
Carfilzomib Mitigates Lipopolysaccharide/D-Galactosamine/Dimethylsulfoxide-Induced Acute Liver Failure in Mice
 , , , , ,
, , , , ,
Abstract
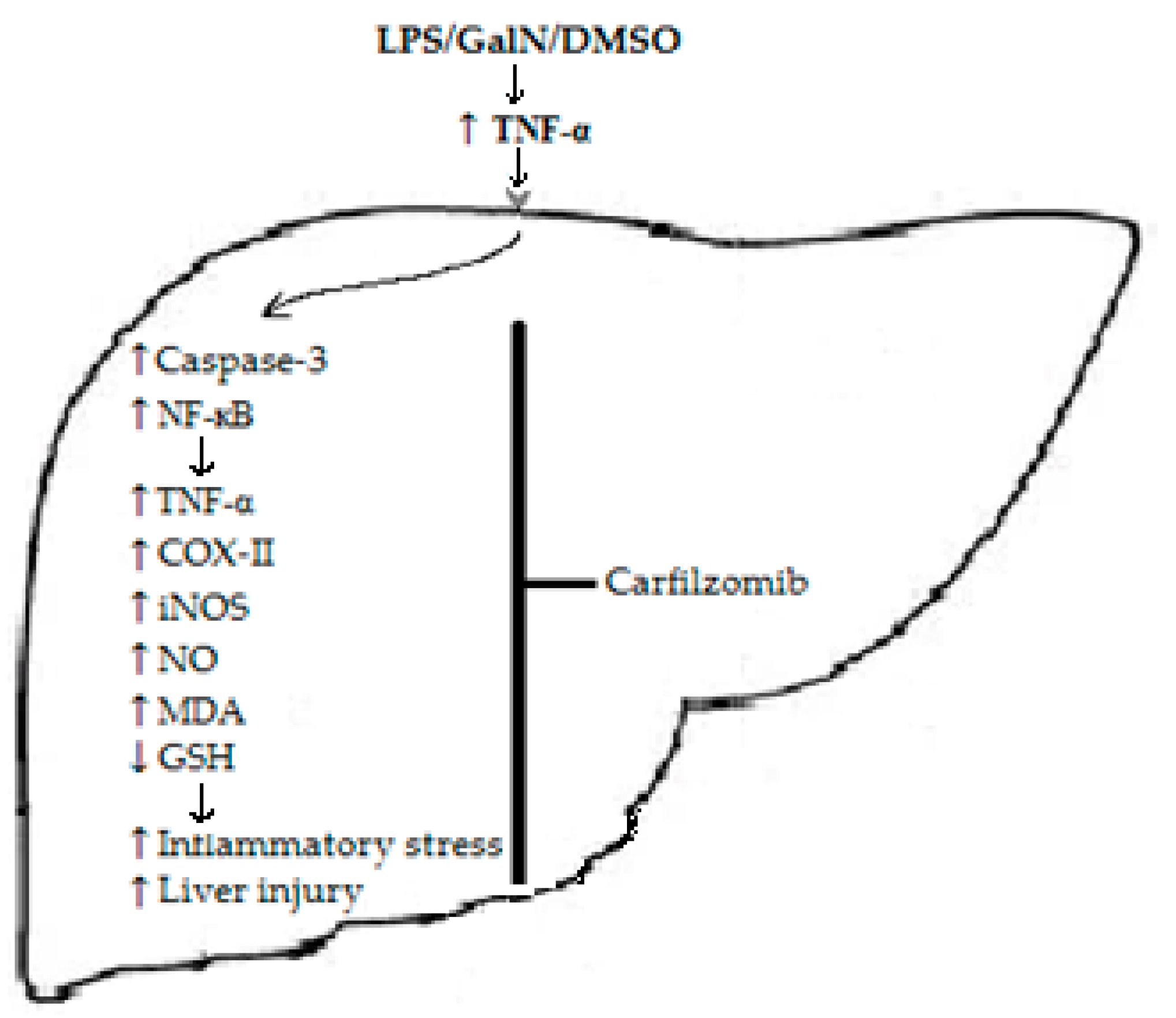
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.3. Serum Separation and Liver Preparations
2.4. Biochemical Analysis
2.5. Gene Expression
2.6. Western Blot
2.7. Histopathology
2.8. Statistical Analysis
3. Results
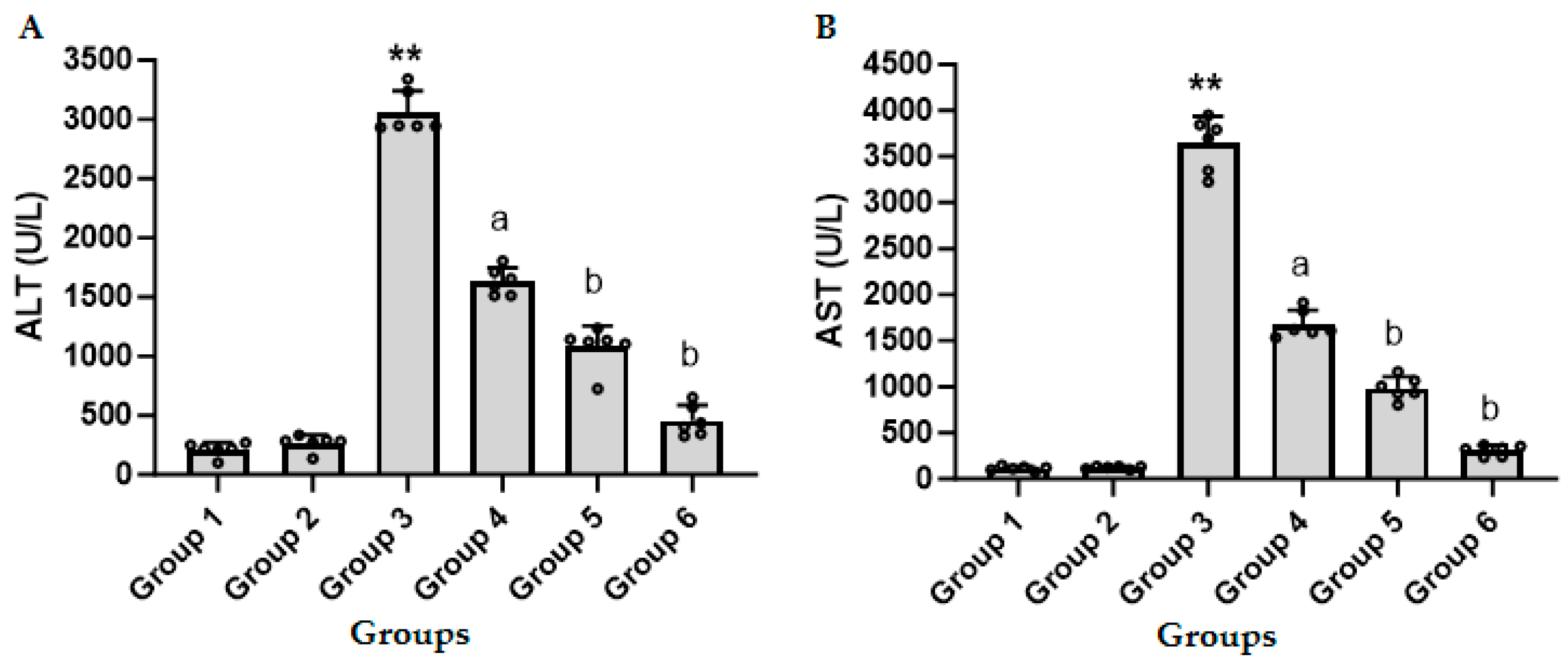
3.1. CFZ Decreases the LPS/GalN/DMSO-Induced Increase in ALT and AST in Serum
3.2. CFZ Decreases the LPS/GalN/DMSO-Induced Increase in Serum TNF-α, Hepatic NF-кB and Hepatic Caspase 3 Activity
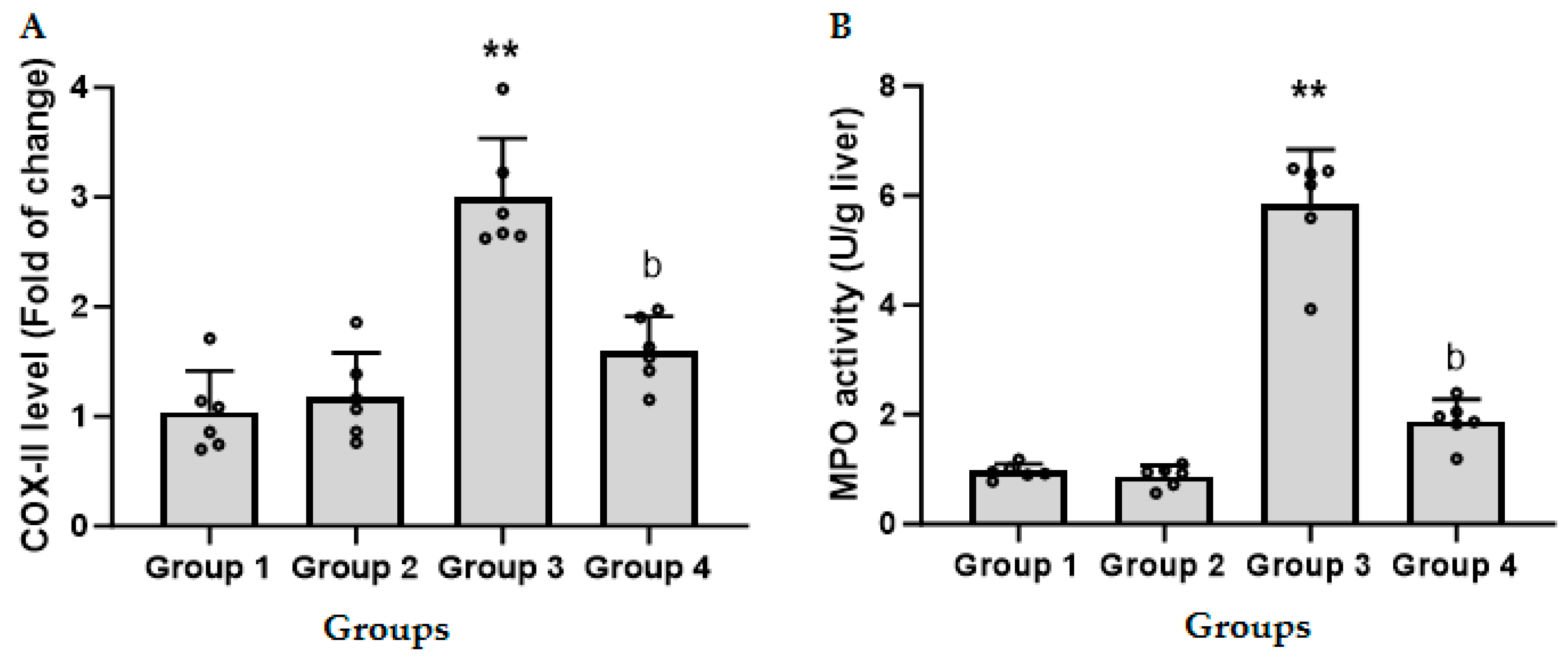
3.3. CFZ Decreases the LPS/GalN/DMSO-Induced Increase in Hepatic COX-II
3.4. CFZ Ameliorates the LPS/GalN/DMSO-Induced Neutrophil Recruitment
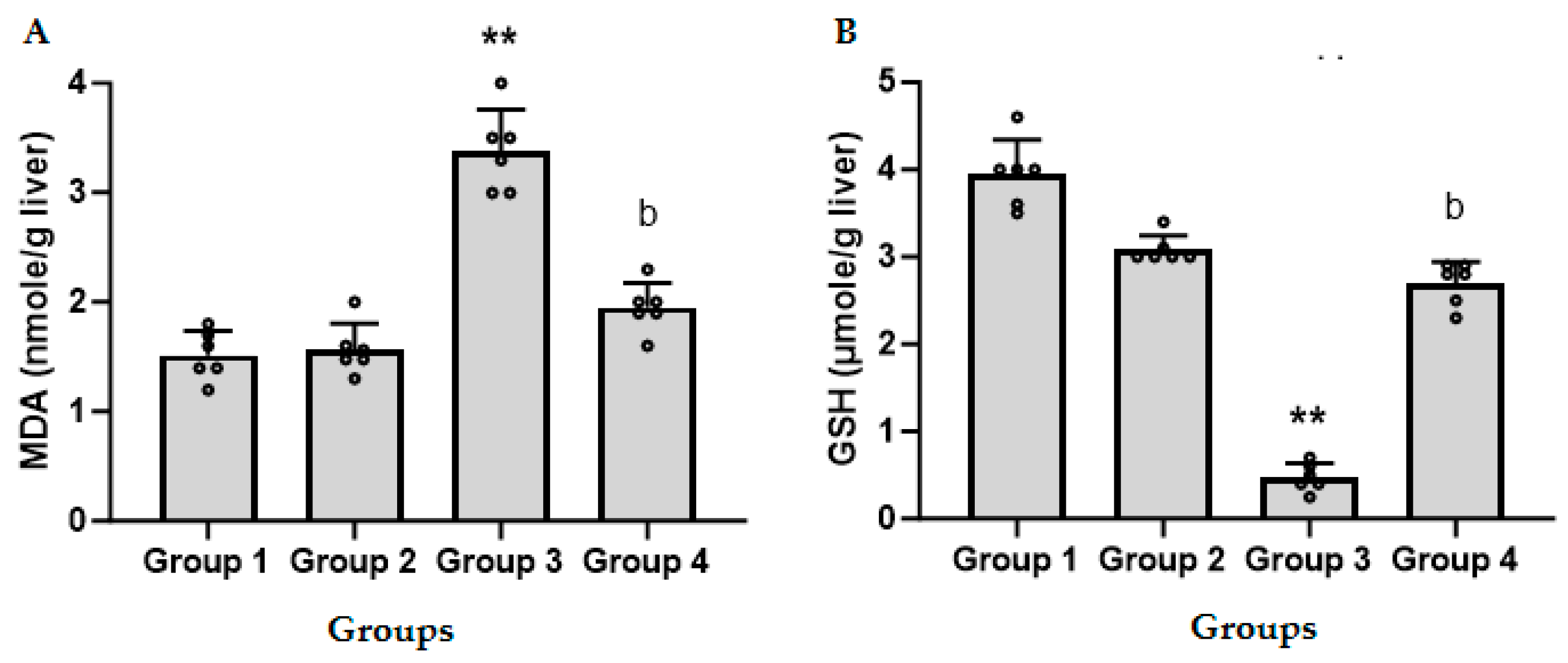
3.5. CFZ Ameliorates the LPS/GalN/DMSO-Induced Altered Nitrosative and Oxidative Stress
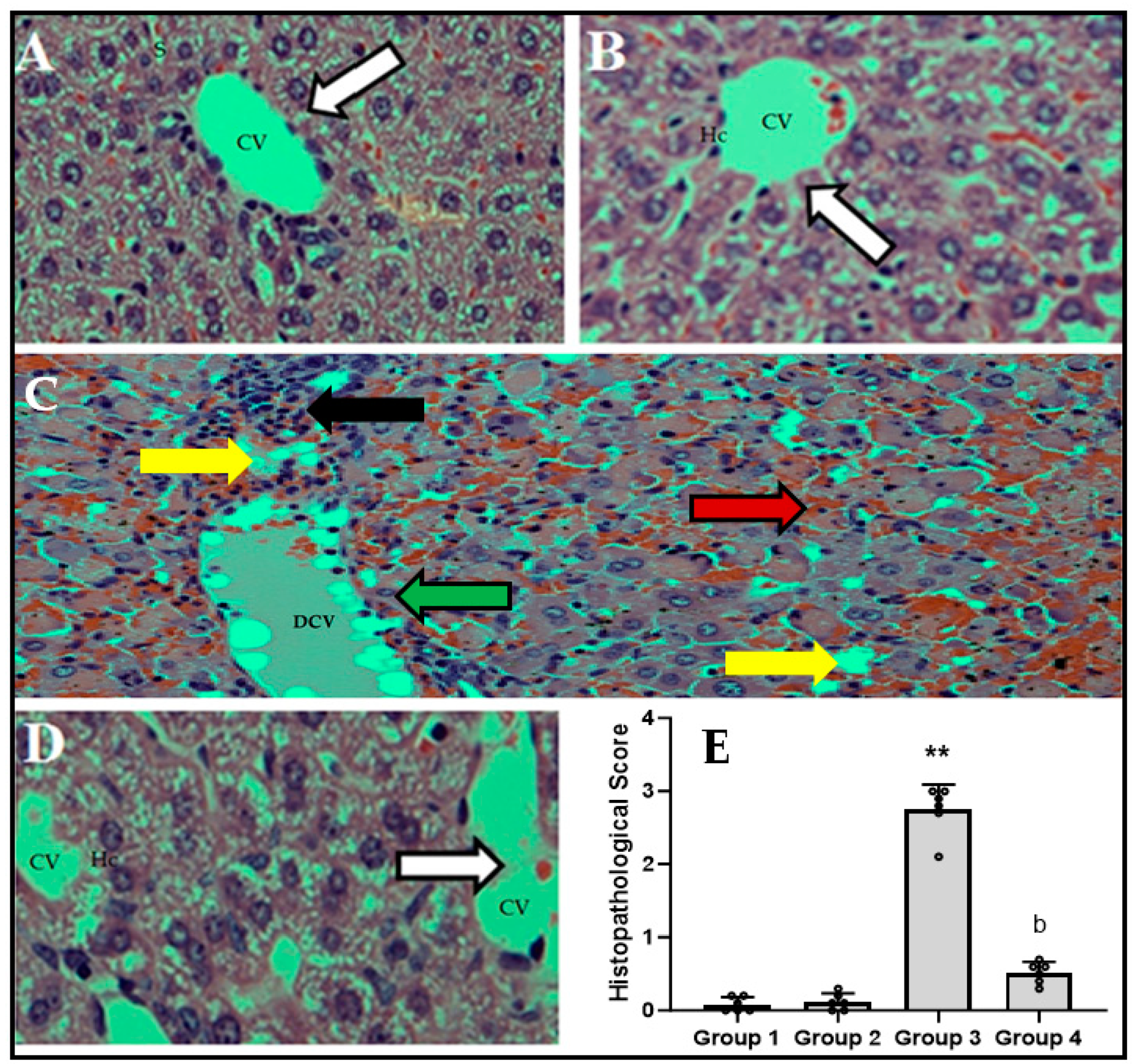
3.6. CFZ Decreases LPS/GalN/DMSO-Caused-Histopathological Changes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sowa, J.P.; Gerken, G.; Canbay, A. Acute Liver Failure—It’s Just a Matter of Cell Death. Dig. Dis. 2016, 34, 423–428. [Google Scholar] [CrossRef]
- Maes, M.; Vinken, M.; Jaeschke, H. Experimental models of hepatotoxicity related to acute liver failure. Toxicol. Appl. Pharmacol. 2016, 290, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Jaeschke, H.; Hasegawa, T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006, 26, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Smith, C.W. Mechanisms of neutrophil-induced parenchymal cell injury. J. Leukoc. Biol. 1997, 61, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Mohler, K.M.; Sleath, P.R.; Fitzner, J.N.; Cerretti, D.P.; Alderson, M.; Kerwar, S.S.; Torranee, D.S.; Otten-Evans, C.; Greenstreet, T.; Weerawarna, K. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 1994, 370, 218. [Google Scholar] [CrossRef]
