

Disease Mechanisms and Therapeutic Approaches in SMARD1—Insights from Animal Models and Cell Models

Abstract

1. Introduction
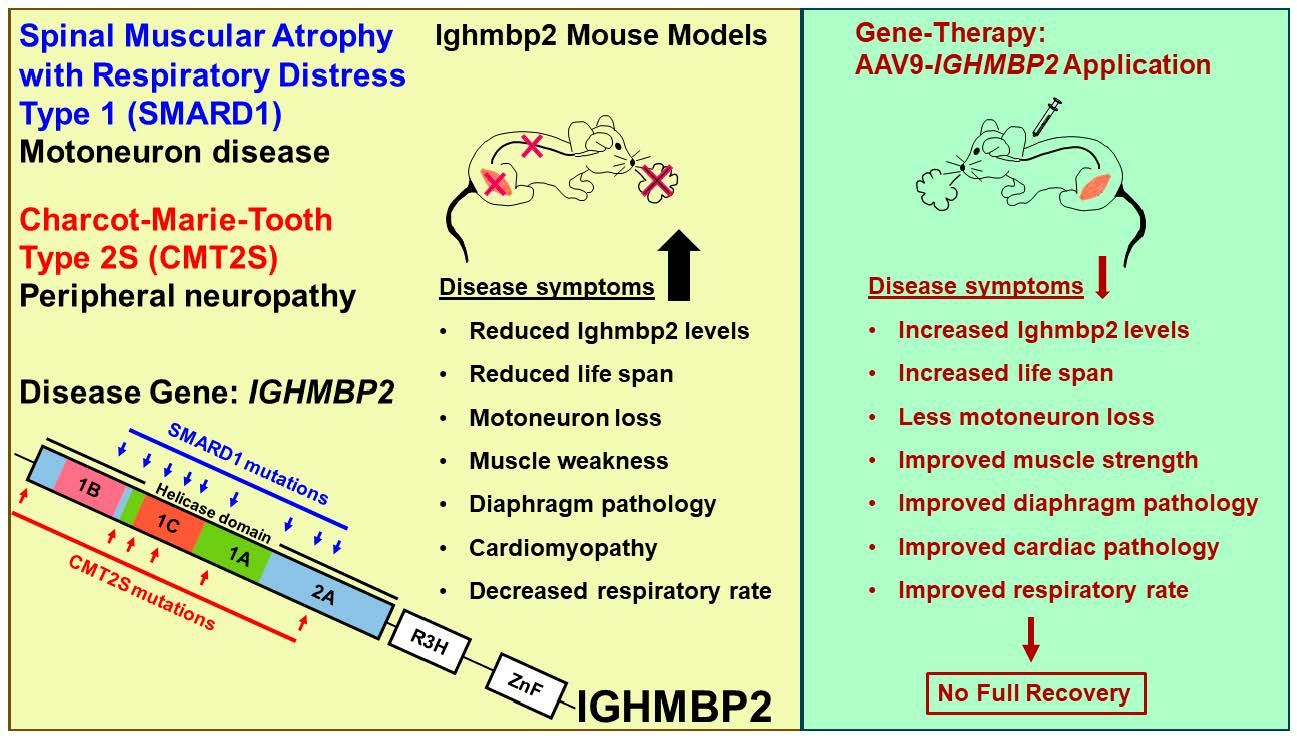
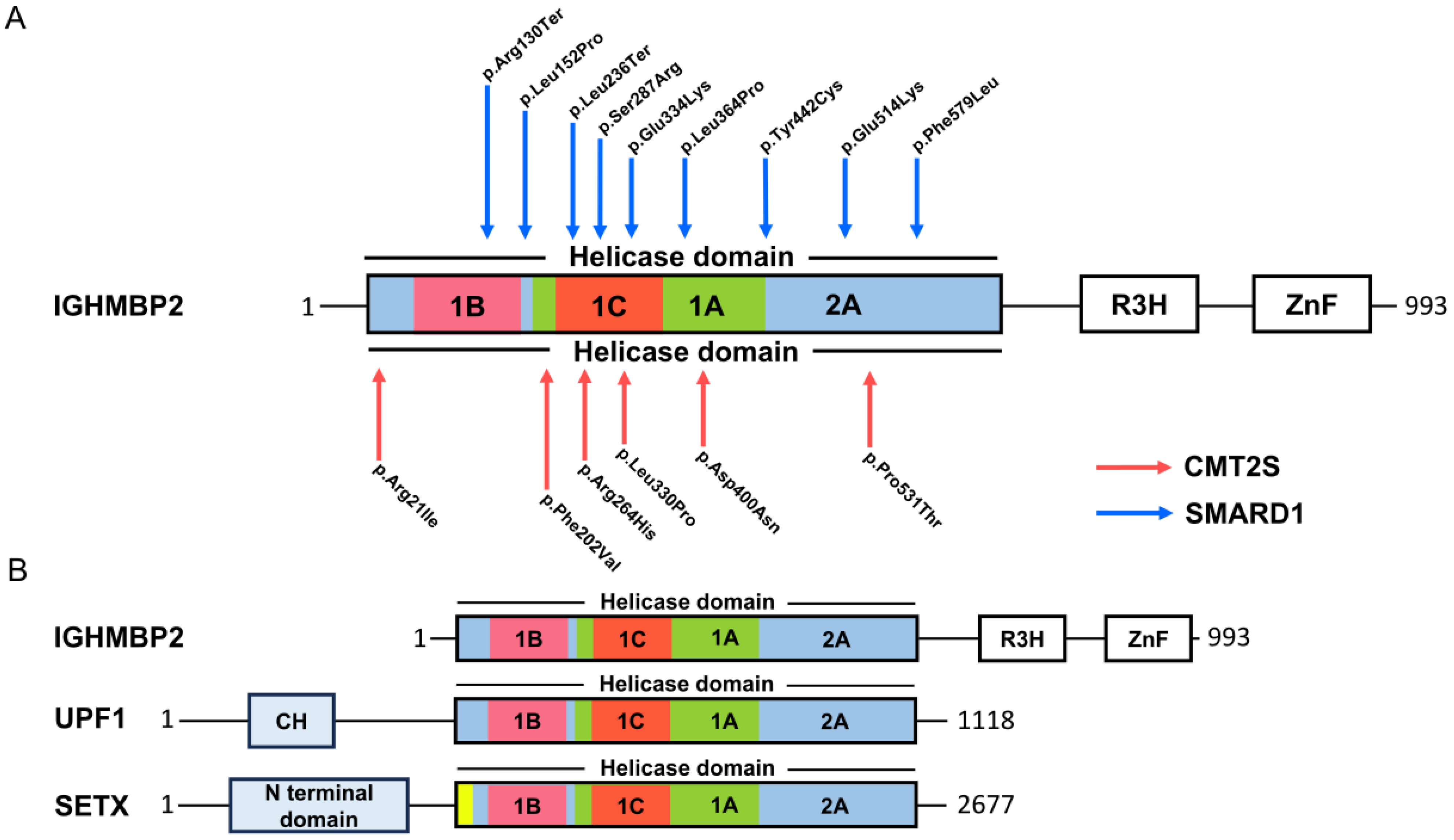
1.1. Spinal Muscular Atrophy with Respiratory Distress Type 1 (SMARD1)
1.2. Yeast Assay Analysis of Single IGHMBP2 Mutations
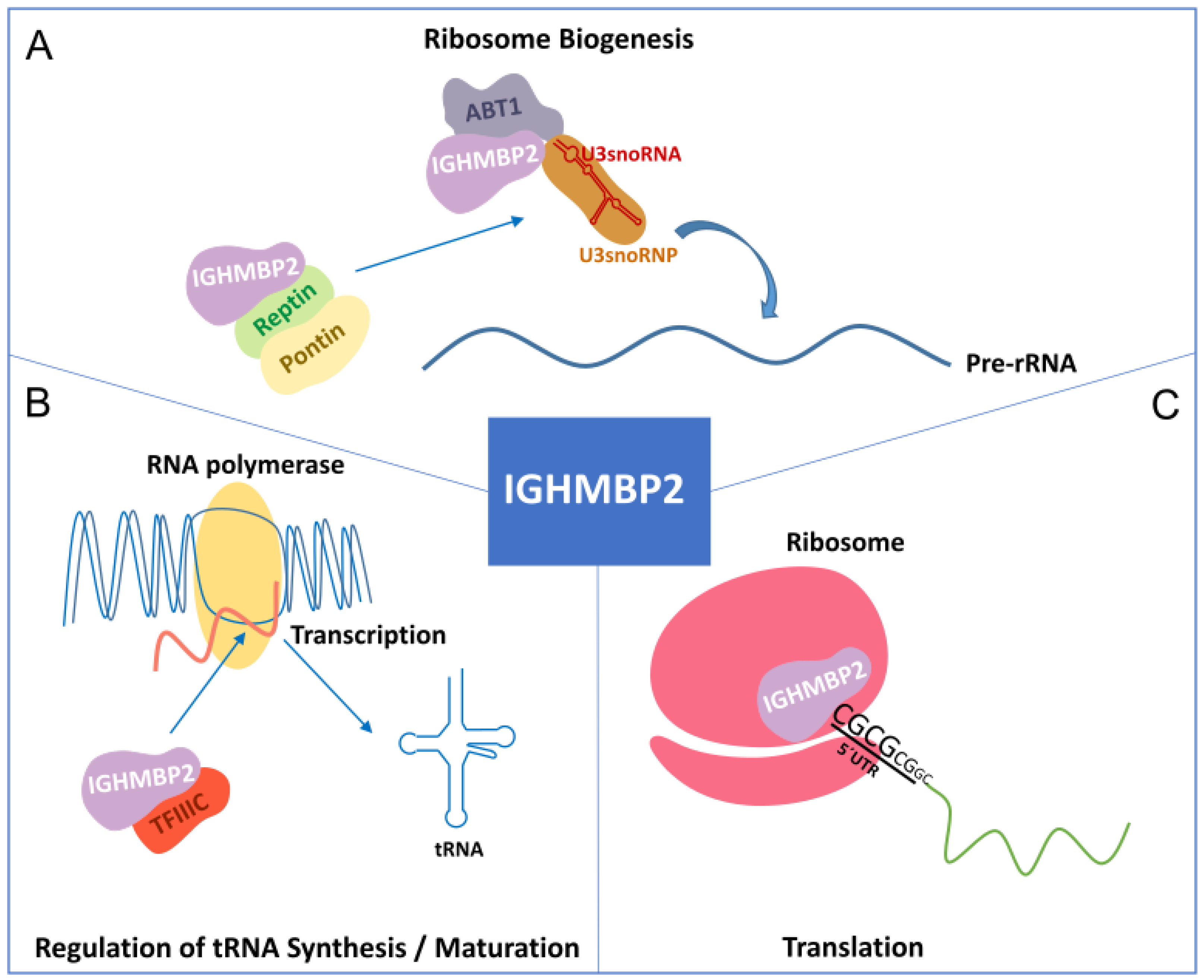
2. IGHMBP2 Function and Modifiers of IGHMBP2 Deficiency
2.1. Modifiers of IGHMBP2 Deficiency
2.2. Function of IGHMBP2
3. Mouse Models of SMARD1 and Peripheral Neuropathies
3.1. The Nmd2J Mouse as a Model for SMARD1
3.2. A SMARD1 Mouse with Respiratory Distress
3.3. Ighmbp2 Mouse Models Recapitulating Peripheral Neuropathies
4. Insights into Cellular and Functional Dysregulation
4.1. Cellular Dysregulations in Motoneurons
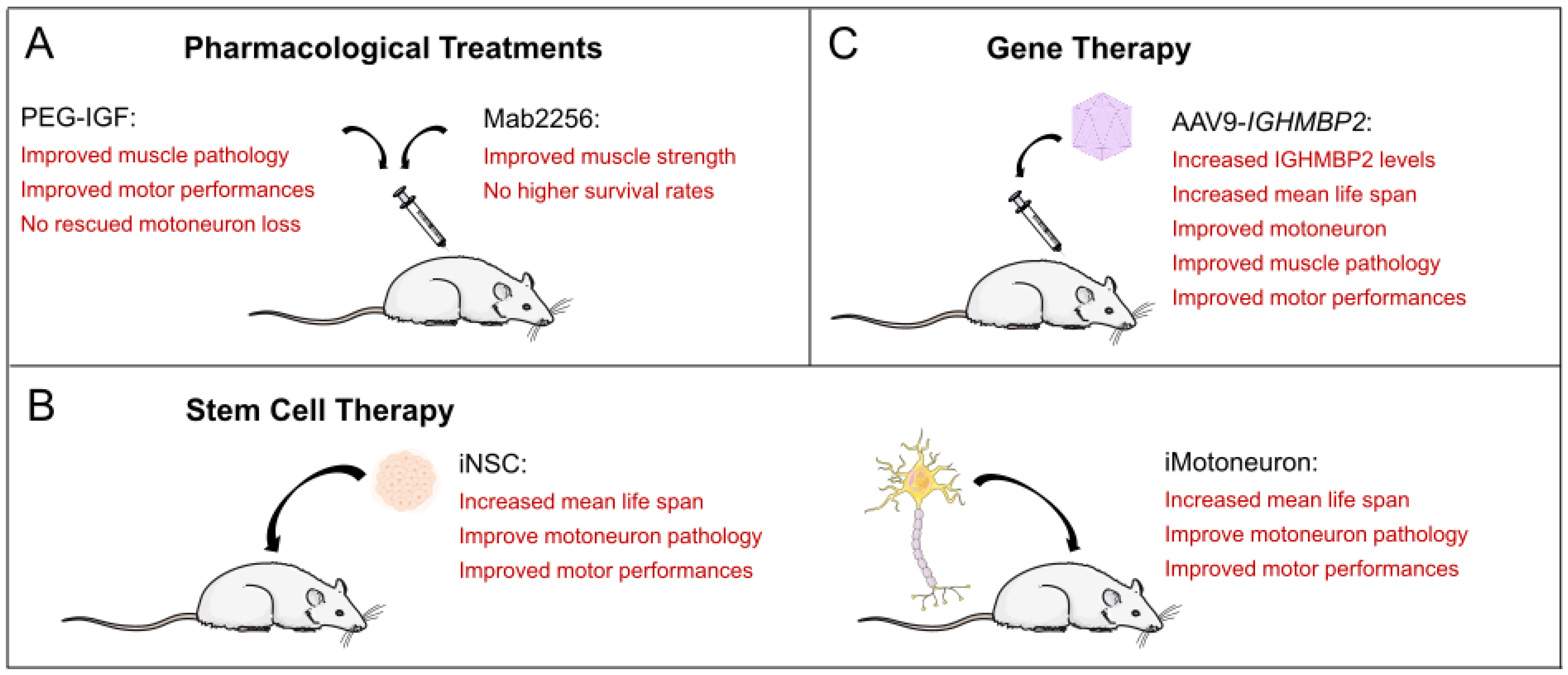
4.2. Cellular Dysregulation Due to Reduced Growth Factor Release
5. Stem Cell Therapy in SMARD1 Mouse Models
5.1. Transplantation of Mouse Stem Cells and Mouse Motoneurons
5.2. Transplantation of Human iPSC-Derived NSCs
6. Gene Therapy in Mouse Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Phenotype of Untreated Mice | Phenotype after AAV9-IGHMBP2 Application |
|---|---|---|
| Ighmbp2nmd−2J/J (Nmd2J) Spontaneous point mutation in intron 4 (c.39A->G) in C57BL/6 background Cox et al., 1998 [33] Grohmann et al., 2004 [35] | IV, 5 × 1011 vg per pub at P1 by Nizzardo et al., 2015 [63] | |
| 20–30% remaining Ighmbp2 | Two-fold increase in IGHMBP2 levels. Increased mean life span. | |
| Survival ranges from 6 to 10 weeks up to 11 months. Lower body weight. | Increased weight but not reaching wild-type level. Preservation of motor units and restored NMJ function. | |
| Motoneuron loss, loss of NMJ function. Affected muscle fibers. | Improved muscle fiber morphology. Neuromuscular function improved but did not reach wild-type levels. | |
| Cardiomyopathy is present in later stages of the disease. | Slightly improved cardiac hypertrophy. | |
