxicol. 2016, 12, 54–57. [Google Scholar] [CrossRef]
- Olsen, Y. What Is Addiction? History, Terminology, and Core Concepts. Med. Clin. N. Am. 2022, 106, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.L. Addiction and brain reward and antireward pathways. Adv. Psychosom. Med. 2011, 30, 22–60. [Google Scholar] [CrossRef] [PubMed]
- Iovino, M.; Messana, T.; Lisco, G.; Mariano, F.; Giagulli, V.A.; Guastamacchia, E.; De Pergola, G.; Triggiani, V. Neuroendocrine Modulation of Food Intake and Eating Behavior. Endocr. Metab. Immune Disord. Drug Targets 2022, 22, 1252–1262. [Google Scholar] [CrossRef]
- Narayanan, N.S.; Guarnieri, D.J.; DiLeone, R.J. Metabolic hormones, dopamine circuits, and feeding. Front. Neuroendocrinol. 2010, 31, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Hauck, C.; Cook, B.; Ellrott, T. Food addiction, eating addiction and eating disorders. Proc. Nutr. Soc. 2020, 79, 103–112. [Google Scholar] [CrossRef]
- Novelle, M.G.; Diéguez, C. Food Addiction and Binge Eating: Lessons Learned from Animal Models. Nutrients 2018, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.J.; Lingford-Hughes, A.; Erritzoe, D.; Stokes, P.R. The dopamine theory of addiction: 40 years of highs and lows. Nat. Rev. Neurosci. 2015, 16, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Constant, A.; Moirand, R.; Thibault, R.; Val-Laillet, D. Meeting of Minds around Food Addiction: Insights from Addiction Medicine, Nutrition, Psychology, and Neurosciences. Nutrients 2020, 12, 3564. [Google Scholar] [CrossRef]
- Römer, S.S.; Bliokas, V.; Teo, J.T.; Thomas, S.J. Food addiction, hormones and blood biomarkers in humans: A systematic literature review. Appetite 2023, 183, 106475. [Google Scholar] [CrossRef]
- Pak, K.; Seok, J.W.; Lee, M.J.; Kim, K.; Kim, I.J. Dopamine receptor and dopamine transporter in obesity: A meta-analysis. Synapse 2023, 77, e22254. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Baler, R.D. Reward, dopamine and the control of food intake: Implications for obesity. Trends Cogn. Sci. 2011, 15, 37–46. [Google Scholar] [CrossRef]
- Morsali, M.; Poorolajal, J.; Shahbazi, F.; Vahidinia, A.; Doosti-Irani, A. Diet Therapeutics Interventions for Obesity: A Systematic Review and Network Meta-Analysis. J. Res. Health Sci. 2021, 21, e00521. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; El Ghoch, M.; Colao, A.; Hassapidou, M.; Yumuk, V.; Busetto, L.; Obesity Management Task Force (OMTF) of the European Association for the Study of Obesity (EASO). European Guidelines for Obesity Management in Adults with a Very Low-Calorie Ketogenic Diet: A Systematic Review and Meta-Analysis. Obes. Facts 2021, 14, 222–245. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, L.J.; Veronese, N.; Di Bella, G.; Cusumano, C.; Parisi, A.; Tagliaferri, F.; Ciriminna, S.; Barbagallo, M. Mediterranean diet in the management and prevention of obesity. Exp. Gerontol. 2023, 174, 112121. [Google Scholar] [CrossRef]
- Franz, M.J.; VanWormer, J.J.; Crain, A.L.; Boucher, J.L.; Histon, T.; Caplan, W.; Bowman, J.D.; Pronk, N.P. Weight-loss outcomes: A systematic review and meta-analysis of weight-loss clinical trials with a minimum 1-year follow-up. J. Am. Diet. Assoc. 2007, 107, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- van Baak, M.A.; Mariman, E.C.M. Dietary Strategies for Weight Loss Maintenance. Nutrients 2019, 11, 1916. [Google Scholar] [CrossRef] [PubMed]
- Flore, G.; Preti, A.; Carta, M.G.; Deledda, A.; Fosci, M.; Nardi, A.E.; Loviselli, A.; Velluzzi, F. Weight Maintenance after Dietary Weight Loss: Systematic Review and Meta-Analysis on the Effectiveness of Behavioural Intensive Intervention. Nutrients 2022, 14, 1259. [Google Scholar] [CrossRef] [PubMed]
- Blomain, E.S.; Dirhan, D.A.; Valentino, M.A.; Kim, G.W.; Waldman, S.A. Mechanisms of Weight Regain following Weight Loss. ISRN Obes. 2013, 2013, 210524. [Google Scholar] [CrossRef] [PubMed]
- van Baak, M.A.; Mariman, E.C.M. Mechanisms of weight regain after weight loss—The role of adipose tissue. Nat. Rev. Endocrinol. 2019, 15, 274–287. [Google Scholar] [CrossRef]