- Masson, M.J.; Carpenter, L.D.; Graf, M.L.; Pohl, L.R. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology 2008, 48, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.; Nagi, M.N.; Alhareth, D.Y.; Al-Hamamah, M.A.; Mahmoud, M.A.; Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Harisa, G.I.; et al. Crosstalk of TNF-α, IFN-γ, NF-kB, STAT1 and redox signaling in lipopolysaccharide/d-galactosamine/dimethylsulfoxide-induced fulminant hepatic failure in mice. Saudi Pharm. J. 2023, 31, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29 (Suppl. 1), 3–9. [Google Scholar] [CrossRef] [PubMed]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef]
- Bardag-Gorce, F. Proteasome inhibitor treatment in alcoholic liver disease. World J. Gastroenterol. 2011, 17, 2558–2562. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Sawada, H.; Izumi, Y.; Kume, T.; Katsuki, H.; Shimohama, S.; Akaike, A. Proteasome inhibition induces glutathione synthesis and protects cells from oxidative stress: Relevance to Parkinson disease. J. Biol. Chem. 2007, 282, 4364–4372. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Wilck, N.; Meiners, S.; Ludwig, A.; Baumann, G.; Stangl, K.; Stangl, V. Proteasome inhibition prevents experimentally-induced endothelial dysfunction. Life Sci. 2009, 84, 929–934. [Google Scholar] [CrossRef]
- Williams, A.J.; Hale, S.L.; Moffett, J.R.; Dave, J.R.; Elliott, P.J.; Adams, J.; Tortella, F.C. Delayed treatment with MLN519 reduces infarction and associated neurologic deficit caused by focal ischemic brain injury in rats via antiinflammatory mechanisms involving nuclear factor-kappaB activation, gliosis, and leukocyte infiltration. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2003, 23, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Takaoka, M.; Shibata, A.; Ohkita, M.; Matsumura, Y. Preventive effect of lactacystin, a selective proteasome inhibitor, on ischemic acute renal failure in rats. J. Pharmacol. Exp. Ther. 2001, 298, 501–507. [Google Scholar] [PubMed]
- Park, W.J.; Kim, S.Y.; Kim, Y.R.; Park, J.W. Bortezomib alleviates drug-induced liver injury by regulating CYP2E1 gene transcription. Int. J. Mol. Med. 2016, 37, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.; Algfeley, S.G.; Al-Hosaini, K.A.; Korashy, H.M.; Imam, F.; Nagi, M.N. Therapeutic potential of carfilzomib, an irreversible proteasome inhibitor, against acetaminophen-induced hepatotoxicity in mice. J. Biochem. Mol. Toxicol. 2017, 31, e21877. [Google Scholar] [CrossRef]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. [Google Scholar] [CrossRef]
- Chen, Q.; Lu, X.; Zhang, X. Noncanonical NF-κB Signaling Pathway in Liver Diseases. J. Clin. Transl. Hepatol. 2021, 9, 81–89. [Google Scholar] [CrossRef]
- VPalombella, J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-κB. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Hu, J.J.; Wang, H.; Pan, C.W.; Lin, M.X. Isovitexin alleviates liver injury induced by lipopolysaccharide/d-galactosamine by activating Nrf2 and inhibiting NF-κB activation. Microb. Pathog. 2018, 119, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.M. Abatement by naringin of lomefloxacin-induced genomic instability in mice. Mutagenesis 2008, 23, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Harisa, G.I.; Mariee, A.D.; Abo-Salem, O.M.; Attiaa, S.M. Erythrocyte nitric oxide synthase as a surrogate marker for mercury-induced vascular damage: The modulatory effects of naringin. Environ. Toxicol. 2014, 29, 1314–1322. [Google Scholar] [CrossRef]
