

Exploring the Disease-Associated Microglia State in Amyotrophic Lateral Sclerosis
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Methods
2.1. Study Design
2.2. Spinal Cord Samples
2.3. Neuropathological Examination
2.4. RNA Isolation from Frozen Spinal Cord Samples
2.5. Reverse Transcription and Gene mRNA Expression Analysis via RT-qPCR
2.6. Statistical Data Analysis
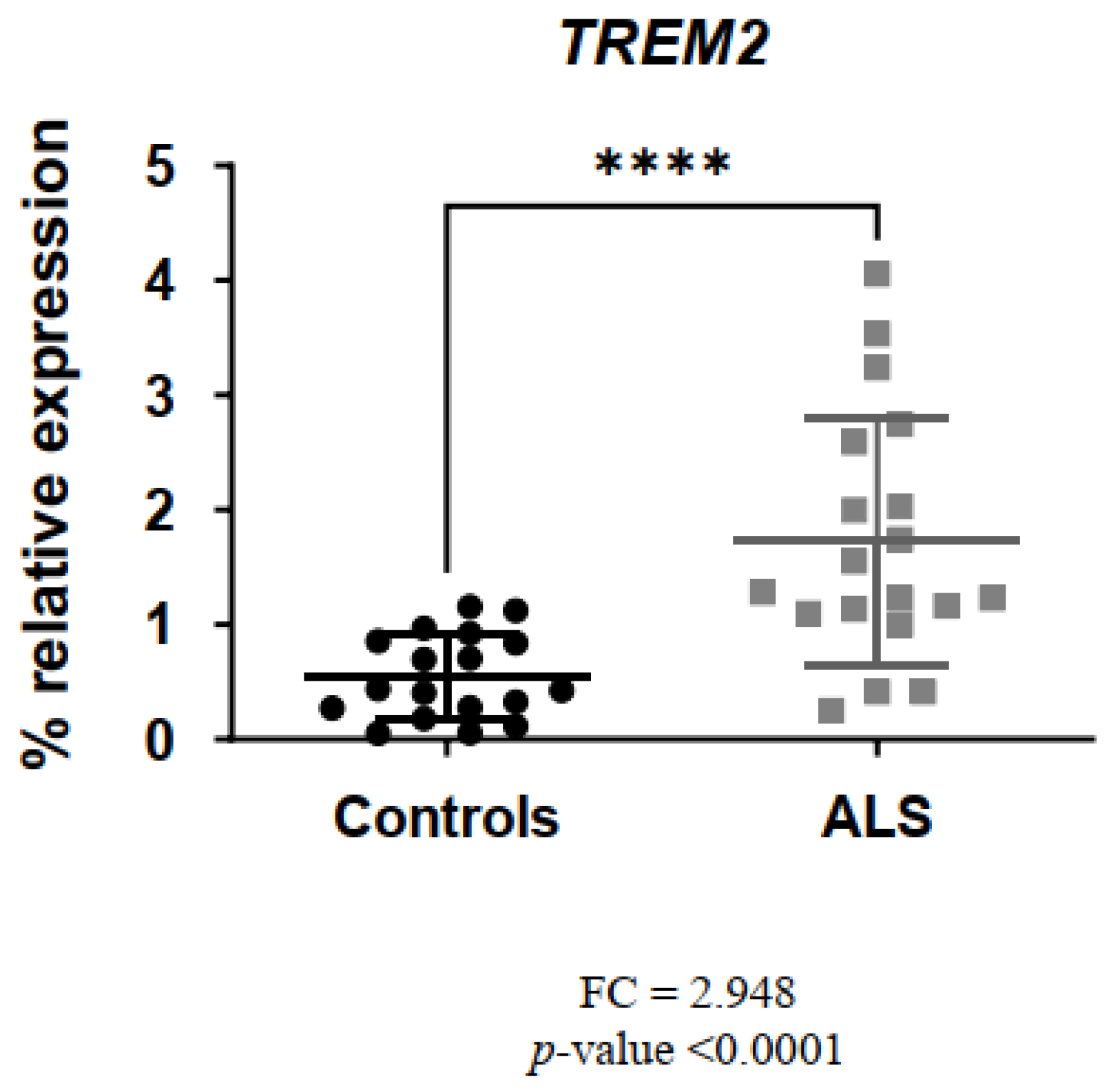
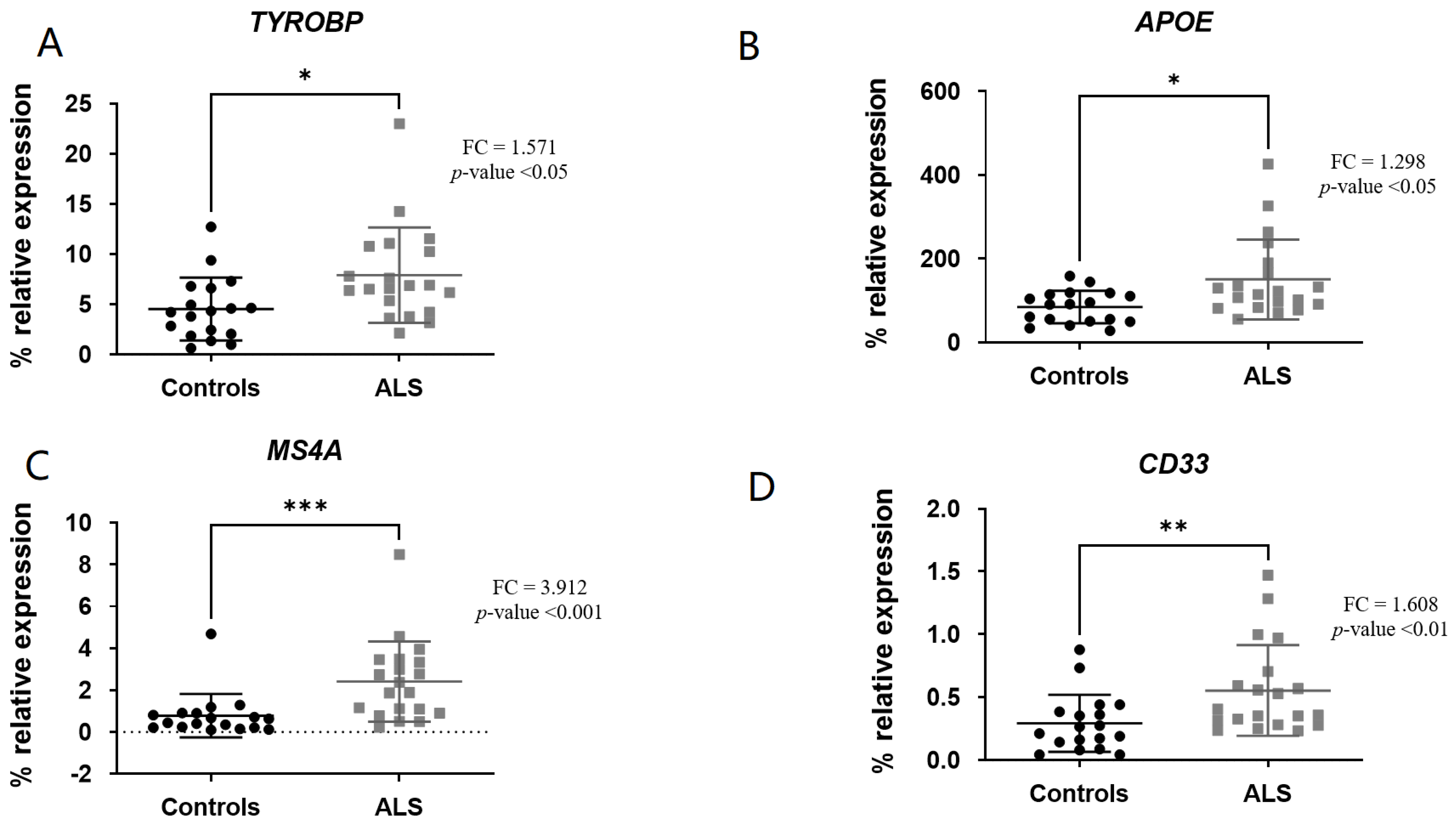
3. Results
3.1. Sample Composition
3.2. Comparison of Gene Expression Levels
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflammation 2021, 18, 258. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Friedman, B.A.; Dejanovic, B.; Sheng, M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Eggen, B.J.L.; Raj, D.; Hanisch, U.K.; Boddeke, H.W.G.M. Microglial phenotype and adaptation. J. Neuroimmune Pharmacol. 2013, 8, 807–823. [Google Scholar] [CrossRef]
- Schwabenland, M.; Brück, W.; Priller, J.; Stadelmann, C.; Lassmann, H.; Prinz, M. Analyzing microglial phenotypes across neuropathologies: A practical guide. Acta Neuropathol. 2021, 142, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Xu, Y.J.; Au, N.P.B.; Ma, C.H.E. Functional and Phenotypic Diversity of Microglia: Implication for Microglia-Based Therapies for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 896852. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- FFriedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Colonna, M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 2021, 218, 1–10. [Google Scholar] [CrossRef]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Smajić, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; E Doykan, C.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Walker, D.G.; Whetzel, A.M.; Serrano, G.; Sue, L.I.; Beach, T.G.; Lue, L.F. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 2015, 36, 571–582. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Pottier, C.; Ravenscroft, T.A.; Brown, P.H.; Finch, N.A.; Baker, M.; Parsons, M.; Asmann, Y.W.; Ren, Y.; Christopher, E.; Levitch, D.; et al. TYROBP genetic variants in early-onset Alzheimer’s disease. Neurobiol. Aging 2016, 48, 222.e9–222.e15. [Google Scholar] [CrossRef] [PubMed]
- Deming, Y.; Filipello, F.; Cignarella, F.; Cantoni, C.; Hsu, S.; Mikesell, R.; Li, Z.; Del-Aguila, J.L.; Dube, U.; Farias, F.G.; et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 2019, 11, eaau2291. [Google Scholar] [CrossRef]