- Busetto, L.; Bettini, S.; Makaronidis, J.; Roberts, C.A.; Halford, J.C.G.; Batterham, R.L. Mechanisms of weight regain. Eur. J. Intern. Med. 2021, 93, 3–7. [Google Scholar] [CrossRef] [PubMed]
- van Galen, K.A.; Schrantee, A.; Ter Horst, K.W.; la Fleur, S.E.; Booij, J.; Constable, R.T.; Schwartz, G.J.; DiLeone, R.J.; Serlie, M.J. Brain responses to nutrients are severely impaired and not reversed by weight loss in humans with obesity: A randomized crossover study. Nat. Metab. 2023, 5, 1059–1072. [Google Scholar] [CrossRef]
- Geisler, C.E.; Hayes, M.R. Metabolic hormone action in the VTA: Reward-directed behavior and mechanistic insights. Physiol. Behav. 2023, 268, 114236. [Google Scholar] [CrossRef] [PubMed]
- Rezitis, J.; Herzog, H.; Ip, C.K. Neuropeptide Y interaction with dopaminergic and serotonergic pathways: Interlinked neurocircuits modulating hedonic eating behaviours. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 113, 110449. [Google Scholar] [CrossRef] [PubMed]
- Abizaid, A. Ghrelin and dopamine: New insights on the peripheral regulation of appetite. J. Neuroendocrinol. 2009, 21, 787–793. [Google Scholar] [CrossRef]
- Kawahara, Y.; Kawahara, H.; Kaneko, F.; Yamada, M.; Nishi, Y.; Tanaka, E.; Nishi, A. Peripherally administered ghrelin induces bimodal effects on the mesolimbic dopamine system depending on food-consumptive states. Neuroscience 2009, 161, 855–864. [Google Scholar] [CrossRef]
- Kawahara, Y.; Kaneko, F.; Yamada, M.; Kishikawa, Y.; Kawahara, H.; Nishi, A. Food reward-sensitive interaction of ghrelin and opioid receptor pathways in mesolimbic dopamine system. Neuropharmacology 2013, 67, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Skibicka, K.P.; Shirazi, R.H.; Hansson, C.; Dickson, S.L. Ghrelin interacts with neuropeptide Y Y1 and opioid receptors to increase food reward. Endocrinology 2012, 153, 1194–1205. [Google Scholar] [CrossRef]
- Perez-Bonilla, P.; Santiago-Colon, K.; Leinninger, G.M. Lateral hypothalamic area neuropeptides modulate ventral tegmental area dopamine neurons and feeding. Physiol. Behav. 2020, 223, 112986. [Google Scholar] [CrossRef]
- Vallöf, D.; Kalafateli, A.L.; Jerlhag, E. Brain region specific glucagon-like peptide-1 receptors regulate alcohol-induced behaviors in rodents. Psychoneuroendocrinology 2019, 103, 284–295. [Google Scholar] [CrossRef]
- Sørensen, G.; Reddy, I.A.; Weikop, P.; Graham, D.L.; Stanwood, G.D.; Wortwein, G.; Galli, A.; Fink-Jensen, A. The glucagon-like peptide 1 (GLP-1) receptor agonist exendin-4 reduces cocaine self-administration in mice. Physiol. Behav. 2015, 149, 262–268. [Google Scholar] [CrossRef]
- Falk, S.; Petersen, J.; Svendsen, C.; Romero-Leguizamón, C.R.; Jørgensen, S.H.; Krauth, N.; Ludwig, M.Q.; Lundø, K.; Roostalu, U.; Skovbjerg, G.; et al. GLP-1 and nicotine combination therapy engages hypothalamic and mesolimbic pathways to reverse obesity. Cell Rep. 2023, 42, 112466. [Google Scholar] [CrossRef] [PubMed]
- Mietlicki-Baase, E.G.; Ortinski, P.I.; Reiner, D.J.; Sinon, C.G.; McCutcheon, J.E.; Pierce, R.C.; Roitman, M.F.; Hayes, M.R. Glucagon-like peptide-1 receptor activation in the nucleus accumbens core suppresses feeding by increasing glutamatergic AMPA/kainate signaling. J. Neurosci. 2014, 34, 6985–6992. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U. Reuptake inhibitors of dopamine, noradrenaline, and serotonin. In Appetite Control; Springer: Berlin/Heidelberg, Germany, 2012; pp. 339–347. [Google Scholar] [CrossRef]
- Billes, S.K.; Cowley, M.A. Inhibition of dopamine and norepinephrine reuptake produces additive effects on energy balance in lean and obese mice. Neuropsychopharmacology 2007, 32, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Billes, S.K.; Cowley, M.A. Catecholamine reuptake inhibition causes weight loss by increasing locomotor activity and thermogenesis. Neuropsychopharmacology 2008, 33, 1287–1297. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, M.; Ren, L.; Zhong, X.; Ding, Y.; Liu, T.; Liu, Z.; Yang, X.; Cui, L.; Yang, L.; Fan, Y.; et al. D2-Like Receptors Mediate Dopamine-Inhibited Insulin Secretion via Ion Channels in Rat Pancreatic β-Cells. Front. Endocrinol. 2020, 11, 152. [Google Scholar] [CrossRef]