- Bradley, P.P.; Priebat, D.A.; Christensen, R.D.; Rothstein, G. Measurement of cutaneous inflammation: Estimation of neutrophil content with an enzyme marker. J. Investig. Dermatol. 1982, 78, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamamah, M.A.; Alotaibi, M.R.; Ahmad, S.F.; Ansari, M.A.; Attia, M.S.M.; Nadeem, A.; Bakheet, S.A.; Sobeai, H.M.A.; Attia, S.M. Genetic and epigenetic alterations induced by the small-molecule panobinostat: A mechanistic study at the chromosome and gene levels. DNA Repair. 2019, 78, 70–80. [Google Scholar] [CrossRef]
- Attia, S.M.; Ahmad, S.F.; Harisa, G.I.; Mansour, A.M.; El Sayed, S.M.; Bakheet, S.A. Wogonin attenuates etoposide-induced oxidative DNA damage and apoptosis via suppression of oxidative DNA stress and modulation of OGG1 expression. Food Chem. Toxicol. 2013, 59, 724–730. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Zoheir, K.M.; Ansari, M.A.; Korashy, H.M.; Bakheet, S.A.; Ashour, A.E.; Al-Shabanah, O.A.; Al-harbi, M.M.; Attia, S.M. The role of poly(ADP-ribose) polymerase-1 inhibitor in carrageenan-induced lung inflammation in mice. Mol. Immunol. 2015, 63, 394–405. [Google Scholar] [CrossRef]
- Attia, S.M.; Alshahrani, A.Y.; Al-Hamamah, M.A.; Attia, M.M.; Saquib, Q.; Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A. Dexrazoxane Averts Idarubicin-Evoked Genomic Damage by Regulating Gene Expression Profiling Associated With the DNA Damage-Signaling Pathway in BALB/c Mice. Toxicol. Sci. 2017, 160, 161–172. [Google Scholar] [CrossRef]
- Rosner, B. Fundamentals of Biostatistics, 7th ed.; Brooks/Cole; Cengage Learning: Boston, MA, USA, 2011. [Google Scholar]