- Yamamoto, T.; Murayama, S.; Takao, M.; Isa, T.; Higo, N. Expression of secreted phosphoprotein 1 (osteopontin) in human sensorimotor cortex and spinal cord: Changes in patients with amyotrophic lateral sclerosis. Brain Res. 2017, 1655, 168–175. [Google Scholar] [CrossRef]
- García-Merino, I.M.; Consuegra, I.; Jiménez, J.L.; Muñoz-Fernández, M.Á. Specific legislation on biobanks in Spain. Biopreserv Biobank. 2015, 13, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Jericó, I.; Vicuña-Urriza, J.; Blanco-Luquin, I.; Macias, M.; Martinez-Merino, L.; Roldán, M.; Rojas-Garcia, R.; Pagola-Lorz, I.; Carbayo, A.; De Luna, N.; et al. Profiling TREM2 expression in amyotrophic lateral sclerosis. Brain Behav. Immun. 2023, 109, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Bell, J.E.; Alafuzoff, I.; Al-Sarraj, S.; Arzberger, T.; Bogdanovic, N.; Budka, H.; Dexter, D.T.; Falkai, P.; Ferrer, I.; Gelpi, E.; et al. Management of a twenty-first century brain bank: Experience in the BrainNet Europe consortium. Acta Neuropathol. 2008, 115, 497–507. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef]
- Ruan, C.; Elyaman, W. A New Understanding of TMEM119 as a Marker of Microglia. Front. Cell Neurosci. 2022, 16, 902372. [Google Scholar] [CrossRef]
- Young, K.F.; Gardner, R.; Sariana, V.; Whitman, S.A.; Bartlett, M.J.; Falk, T.; Morrison, H.W. Can quantifying morphology and TMEM119 expression distinguish between microglia and infiltrating macrophages after ischemic stroke and reperfusion in male and female mice? J. Neuroinflammation. 2021, 18, 1–15. [Google Scholar] [CrossRef]
- Takahashi, K. Microglial heterogeneity in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2023, 82, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Tan, C.C.; Hou, X.H.; Cao, X.P.; Tan, L.; Yu, J.T. TREM2 Variants and Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2019, 68, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhao, S.; Bosco, D.B.; Nguyen, A.; Wu, L.J. Microglial TREM2 in amyotrophic lateral sclerosis. Dev. Neurobiol. 2022, 82, 125–137. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of AD. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Haure-Mirande, J.V.; Audrain, M.; Ehrlich, M.E.; Gandy, S. Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol. Neurodegener. 