- Uefune, F.; Aonishi, T.; Kitaguchi, T.; Takahashi, H.; Seino, S.; Sakano, D.; Kume, S. Dopamine Negatively Regulates Insulin Secretion Through Activation of D1-D2 Receptor Heteromer. Diabetes 2022, 71, 1946–1961. [Google Scholar] [CrossRef] [PubMed]
- Aslanoglou, D.; Bertera, S.; Sánchez-Soto, M.; Benjamin Free, R.; Lee, J.; Zong, W.; Xue, X.; Shrestha, S.; Brissova, M.; Logan, R.W.; et al. Dopamine regulates pancreatic glucagon and insulin secretion via adrenergic and dopaminergic receptors. Transl. Psychiatry 2021, 11, 59. [Google Scholar] [CrossRef]
- Farino, Z.J.; Morgenstern, T.J.; Maffei, A.; Quick, M.; De Solis, A.J.; Wiriyasermkul, P.; Freyberg, R.J.; Aslanoglou, D.; Sorisio, D.; Inbar, B.P.; et al. New roles for dopamine D2 and D3 receptors in pancreatic beta cell insulin secretion. Mol. Psychiatry 2020, 25, 2070–2085. [Google Scholar] [CrossRef]
- Mietlicki-Baase, E.G.; Ortinski, P.I.; Rupprecht, L.E.; Olivos, D.R.; Alhadeff, A.L.; Pierce, R.C.; Hayes, M.R. The food intake-suppressive effects of glucagon-like peptide-1 receptor signaling in the ventral tegmental area are mediated by AMPA/kainate receptors. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1367–E1374. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://diabetesjournals.org/care/article/46/Supplement_1/S140/148057/9-Pharmacologic-Approaches-to-Glycemic-Treatment (accessed on 25 September 2023).
- Available online: https://diabetesjournals.org/care/article/34/4/789/38811/Bromocriptine-A-Sympatholytic-D2-Dopamine-Agonist (accessed on 25 September 2023).
- Aslanoglou, D.; Bertera, S.; Friggeri, L.; Sánchez-Soto, M.; Lee, J.; Xue, X.; Logan, R.W.; Lane, J.R.; Yechoor, V.K.; McCormick, P.J.; et al. Dual pancreatic adrenergic and dopaminergic signaling as a therapeutic target of bromocriptine. iScience 2022, 25, 104771. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Cincotta, A.H.; O’Connor, C.M.; Ezrokhi, M.; Rutty, D.; Ma, Z.J.; Scranton, R.E. Randomized Clinical Trial of Quick-Release Bromocriptine Among Patients With Type 2 Diabetes on Overall Safety and Cardiovascular Outcomes. Diabetes Care 2016, 33, 1503–1508, Erratum in Diabetes Care 2016, 39, 1846. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Shih, L.C. Parkinson’s disease—Current treatment. Curr. Opin. Neurol. 2023, 36, 302–308. [Google Scholar] [CrossRef]
- Dietrichs, E.; Alves, G.; Benjaminsen, E.; Johansen, K.K.; Tysnes, O.B. Treatment of motor symptoms in Parkinson’s disease. Tidsskr. Nor Laegeforen. 2023, 143. [Google Scholar] [CrossRef]
- Yamamoto, M. Do dopamine agonists provide neuroprotection? Neurology 1998, 51 (Suppl. S2), S10–S12. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Woitalla, D.; Buhmann, C.; Hilker-Roggendorf, R.; Höglinger, G.; Koschel, J.; Müller, T.; Weise, D. Role of dopamine agonists in Parkinson’s disease therapy. J. Neural Transm. 2023, 130, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.D.; Marsh, L. Impulse control disorders and compulsive behaviors associated with dopaminergic therapies in Parkinson disease. Neurol. Clin. Pract. 2012, 2, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Kozuka, C.; Okamoto, S.; Yonamine, M.; Tanaka, H.; Shimabukuro, M. Brown rice-specific γ-oryzanol as a promising prophylactic avenue to protect against diabetes mellitus and obesity in humans. J. Diabetes Investig. 2019, 10, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, C.; Yabiku, K.; Sunagawa, S.; Ueda, R.; Taira, S.; Ohshiro, H.; Ikema, T.; Yamakawa, K.; Higa, M.; Tanaka, H.; et al. Brown rice and its component, γ-oryzanol, attenuate the preference for high-fat diet by decreasing hypothalamic endoplasmic reticulum stress in mice. Diabetes 2012, 61, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, C.; Sunagawa, S.; Ueda, R.; Higa, M.; Tanaka, H.; Shimizu-Okabe, C.; Ishiuchi, S.; Takayama, C.; Matsushita, M.; Tsutsui, M.; et al. γ-Oryzanol protects pancreatic β-cells against endoplasmic reticulum stress in male mice. Endocrinology 2015, 156, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, C.; Sunagawa, S.; Ueda, R.; Higa, M.; Ohshiro, Y.; Tanaka, H.; Shimizu-Okabe, C.; Takayama, C.; Matsushita, M.; Tsutsui, M.; et al. A novel insulinotropic mechanism of whole grain-derived γ-oryzanol via the suppression of local dopamine D2 receptor signalling in mouse islet. Br. J. Pharmacol. 2015, 172, 4519–4534. [Google Scholar] [CrossRef]