| Ighmbp2nmd−2J/J (Nmd2J) Spontaneous point mutation in intron 4 (c.39A->G) in C57BL/6 background Cox et al., 1998 [33] Grohmann et al., 2004 [35] | ICV, 1.25 × 1011 vg per pub at P2,3 by Shababi et al., 2016 [47] and 2018 [64] | |
| 20–30% remaining Ighmbp2. | 30% increase in IGHMBP2 levels at 30 days. | |
| Survival ranges from 6 to 10 weeks up to 11 months. Lower body weight. | Increased survival to 11–12 months. Increased body weight but not reaching wild-type levels. | |
| Motor unit loss and loss of NMJ function. Affected muscle fibers. | Rescued loss of motoneurons and motor axons and increased motor performance, but not reaching wild-type levels. | |
| Degeneration of the diaphragm. | Improved muscle pathology in hind limbs and diaphragm. | |
| Cardiomyopathy is present in later stages of the disease. | Improved cardiac pathology. | |
| Ighmbp2nmd−2J/J (Nmd2J) Spontaneous point mutation in intron 4 (c.39A->G) in C57BL/6 background Cox et al., 1998 [33] Grohmann et al., 2004 [35] | IV, 1.25 × 1011 vg per pub at P2 by Shababi et al., 2018 [64] | |
| 20–30% remaining Ighmbp2. | Lower IGHMBP2 levels than ICV-treated Nmd2J. | |
| Survival ranges from 6 to 10 weeks up to 11 months. Lower body weight. | Equal survival rate as with ICV-injected Nmd2J. | |
| Motor unit loss and loss of NMJ function. | No rescue of hind limb contracture and motor function. Gastrocnemius muscle and NMJ defects are not rescued. | |
| Degeneration of the diaphragm. Cardiomyopathy is present in the later stages of the disease. | Improved diaphragm and cardiac pathology comparable to ICV-injected Nmd2J. | |
| FVB/NJ-Ighmbp2nmd/nmd Point mutation in intron 4 (c.39A->G) introduced by CRISPR/Cas9 in congenic FVB/NJ background Shababi et al., 2019 [48] | Low-dose ICV, 1.25 × 1011 vg per pub at P2, High-dose ICV, 2.5 × 1011 vg per pub at P2,3 by Shababi et al., 2021 [65] | |
| Life span of 18–21 days. | Low dose: increased survival up to 80 days. High dose: increased survival beyond 100 days. | |