2022, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; DiVito, J.; Ionita, I.; et al. Genome-wide Association Analysis Reveals Putative Alzheimer’s Disease Susceptibility Loci in Addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 2019, 15, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Eskandari-Sedighi, G.; Jung, J.; Macauley, M.S. CD33 isoforms in microglia and Alzheimer’s disease: Friend and foe. Mol. Aspects Med. 2023, 90, 101111. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, C.; Carrasquillo, M.M. UKPMC Funders Group Author Manuscript and CD2AP are associated with Alzheimer ’ s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236. [Google Scholar] [CrossRef]
- Tortora, F.; Rendina, A.; Angiolillo, A.; Di Costanzo, A.; Aniello, F.; Donizetti, A.; Febbraio, F.; Vitale, E. CD33 rs2455069 SNP: Correlation with Alzheimer’s Disease and Hypothesis of Functional Role. Int. J. Mol. Sci. 2022, 23, 3629. [Google Scholar] [CrossRef]
- Schwabe, T.; Srinivasan, K.; Rhinn, H. Shifting paradigms: The central role of microglia in Alzheimer’s disease. Neurobiol. Dis. 2020, 143, 104962. [Google Scholar] [CrossRef]
- Xie, Y.; Luo, X.; He, H.; Tang, M. Novel Insight Into the Role of Immune Dysregulation in Amyotrophic Lateral Sclerosis Based on Bioinformatic Analysis. Front. Neurosci. 2021, 15, 657465. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St. Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1–42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Sánchez-Ruiz de Gordoa, J.; Erro, M.E.; Vicuña-Urriza, J.; Zelaya, M.V.; Tellechea, P.; Acha, B.; Zueco, S. Microglia-Related Gene Triggering Receptor Expressed in Myeloid Cells 2 (TREM2) Is Upregulated in the Substantia Nigra of Progressive Supranuclear Palsy. Mov. Disord. 2020, 35, 885–890. [Google Scholar] [CrossRef]
- Ma, J.; Yu, J.T.; Tan, L. MS4A Cluster in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 1240–1248. [Google Scholar] [CrossRef]
- Hur, E.M.; Youssef, S.; Haws, M.E.; Zhang, S.Y.; Sobel, R.A.; Steinman, L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat. Immunol. 2007, 8, 74–83. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Cunha, M.I.; Su, M.; Cantuti-Castelvetri, L.; Müller, S.A.; Schifferer, M.; Djannatian, M.; Alexopoulos, I.; van der Meer, F.; Winkler, A.; van Ham, T.J.; et al. Pro-inflammatory activation following demyelination is required for myelin clearance and oligodendrogenesis. J. Exp. Med. 2020, 217, e20191390. [Google Scholar] [CrossRef]