- Farghali, H.; Cerný, D.; Kameníková, L.; Martínek, J.; Horínek, A.; Kmonícková, E.; Zídek, Z. Resveratrol attenuates lipopolysaccharide-induced hepatitis in D-galactosamine sensitized rats: Role of nitric oxide synthase 2 and heme oxygenase-1. Nitric Oxide 2009, 21, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, W.; Dong, S.; Song, L.; Zhao, S.; Wu, C.; Wang, X.; Liu, F.; Xie, J.; Wang, J. Protective effects of sea buckthorn polysaccharide extracts against LPS/d-GalN-induced acute liver failure in mice via suppressing TLR4-NF-κB signaling. J. Ethnopharmacol. 2015, 176, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.T.; Liu, W.; Xue, C.R.; Wu, S.X.; Chen, W.N.; Lin, X.J.; Lin, X. AKT activator SC79 protects hepatocytes from TNF-alpha-mediated apoptosis and alleviates d-Gal/LPS-induced liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G387–G396. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Gong, X.; Kuang, G.; Jiang, R.; Chen, R.; Wan, J. Sesamin ameliorates lipopolysaccharide/d-galactosamine-induced fulminant hepatic failure by suppression of Toll-like receptor 4 signaling in mice. Biochem. Biophys. Res. Commun. 2015, 461, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Zhao, L.; Shi, M.; Wei, Z.; Yang, Z.; Guo, C.; Fu, Y. Costunolide protects lipopolysaccharide/d-galactosamine-induced acute liver injury in mice by inhibiting NF-kappaB signaling pathway. J. Surg. Res. 2017, 220, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Fisher, M.A.; Lawson, J.A.; Simmons, C.A.; Farhood, A.; Jones, D.A. Activation of caspase 3 (CPP32)-like proteases is essential for TNF-alpha-induced hepatic parenchymal cell apoptosis and neutrophil-mediated necrosis in a murine endotoxin shock model. J. Immunol. 1998, 160, 3480–3486. [Google Scholar] [CrossRef]
- Wang, H.; Xu, D.X.; Lv, J.W.; Ning, H.; Wei, W. Melatonin attenuates lipopolysaccharide (LPS)-induced apoptotic liver damage in D-galactosamine-sensitized mice. Toxicology 2007, 237, 49–57. [Google Scholar] [CrossRef]