| Lower body weight. | Low dose: weight gain, but not reaching wild-type levels. High dose: weight gain, but not reaching wild-type levels. | |
| Severe muscle weakness in the hind limbs. | Low dose: Rotarod performances improved to wild-type levels. Grip strength improved, but not to wild type level. High dose: Rotarod performances improved to wild-type levels. Grip strength improved, but not to wild-type levels. | |
| Reduced muscle fiber areas. | High dose: Muscle fiber pathology improved, but not to wild-type levels. | |
| Reduced NMJ innervation. | High dose: NMJ innervation improved to wild-type levels. | |
| Reduced motoneuron number and area. | High dose: motoneuron number and area are improved to wild-type levels. | |
| FVB/NJ-Ighmbp2D564N Missense mutation D564N introduced by CRISPR/Cas9 in FVB/NJ background Smith et al., 2022 [49] | ICV 5 × 1011 vg per pub at P2 by Smith et al., 2022 [49] | |
| Ighmbp2 level is similar to the wild-type control. | Ighmbp2/IGHMBP2 level is similar to the wild-type control. | |
| Lifespan of 12–22 days. Lower body weight. | 5- to 10-times increased survival rate and weight gain, but not reaching wild-type levels. | |
| Muscle weakness in the hindlimbs. | Improved motor function (time-to-right, grip strength, hind limb splay, grip strength, and rotarod), but not reaching wild-type levels. | |
| Reduced motoneuron numbers and size. Decreased number of innervated endplates. | Increased motoneuron number, but not reaching wild-type levels. Increased innervated endplates, reaching wild-type levels. | |
| Reduced muscle fiber size. | Increased muscle fiber size, but not reaching wild-type levels. | |
| Decreased respiratory rate under normoxic conditions. | Improved respiratory frequency and number of apneas and erratic breathing under normoxic and hypercapnia + hypoxia conditions, similar to wild-type levels. |
6.1. AAV9-IGHMBP2 Application to the Nmd2J Model
6.2. AAV9-IGHMBP2 Application to the FVBIghmbp2nmd/nmd Model
6.3. AAV9-IGHMBP2 Application to a SMARD1 Mouse Model with Respiratory Distress