- DeTora, L.M.; Toroser, D.; Sykes, A.; Vanderlinden, C.; Plunkett, F.J.; Lane, T.; Hanekamp, E.; Dormer, L.; DiBiasi, F.; Bridges, D.; et al. Good Publication Practice (GPP) Guidelines for Company-Sponsored Biomedical Research: 2022 Update. Ann. Intern. Med. 2022, 175, 1298–1304. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| ALS | Controls | p-Value | |
|---|---|---|---|
| Spinal cord donors | |||
| n | 20 | 18 | |
| Age (years) (SD) 1 | 70.1 (±13.79) | 73.3 (±14.29) | 0.491 |
| Sex (F/M) | 12/8 | 6/12 | 0.093 |
| PMI 2 (h); median (range) | 6.7 (3–12) | 6.9 (2–16) | 0.734 |
| ID | Accesion Number 1 | Amplicon Size (bp 2) | Tm | Forward Primer | Tm2 3 | Reverse Primer |
|---|---|---|---|---|---|---|
| TREM2 | NM_018965 | 116 | 61.0 | CTGCTCATCTTACTCTTTGTCAC | 62.3 | CAGTGCTTCATGGAGTCATAGG |
| TYROBP | NM_003332 | 146 | 61.6 | CTGGCCGTGTACTTCCTG | 62 | GTGTTGAGGTCGCTGTAGAC |
| APOE | NM_000041 | 143 | 62.2 | TTGCTGGTCACATTCCTGG | 62.2 | AGGTAATCCCAAAAGCGACC |
| MS4A | NM_148975 | 148 | 61.9 | TCTTGAAGGGAGAACCCAAAG | 62 | CCCCAAATTGTGTACCCGATA |
| CD33 | NM_001772 | 132 | 61.9 | TGTCAGGTGAAGTTCGCTG | 61.8 | TGCTCTGGTCTCTTGTTTCC |
| TMEM119 | NM_181724 | 111 | 61.7 | CCACTCTCGCTCCATTCG | 61.6 | CAGCAACAGAAGGATGAGGA |
| LPL | NM_000237 | 129 | 61.9 | AAAGTGTCTCATTTGCAGAAAGG | 61.9 | CACAGAATTCACATGCCGTTC |
| SPP1 | NM_001040058 | 148 | 61.8 | GTCCCCACAGTAGACACATATG | 62.1 | TCAACTCCTCGCTTTCCATG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jauregui, C.; Blanco-Luquin, I.; Macías, M.; Roldan, M.; Caballero, C.; Pagola, I.; Mendioroz, M.; Jericó, I. Exploring the Disease-Associated Microglia State in Amyotrophic Lateral Sclerosis. Biomedicines 2023, 11, 2994. https://doi.org/10.3390/biomedicines11112994
Jauregui C, Blanco-Luquin I, Macías M, Roldan M, Caballero C, Pagola I, Mendioroz M, Jericó I. Exploring the Disease-Associated Microglia State in Amyotrophic Lateral Sclerosis. Biomedicines. 2023; 11(11):2994. https://doi.org/10.3390/biomedicines11112994
Chicago/Turabian StyleJauregui, Carlota, Idoia Blanco-Luquin, Mónica Macías, Miren Roldan, Cristina Caballero, Inma Pagola, Maite Mendioroz, and Ivonne Jericó. 2023. "Exploring the Disease-Associated Microglia State in Amyotrophic Lateral Sclerosis" Biomedicines 11, no. 11: 2994. https://doi.org/10.3390/biomedicines11112994
APA StyleJauregui, C., Blanco-Luquin, I., Macías, M., Roldan, M., Caballero, C., Pagola, I., Mendioroz, M., & Jericó, I. (2023). Exploring the Disease-Associated Microglia State in Amyotrophic Lateral Sclerosis. Biomedicines, 11(11), 2994. https://doi.org/10.3390/biomedicines11112994