- Araujo, S.M.; de Paula, M.T.; Poetini, M.R.; Meichtry, L.; Bortolotto, V.C.; Zarzecki, M.S.; Jesse, C.R.; Prigol, M. Effectiveness of γ-oryzanol in reducing neuromotor deficits, dopamine depletion and oxidative stress in a Drosophila melanogaster model of Parkinson’s disease induced by rotenone. Neurotoxicology 2015, 51, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, P. The Beneficial Effect of Rice Bran Extract Against Rotenone-Induced Experimental Parkinson’s Disease in Rats. Curr. Mol. Pharmacol. 2021, 14, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Chern, Y. AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J. Neurogenet. 2015, 29, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson’s Disease. J. Park. Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.K.; Go, J.; Park, H.Y.; Choi, Y.K.; Seo, Y.J.; Choi, J.H.; Rhee, M.; Lee, T.G.; Lee, C.H.; Kim, K.S. Metformin regulates astrocyte reactivity in Parkinson’s disease and normal aging. Neuropharmacology 2020, 175, 108173. [Google Scholar] [CrossRef]
- Available online: https://www.ncbi.nlm.nih.gov/gene/586 (accessed on 5 September 2023).
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Shaikh, M.F.; Othman, I. Emerging neuroprotective effect of metformin in Parkinson’s disease: A molecular crosstalk. Pharmacol. Res. 2020, 152, 104593. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A Future Therapy for Neurodegenerative Diseases: Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.H.; Chang, Y.L.; Gau, S.Y.; Tsai, T.H.; Lee, C.Y. Dose-Response Association of Metformin with Parkinson’s Disease Odds in Type 2 Diabetes Mellitus. Pharmaceutics 2022, 14, 946. [Google Scholar] [CrossRef]
- Alrouji, M.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Ashour, N.A.; Jabir, M.S.; Negm, W.A.; Batiha, G.E. Metformin role in Parkinson’s disease: A double-sword effect. Mol. Cell. Biochem. 2023. [Google Scholar] [CrossRef]
- Moore, E.; Mander, A.; Ames, D.; Carne, R.; Sanders, K.; Watters, D. Cognitive impairment and vitamin B12: A review. Int. Psychogeriatr. 2012, 24, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M. Cardiovascular benefits and safety profile of acarbose therapy in prediabetes and established type 2 diabetes. Cardiovasc. Diabetol. 2007, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, H. MicroRNAs, Parkinson’s Disease, and Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 2953. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, S.; Landreth, G.E. Antiinflammatory properties of PPARgamma agonists following ischemia. Drug News Perspect. 2004, 17, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Tjokroprawiro, A. New approach in the treatment of T2DM and metabolic syndrome (focus on a novel insulin sensitizer). Acta Med. Indones. 2006, 38, 160–166. [Google Scholar]
- Kapadia, R.; Yi, J.H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 2008, 13, 1813–1826. [Google Scholar] [CrossRef]
- Culman, J.; Nguyen-Ngoc, M.; Glatz, T.; Gohlke, P.; Herdegen, T.; Zhao, Y. Treatment of rats with pioglitazone in the reperfusion phase of focal cerebral ischemia: A preclinical stroke trial. Exp. Neurol. 2012, 238, 243–253. [Google Scholar] [CrossRef]
- White, A.T.; Murphy, A.N. Administration of thiazolidinediones for neuroprotection in ischemic stroke: A pre-clinical systematic review. J. Neurochem. 2010, 115, 845–853. [Google Scholar] [CrossRef]