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jablonka, S.; Hennlein, L.; Sendtner, M. Therapy development for spinal muscular atrophy: Perspectives for muscular dystrophies and neurodegenerative disorders. Neurol. Res. Pract. 2022, 4, 2. [Google Scholar] [CrossRef]
- Rudnik Schoneborn, S.; Forkert, R.; Hahnen, E.; Wirth, B.; Zerres, K. Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: Further delineation on the basis of SMN gene deletion findings. Neuropediatrics 1996, 27, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Mellins, R.B.; Hays, A.P.; Gold, A.P.; Berdon, W.E.; Bowdler, J.D. Respiratory distress as the initial manifestation of Werdnig-Hoffmann disease. Pediatrics 1974, 53, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, A.; Guenther, U.P.; Jankowsky, E. The RNA helicase database. Nucleic Acids Res. 2011, 39, D338–D341. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xing, J.; Shi, Y.; Yuan, E. Exploring the relationship between IGHMBP2 gene mutations and spinal muscular atrophy with respiratory distress type 1 and Charcot-Marie-Tooth disease type 2S: A systematic review. Front. Neurosci. 2023, 17, 1252075. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, K.; Schuelke, M.; Diers, A.; Hoffmann, K.; Lucke, B.; Adams, C.; Bertini, E.; Leonhardt-Horti, H.; Muntoni, F.; Ouvrier, R.; et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 2001, 29, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, K.; Varon, R.; Stolz, P.; Schuelke, M.; Janetzki, C.; Bertini, E.; Bushby, K.; Muntoni, F.; Ouvrier, R.; Van Maldergem, L.; et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann. Neurol. 2003, 54, 719–724. [Google Scholar] [CrossRef]
- Rudnik-Schoneborn, S.; Stolz, P.; Varon, R.; Grohmann, K.; Schachtele, M.; Ketelsen, U.P.; Stavrou, D.; Kurz, H.; Hubner, C.; Zerres, K. Long-term observations of patients with infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Neuropediatrics 2004, 35, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Saladini, M.; Nizzardo, M.; Govoni, A.