- Xu, L.; Zheng, X.; Wang, Y.; Fan, Q.; Zhang, M.; Li, R.; Ye, J.; Wu, X.; Zhao, W.; Zhang, Y. Berberine protects acute liver failure in mice through inhibiting inflammation and mitochondria-dependent apoptosis. Eur. J. Pharmacol. 2018, 819, 161–168. [Google Scholar] [CrossRef]
- Li, J.; Chen, B.; Zhong, L.; Gao, F.; Zhu, H.; Wang, F. AMP-activated protein kinase agonist N6-(3-hydroxyphenyl)adenosine protects against fulminant hepatitis by suppressing inflammation and apoptosis. Cell Death Dis. 2018, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Chen, C.; Yu, H.; Diao, H.; Shi, C.; Wang, Y.; Li, G.; Shi, M. Oridonin ameliorates lipopolysaccharide/D-galactosamine-induced acute liver injury in mice via inhibition of apoptosis. Am. J. Transl. Res. 2017, 9, 4271–4279. [Google Scholar] [PubMed]
- Luo, M.; Zhao, A.; Li, J.; Chen, Y.; Tian, D.; Wang, C.; Hu, Z.; Gao, J. Acute liver injury attenuation of a novel recombinant sTNFR through blocking hepatic apoptosis. Immunopharmacol. Immunotoxicol. 2015, 37, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Li, G.; Lim, K.; DeFrances, M.C.; Gandhi, C.R.; Wu, T. Transgenic expression of cyclooxygenase-2 in hepatocytes accelerates endotoxin-induced acute liver failure. J. Immunol. 2008, 181, 8027–8035. [Google Scholar] [CrossRef]
- Grattagliano, I.; Calamita, G.; Cocco, T.; Wang, D.Q.; Portincasa, P. Pathogenic role of oxidative and nitrosative stress in primary biliary cirrhosis. World J. Gastroenterol. 2014, 20, 5746–5759. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Yang, Y.; Huang, L.; Zhang, Q.; Wan, R.-Z.; Zhang, P.; Zhang, B. Antioxidation, anti-inflammation and anti-apoptosis by paeonol in LPS/d-GalN-induced acute liver failure in mice. Int. Immunopharmacol. 2017, 46, 124–132. [Google Scholar] [CrossRef]