- Tanaka, R.; Yamashiro, K.; Okuma, Y.; Shimura, H.; Nakamura, S.; Ueno, Y.; Tanaka, Y.; Miyamoto, N.; Tomizawa, Y.; Nakahara, T.; et al. Effects of Pioglitazone for Secondary Stroke Prevention in Patients with Impaired Glucose Tolerance and Newly Diagnosed Diabetes: The J-SPIRIT Study. J. Atheroscler. Thromb. 2015, 22, 1305–1316. [Google Scholar] [CrossRef]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M.; Guarino, P.D.; Lovejoy, A.M.; Peduzzi, P.N.; Conwit, R.; et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef]
- de Jong, M.; van der Worp, H.B.; van der Graaf, Y.; Visseren, F.L.J.; Westerink, J. Pioglitazone and the secondary prevention of cardiovascular disease. A meta-analysis of randomized-controlled trials. Cardiovasc. Diabetol. 2017, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Jojo, G.M.; Kuppusamy, G. Scope of new formulation approaches in the repurposing of pioglitazone for the management of Alzheimer’s disease. J. Clin. Pharm. Ther. 2019, 44, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, M.Y.; Taheri, N.; Opulencia, M.J.C.; Bokov, D.O.; Abdullaev, S.Y.; Gholamrezapour, M.; Heidari, M.; Bazmandegan, G. Neuroprotective and Anti-inflammatory Effects of Pioglitazone on Traumatic Brain Injury. Mediat. Inflamm. 2022, 2022, 9860855. [Google Scholar] [CrossRef] [PubMed]
- Alhowail, A.; Alsikhan, R.; Alsaud, M.; Aldubayan, M.; Rabbani, S.I. Protective Effects of Pioglitazone on Cognitive Impairment and the Underlying Mechanisms: A Review of Literature. Drug Des. Dev. Ther. 2022, 16, 2919–2931. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Dehmer, T.; Heneka, M.T.; Sastre, M.; Dichgans, J.; Schulz, J.B. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J. Neurochem. 2004, 88, 494–501. [Google Scholar] [CrossRef]
- Randy, L.H.; Guoying, B. Agonism of Peroxisome Proliferator Receptor-Gamma may have Therapeutic Potential for Neuroinflammation and Parkinson’s Disease. Curr. Neuropharmacol. 2007, 5, 35–46. [Google Scholar] [CrossRef]
- Carta, A.R.; Pisanu, A. Modulating microglia activity with PPAR-γ agonists: A promising therapy for Parkinson’s disease? Neurotox. Res. 2013, 23, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.K.; Alexander, R.C.; Welsh-Bohmer, K.A.; Culp, M.; Chiang, C.; O’Neil, J.; Evans, R.M.; Harrigan, P.; Plassman, B.L.; Burke, J.R.; et al. Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): A prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Burns, D.K.; Gottschalk, W.K. Reassessment of Pioglitazone for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 666958. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Simuni, T.; Elm, J.; Clark-Matott, J.; Graebner, A.K.; Baker, L.; Dunlop, S.R.; Emborg, M.; Kamp, C.; Morgan, J.C.; et al. Peripheral Biomarkers of Parkinson’s Disease Progression and Pioglitazone Effects. J. Park. Dis. 2015, 5, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Singh, A.; Baxi, H.; Taylor, B.; Burgess, J.; Antony, B. Thiazolidinedione use is associated with reduced risk of Parkinson’s disease in patients with diabetes: A meta-analysis of real-world evidence. Neurol. Sci. 2020, 41, 3697–3703. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tao, Y.; Li, J.; Kang, M. Pioglitazone use is associated with reduced risk of Parkinson’s disease in patients with diabetes: A systematic review and meta-analysis. J. Clin. Neurosci. 2022, 106, 154–158. [Google Scholar] [CrossRef]
- Sunnarborg, K.; Tiihonen, M.; Huovinen, M.; Koponen, M.; Hartikainen, S.; Tolppanen, A.M. Association between different diabetes medication classes and risk of Parkinson’s disease in people with diabetes. Pharmacoepidemiol. Drug Saf. 2022, 31, 875–882. [Google Scholar] [CrossRef] [PubMed]
- NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803, Erratum in Lancet Neurol. 2015, 14, 979. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Gallwitz, B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 479–486. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016, 4, 525–536. [Google Scholar] [CrossRef]
- Shyamaladevi, B.; Dash, I.; Badrachalam, R.; Krishnan, M.; Panneerselvam, A.; Undru, S. An update on diagnosis and therapeutics for type-2 diabetes mellitus. Bioinformation 2023, 19, 295–298. [Google Scholar] [CrossRef]
- Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Pariyar, R.; Bastola, T.; Lee, D.H.; Seo, J. Neuroprotective Effects of the DPP4 Inhibitor Vildagliptin in In Vivo and In Vitro Models of Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 2388. [Google Scholar] [CrossRef] [PubMed]
- Badawi, G.A.; Abd El Fattah, M.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology 2017, 25, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Li, C.; Li, M.; Ma, L. Sitagliptin rescues memory deficits in Parkinsonian rats via upregulating BDNF to prevent neuron and dendritic spine loss. Neurol Res. 2018, 40, 736–743. [Google Scholar] [CrossRef]
- Abhangi, K.V.; Patel, J.I. Neuroprotective effects of linagliptin in a rotenone-induced rat model of Parkinson’s disease. Indian J. Pharmacol. 2022, 54, 46–50. [Google Scholar] [CrossRef]
- Yu, H.Y.; Sun, T.; Wang, Z.; Li, H.; Xu, D.; An, J.; Wen, L.L.; Li, J.Y.; Li, W.; Feng, J. Exendin-4 and linagliptin attenuate neuroinflammation in a mouse model of Parkinson’s disease. Neural Regen. Res. 2023, 18, 1818–1826. [Google Scholar] [CrossRef]
- Nassar, N.N.; Al-Shorbagy, M.Y.; Arab, H.H.; Abdallah, D.M. Saxagliptin: A novel antiparkinsonian approach. Neuropharmacology 2015, 89, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Turnes, J.M.; Bassani, T.B.; Souza, L.C.; Vital, M.A.B.F. Ineffectiveness of saxagliptin as a neuroprotective drug in 6-OHDA-lesioned rats. J. Pharm. Pharmacol. 2018, 70, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Wirdefeldt, K.; Yin, L.; Fang, F.; Markaki, I.; Efendic, S.; Ludvigsson, J.F. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors-A nationwide case-control study. Mov. Disord. 2016, 31, 1422–1423. [Google Scholar] [CrossRef]
- Brauer, R.; Wei, L.; Ma, T.; Athauda, D.; Girges, C.; Vijiaratnam, N.; Auld, G.; Whittlesea, C.; Wong, I.; Foltynie, T. Diabetes medications and risk of Parkinson’s disease: A cohort study of patients with diabetes. Brain 2020, 143, 3067–3076. [Google Scholar] [CrossRef]
- Lietzau, G.; Magni, G.; Kehr, J.; Yoshitake, T.; Candeias, E.; Duarte, A.I.; Pettersson, H.; Skogsberg, J.; Abbracchio, M.P.; Klein, T.; et al. Dipeptidyl peptidase-4 inhibitors and sulfonylureas prevent the progressive impairment of the nigrostriatal dopaminergic system induced by diabetes during aging. Neurobiol. Aging 2020, 89, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Chung, S.J.; Yoo, H.S.; Hong, N.; Jung, J.H.; Baik, K.; Lee, Y.H.; Sohn, Y.H.; Lee, P.H. Beneficial effects of dipeptidyl peptidase-4 inhibitors in diabetic Parkinson’s disease. Brain 2021, 144, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhang, X.; Li, P.; Wang, M.; Yan, L.; Bao, Z.; Liu, Q. Association Between Diabetes Medications and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 678649. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Wu, S.L.; Chen, T.C.; Chuang, C.S. Antidiabetic Agents for Treatment of Parkinson’s Disease: A Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 17, 4805. [Google Scholar] [CrossRef]
- Khalaf, M.M.; El-Sayed, M.M.; Kandeil, M.A.; Ahmed, S. A novel protective modality against rotenone-induced Parkinson’s disease: A pre-clinical study with dulaglutide. Int. Immunopharmacol. 2023, 119, 110170. [Google Scholar] [CrossRef]
- Karagiannis, T.; Avgerinos, I.; Liakos, A.; Del Prato, S.; Matthews, D.R.; Tsapas, A.; Bekiari, E. Management of type 2 diabetes with the dual GIP/GLP-1 receptor agonist tirzepatide: A systematic review and meta-analysis. Diabetologia 2022, 65, 1251–1261. [Google Scholar] [CrossRef]
- De Mesquita, Y.L.L.; Pera Calvi, I.; Reis Marques, I.; Almeida Cruz, S.; Padrao, E.M.H.; Carvalho, P.E.P.; da Silva, C.H.A.; Cardoso, R.; Moura, F.A.; Rafalskiy, V.V. Efficacy and safety of the dual GIP and GLP-1 receptor agonist tirzepatide for weight loss: A meta-analysis of randomized controlled trials. Int. J. Obes. 2023, 47, 883–892. [Google Scholar] [CrossRef]