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. Spinal muscular atrophy with respiratory distress type 1: Clinical phenotypes, molecular pathogenesis and therapeutic insights. J. Cell Mol. Med. 2020, 24, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Kaindl, A.M.; Guenther, U.P.; Rudnik-Schoneborn, S.; Varon, R.; Zerres, K.; Gressens, P.; Schuelke, M.; Hubner, C.; von Au, K. Distal spinal-muscular atrophy 1 (DSMA1 or SMARD1). Arch. Pediatr. 2008, 15, 1568–1572. [Google Scholar] [CrossRef]
- Eckart, M.; Guenther, U.P.; Idkowiak, J.; Varon, R.; Grolle, B.; Boffi, P.; Van, M.L.; Hubner, C.; Schuelke, M.; von Au, K. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics 2012, 129, e148–e156. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Robb, S.A.; Mohammed, S.; Lillis, S.; Simonds, A.; Manzur, A.Y.; Walter, S.; Wraige, E. Interfamilial phenotypic heterogeneity in SMARD1. Neuromuscul. Disord. 2009, 19, 193–195. [Google Scholar] [CrossRef]
- Blaschek, A.; Glaser, D.; Kuhn, M.; Schroeder, A.S.; Wimmer, C.; Heimkes, B.; Schon, C.; Muller-Felber, W. Early infantile sensory-motor neuropathy with late onset respiratory distress. Neuromuscul. Disord. 2014, 24, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Guenther, U.P.; Handoko, L.; Varon, R.; Stephani, U.; Tsao, C.Y.; Mendell, J.R.; Lutzkendorf, S.; Hubner, C.; von Au, K.; Jablonka, S.; et al. Clinical variability in distal spinal muscular atrophy type 1 (DSMA1): Determination of steady-state IGHMBP2 protein levels in five patients with infantile and juvenile disease. J. Mol. Med. 2009, 87, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.D.; Huang, L.; Tetreault, M.; Majewski, J.; Boycott, K.M.; Bulman, D.E.; Care4Rare Canada, C.; Dyment, D.A.; McMillan, H.J. Autosomal recessive axonal polyneuropathy in a sibling pair due to a novel homozygous mutation in IGHMBP2. Neuromuscul. Disord. 2015, 25, 794–799. [Google Scholar] [CrossRef]