- Hwang, J.-S.; Kwon, M.-Y.; Kim, K.-H.; Lee, Y.; Lyoo, I.K.; Kim, J.E.; Oh, E.-S.; Han, I.-O. Lipopolysaccharide (LPS)-stimulated iNOS induction is increased by glucosamine under normal glucose conditions but is inhibited by glucosamine under high glucose conditions in macrophage cells. J. Biol. Chem. 2017, 292, 1724–1736. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-J.; Lin, Y.-W.; Chuang, C.-Y.; Tsai, H.-C.; Chen, R.-M. Liver nitrosation and inflammation in septic rats were suppressed by propofol via downregulating TLR4/NF-κB-mediated iNOS and IL-6 gene expressions. Life Sci. 2018, 195, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 173–179. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A. Reactive oxygen species in the normal and acutely injured liver. J. Hepatol. 2011, 55, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Su, C.; Fu, J.; Zhang, P.; Jiang, X.; Xu, D.; Hu, L.; Song, E.; Song, Y. Role of alpha-lipoic acid in LPS/d-GalN induced fulminant hepatic failure in mice: Studies on oxidative stress, inflammation and apoptosis. Int. Immunopharmacol. 2014, 22, 293–302. [Google Scholar] [CrossRef]
- Ito, K.; Ozasa, H.; Noda, Y.; Arii, S.; Horikawa, S. Effects of free radical scavenger on acute liver injury induced by d-galactosamine and lipopolysaccharide in rats. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2008, 38, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.G.; Sha, K.H.; Zhang, L.G.; Liu, X.X.; Yang, F.; Cheng, J.Y. Protective effects of alpinetin on lipopolysaccharide/d-Galactosamine-induced liver injury through inhibiting inflammatory and oxidative responses. Microb. Pathog. 2019, 126, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Liu, H.L.; Yu, Z.J.; Huang, Y.H.; Gao, D.; Hao, H.; Liao, S.S.; Xu, F.Y.; Zhou, X.Y. BML-111 Protected LPS/D-GalN-Induced Acute Liver Injury in Rats. Int. J. Mol. Sci. 2016, 17, 1114. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Li, H.; Zhao, Y.; Cai, E.; Zhu, H.; Li, P.; Liu, J. Hepatoprotective effect of α-mangostin against lipopolysaccharide/d-galactosamine-induced acute liver failure in mice. Biomed. Pharmacother. 2018, 106, 896–901. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | GenBank Accession # | Annealing Temperature | Orientation | Sequences (5’ to 3’) |
|---|---|---|---|---|
| COX-II | NM_011198 | 60 °C | Forward Reverse | TGGTGCCTGGTCTGATGATG GTGGTAACCGCTCAGGTGTTG |
| iNOS | NM_010927 | 60 °C | Forward Reverse | CCTGGTACGGGCATTGCT GCTCATGCGGCCTCCTTT |
| GAPDH | NM_008084 | 60 °C | Forward Reverse | TGAAGCAGGCATCTGAGGG CGAAGGTGGAAGAGTGGGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhareth, D.Y.; Alanazi, A.; Alanazi, W.A.; Ansari, M.A.; Nagi, M.N.; Ahmad, S.F.; Attia, M.S.M.; Nadeem, A.; Bakheet, S.A.; Attia, S.M. Carfilzomib Mitigates Lipopolysaccharide/D-Galactosamine/Dimethylsulfoxide-Induced Acute Liver Failure in Mice. Biomedicines 2023, 11, 3098. https://doi.org/10.3390/biomedicines11113098
Alhareth DY, Alanazi A, Alanazi WA, Ansari MA, Nagi MN, Ahmad SF, Attia MSM, Nadeem A, Bakheet SA, Attia SM. Carfilzomib Mitigates Lipopolysaccharide/D-Galactosamine/Dimethylsulfoxide-Induced Acute Liver Failure in Mice. Biomedicines. 2023; 11(11):3098. https://doi.org/10.3390/biomedicines11113098
Chicago/Turabian StyleAlhareth, Dhafer Y., Abdulrazaq Alanazi, Wael A. Alanazi, Mushtaq A. Ansari, Mahmoud N. Nagi, Sheikh F. Ahmad, Mohamed S. M. Attia, Ahmed Nadeem, Saleh A. Bakheet, and Sabry M. Attia. 2023. "Carfilzomib Mitigates Lipopolysaccharide/D-Galactosamine/Dimethylsulfoxide-Induced Acute Liver Failure in Mice" Biomedicines 11, no. 11: 3098. https://doi.org/10.3390/biomedicines11113098