- Lisco, G.; De Tullio, A.; Disoteo, O.; De Geronimo, V.; Piazzolla, G.; De Pergola, G.; Giagulli, V.A.; Jirillo, E.; Guastamacchia, E.; Sabbà, C.; et al. Basal insulin intensification with GLP-1RA and dual GIP and GLP-1RA in patients with uncontrolled type 2 diabetes mellitus: A rapid review of randomized controlled trials and meta-analysis. Front. Endocrinol. 2022, 13, 920541. [Google Scholar] [CrossRef]
- Lv, M.; Xue, G.; Cheng, H.; Meng, P.; Lian, X.; Hölscher, C.; Li, D. The GLP-1/GIP dual-receptor agonist DA5-CH inhibits the NF-κB inflammatory pathway in the MPTP mouse model of Parkinson’s disease more effectively than the GLP-1 single-receptor agonist NLY01. Brain Behav. 2021, 11, e2231. [Google Scholar] [CrossRef]
- Ji, C.; Xue, G.F.; Lijun, C.; Feng, P.; Li, D.; Li, L.; Li, G.; Hölscher, C. A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson’s disease by increasing expression of BNDF. Brain Res. 2016, 1634, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Li, D.; Feng, P.; Li, L.; Xue, G.F.; Li, G.; Hölscher, C. A novel dual GLP-1 and GIP incretin receptor agonist is neuroprotective in a mouse model of Parkinson’s disease by reducing chronic inflammation in the brain. Neuroreport 2016, 27, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C. Glucagon-like peptide 1 and glucose-dependent insulinotropic peptide hormones and novel receptor agonists protect synapses in Alzheimer’s and Parkinson’s diseases. Front. Synaptic Neurosci. 2022, 14, 955258. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Rhea, E.M.; Talbot, K.; Banks, W.A. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2023, 180, 114187, Erratum in Biochem. Pharmacol. 2023, 210, 115474. [Google Scholar] [CrossRef]
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- De Fronzo, R.A.; Hompesch, M.; Kasichayanula, S.; Liu, X.; Hong, Y.; Pfister, M.; Morrow, L.A.; Leslie, B.R.; Boulton, D.W.; Ching, A.; et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 2013, 36, 3169–3176. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef] [PubMed]
- List, J.F.; Whaley, J.M. Glucose dynamics and mechanistic implications of SGLT2 inhibitors in animals and humans. Kidney Int. Suppl. 2011, 79, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- McGuire, D.K.; Shih, W.J.; Cosentino, F.; Charbonnel, B.; Cherney, D.Z.I.; Dagogo-Jack, S.; Pratley, R.; Greenberg, M.; Wang, S.; Huyck, S.; et al. Association of SGLT2 Inhibitors With Cardiovascular and Kidney Outcomes in Patients With Type 2 Diabetes: A Meta-analysis. JAMA Cardiol. 2021, 6, 148–158. [Google Scholar] [CrossRef]
- Lin, K.J.; Wang, T.J.; Chen, S.D.; Lin, K.L.; Liou, C.W.; Lan, M.Y.; Chuang, Y.C.; Chuang, J.H.; Wang, P.W.; Lee, J.J.; et al. Two Birds One Stone: The Neuroprotective Effect of Antidiabetic Agents on Parkinson Disease-Focus on Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors. Antioxidants 2021, 10, 1935. [Google Scholar] [CrossRef]
- Arab, H.H.; Safar, M.M.; Shahin, N.N. Targeting ROS-Dependent AKT/GSK-3β/NF-κB and DJ-1/Nrf2 Pathways by Dapagliflozin Attenuates Neuronal Injury and Motor Dysfunction in Rotenone-Induced Parkinson’s Disease Rat Model. ACS Chem. Neurosci. 2021, 12, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; El-Sayed, M.M.; Kandeil, M.A.; Khalaf, M.M. Empagliflozin attenuates neurodegeneration through antioxidant, anti-inflammatory, and modulation of α-synuclein and Parkin levels in rotenone-induced Parkinson’s disease in rats. Saudi Pharm. J. 2022, 30, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Motawi, T.K.; Al-Kady, R.H.; Abdelraouf, S.M.; Senousy, M.A. Empagliflozin alleviates endoplasmic reticulum stress and augments autophagy in rotenone-induced Parkinson’s disease in rats: Targeting the GRP78/PERK/eIF2α/CHOP pathway and miR-211-5p. Chem. Biol. Interact. 2022, 362, 110002. [Google Scholar] [CrossRef] [PubMed]