- Cottenie, E.; Kochanski, A.; Jordanova, A.; Bansagi, B.; Zimon, M.; Horga, A.; Jaunmuktane, Z.; Saveri, P.; Rasic, V.M.; Baets, J.; et al. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am. J. Hum. Genet. 2014, 95, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Hu, Z.; Mao, X.; Zi, X.; Xia, K.; Tang, B.; Zhang, R. IGHMBP2-related clinical and genetic features in a cohort of Chinese Charcot-Marie-Tooth disease type 2 patients. Neuromuscul. Disord. 2017, 27, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Zhiqiang, L.; Xiaobo, L.; Zhengmao, H.; Shunxiang, H.; Huadong, Z.; Beisha, T.; Ruxu, Z. Clinical and genetic features of Charcot-Marie-Tooth disease patients with IGHMBP2 mutations. Neuromuscul. Disord. 2022, 32, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Pedurupillay, C.R.; Amundsen, S.S.; Baroy, T.; Rasmussen, M.; Blomhoff, A.; Stadheim, B.F.; Orstavik, K.; Holmgren, A.; Iqbal, T.; Frengen, E.; et al. Clinical and molecular characteristics in three families with biallelic mutations in IGHMBP2. Neuromuscul. Disord. 2016, 26, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Schottmann, G.; Jungbluth, H.; Schara, U.; Knierim, E.; Morales Gonzalez, S.; Gill, E.; Seifert, F.; Norwood, F.; Deshpande, C.; von Au, K.; et al. Recessive truncating IGHMBP2 mutations presenting as axonal sensorimotor neuropathy. Neurology 2015, 84, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Hashiguchi, A.; Yoshimura, A.; Yaguchi, H.; Tsuzaki, K.; Ikeda, A.; Wada-Isoe, K.; Ando, M.; Nakamura, T.; Higuchi, Y.; et al. Clinical diversity caused by novel IGHMBP2 variants. J. Hum. Genet. 2017, 62, 599–604. [Google Scholar] [CrossRef]
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.K.; Cao, M.H.; Nguyen, T.T.H.; Le, P.T.; Tran, H.A.; Vu, D.C.; Nguyen, H.T.; Nguyen, M.T.P.; Bui, T.H.; Nguyen, T.B.; et al. A novel IGHMBP2 variant and clinical diversity in Vietnamese SMARD1 and CMT2S patients. Front. Pediatr. 2024, 12, 1165492. [Google Scholar] [CrossRef] [PubMed]