- Ünal, İ.; Cansız, D.; Beler, M.; Sezer, Z.; Güzel, E.; Emekli-Alturfan, E. Sodium-dependent Glucose Co-Transporter-2 Inhibitor Empagliflozin Exerts Neuroprotective Effects in Rotenone-Induced Parkinson’s Disease Model in Zebrafish; Mechanism Involving Ketogenesis and Autophagy. Brain Res. 2023, 15, 148536. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, M.M.; Danazumi, A.U.; Alshammari, M.K.; Bello, R.O.; Alghazwni, M.K.; Alshehri, A.M.; Alshlali, O.M.; Umar, H.I. Repurposing the inhibitors of MMP-9 and SGLT-2 against ubiquitin specific protease 30 in Parkinson’s disease: Computational modelling studies. J. Biomol. Struct. Dyn. 2023, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mui, J.V.; Zhou, J.; Lee, S.; Leung, K.S.K.; Lee, T.T.L.; Chou, O.H.I.; Tsang, S.L.; Wai, A.K.C.; Liu, T.; Wong, W.T.; et al. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors vs. Dipeptidyl Peptidase-4 (DPP4) Inhibitors for New-Onset Dementia: A Propensity Score-Matched Population-Based Study With Competing Risk Analysis. Front Cardiovasc. Med. 2021, 8, 747620. [Google Scholar] [CrossRef]
- Tharmaraja, T.; Ho, J.S.Y.; Sia, C.H.; Lim, N.A.; Chong, Y.F.; Lim, A.Y.L.; Rathakrishnan, R.R.; Yeo, L.L.L.; Sharma, V.K.; Tan, B.Y.Q. Sodium-glucose cotransporter 2 inhibitors and neurological disorders: A scoping review. Ther. Adv. Chronic. Dis. 2022, 13, 20406223221086996. [Google Scholar] [CrossRef]
- Inzucchi, S.; Rosenstock, J.; Umpierrez, G. Type 2 Diabetes and Insulin Secretagogues. J. Clin. Endocrinol. Metab. 2012, 97, 37A. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Iskander, C.; Wang, C.; Xiong, L.Y.; Shah, B.R.; Edwards, J.D.; Kapral, M.K.; Herrmann, N.; Lanctôt, K.L.; Masellis, M.; et al. Association of sulfonylureas with the risk of dementia: A population-based cohort study. J. Am. Geriatr. Soc. 2023, 71, 3059–3070. [Google Scholar] [CrossRef] [PubMed]
- Lechin, F.; van der Dijs, B.; Pardey-Maldonado, B.; Rivera, J.E.; Lechin, M.E.; Baez, S. Amantadine reduces glucagon and enhances insulin secretion throughout the oral glucose tolerance test: Central plus peripheral nervous system mechanisms. Diabetes Metab. Syndr. Obes. 2009, 2, 203–213. [Google Scholar] [CrossRef]
- Sanni, O.; Terre’Blanche, G. Dual A1 and A2A adenosine receptor antagonists, methoxy substituted 2-benzylidene-1-indanone, suppresses intestinal postprandial glucose and attenuates hyperglycaemia in fructose-streptozotocin diabetic rats. BMC Endocr. Disord. 2023, 23, 97. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Roth, B.L. Lorcaserin and pimavanserin: Emerging selectivity of serotonin receptor subtype-targeted drugs. J. Clin. Investig. 2013, 123, 4986–4991. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.M.; Correll, C.U. Increased Metabolic Potential, Efficacy, and Safety of Emerging Treatments in Schizophrenia. CNS Drugs 2023, 37, 545–570. [Google Scholar] [CrossRef]
- Coffey, B.; Jankovic, J.; Claassen, D.O.; Jimenez-Shahed, J.; Gertz, B.J.; Garofalo, E.A.; Stamler, D.A.; Wieman, M.; Savola, J.M.; Gordon, M.F.; et al. Efficacy and Safety of Fixed-Dose Deutetrabenazine in Children and Adolescents for Tics Associated With Tourette Syndrome: A Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2129397. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group; Frank, S.; Testa, C.M.; Stamler, D.; Kayson, E.; Davis, C.; Edmondson, M.C.; Kinel, S.; Leavitt, B.; Oakes, D.; et al. Effect of Deutetrabenazine on Chorea Among Patients With Huntington Disease: A Randomized Clinical Trial. JAMA 2016, 316, 40–50. [Google Scholar] [CrossRef]
- Hainer, V.; Kabrnova, K.; Aldhoon, B.; Kunesova, M.; Wagenknecht, M. Serotonin and norepinephrine reuptake inhibition and eating behavior. Ann. N. Y. Acad. Sci. 2006, 1083, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Tilbrook, D.; Jacob, J.; Parsons, P.; Edwards, C.; Loewen, K.; Kelly, L. Opioid use disorder and type 2 diabetes mellitus: Effect of participation in buprenorphine-naloxone substitution programs on glycemic control. Can. Fam. Physician 2017, 63, e350–e354. [Google Scholar]
- Fagerholm, V.; Haaparanta, M.; Scheinin, M. α2-adrenoceptor regulation of blood glucose homeostasis. Basic Clin. Pharmacol. Toxicol. 2011, 108, 365–370. [Google Scholar] [CrossRef]
- Raingeard, I.; Courtet, P.; Renard, E.; Bringer, J. Naltrexone improves blood glucose control in type 1 diabetic women with severe and chronic eating disorders. Diabetes Care 2004, 27, 847–848. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).