- Rzepnikowska, W.; Kaminska, J.; Kochanski, A. Validation of the Pathogenic Effect of IGHMBP2 Gene Mutations Based on Yeast S. cerevisiae Model. Int. J. Mol. Sci. 2022, 23, 9913. [Google Scholar] [CrossRef] [PubMed]
- Fairman-Williams, M.E.; Guenther, U.P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, E. RNA helicases at work: Binding and rearranging. Trends Biochem. Sci. 2011, 36, 19–29. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989, 17, 4713–4730. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Bowler, M.W.; Lai, T.F.; Song, H. The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1. Nucleic Acids Res. 2012, 40, 11009–11022. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, T.R.; Fukita, Y.; Miyoshi, T.; Shimizu, A.; Honjo, T. Isolation of cDNA encoding a binding protein specific to 5’-phosphorylated single-stranded DNA with G-rich sequences. Nucleic Acids Res. 1993, 21, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Guenther, U.P.; Handoko, L.; Laggerbauer, B.; Jablonka, S.; Chari, A.; Alzheimer, M.; Ohmer, J.; Plottner, O.; Gehring, N.; Sickmann, A.; et al. IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1). Hum. Mol. Genet. 2009, 18, 1288–1300. [Google Scholar] [CrossRef]
- de Planell-Saguer, M.; Schroeder, D.G.; Rodicio, M.C.; Cox, G.A.; Mourelatos, Z. Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum. Mol. Genet. 2009, 18, 2115–2126. [Google Scholar] [CrossRef]
- Vadla, G.P.; Ricardez Hernandez, S.M.; Mao, J.; Garro-Kacher, M.O.; Lorson, Z.C.; Rice, R.P.; Hansen, S.A.; Lorson, C.L.; Singh, K.; Lorson, M.A. ABT1 modifies SMARD1 pathology via interactions with IGHMBP2 and stimulation of ATPase and helicase activity. JCI Insight. 2023, 8, e164608. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.A.; Mahaffey, C.L.; Frankel, W.N. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron 1998, 21, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Vadla, G.P.; Singh, K.; Lorson, C.L.; Lorson, M.A. The contribution and therapeutic implications of IGHMBP2 mutations on IGHMBP2 biochemical activity and ABT1 association. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167091. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, K.; Rossoll, W.; Kobsar, I.; Holtmann, B.; Jablonka, S.; Wessig, C.; Stoltenburg-Didinger, G.; Fischer, U.; Hubner, C.; Martini, R.; et al. Characterization of Ighmbp2 in motor neurons and implications for the pathomechanism in a mouse model of human spinal muscular atrophy with respiratory distress type 1 (SMARD1). Hum. Mol. Genet. 2004, 13, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Surrey, V.; Zoller, C.; Lork, A.A.; Moradi, M.; Balk, S.; Dombert, B.; Saal-Bauernschubert, L.; Briese, M.; Appenzeller, S.; Fischer, U.; et al. Impaired Local Translation of beta-actin mRNA in Ighmbp2-Deficient Motoneurons: Implications for Spinal Muscular Atrophy with respiratory Distress (SMARD1). Neuroscience 2018, 386, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Prusty, A.B.; Hirmer, A.; Sierra-Delgado, J.A.; Huber, H.; Guenther, U.P.; Schlosser, A.; Dybkov, O.; Yildirim, E.; Urlaub, H.; Meyer, K.C.; et al. RNA helicase IGHMBP2 regulates THO complex to ensure cellular mRNA homeostasis. Cell Rep. 2024, 43, 113802. [Google Scholar] [CrossRef]
- Kervestin, S.; Jacobson, A. NMD: A multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 2012, 13, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Schueler, M.; Munschauer, M.; Mastrobuoni, G.; Chen, W.; Kempa, S.; Dieterich, C.; Landthaler, M. MOV10 Is a 5’ to 3’ RNA helicase contributing to UPF1 mRNA target degradation by translocation along 3’ UTRs. Mol. Cell 2014, 54, 573–585. [Google Scholar] [CrossRef]
- Kanaan, J.; Raj, S.; Decourty, L.; Saveanu, C.; Croquette, V.; Le Hir, H. UPF1-like helicase grip on nucleic acids dictates processivity. Nat. Commun. 2018, 9, 3752. [Google Scholar] [CrossRef]
- Singh, A.K.; Choudhury, S.R.; De, S.; Zhang, J.; Kissane, S.; Dwivedi, V.; Ramanathan, P.; Petric, M.; Orsini, L.; Hebenstreit, D.; et al. The RNA helicase UPF1 associates with mRNAs co-transcriptionally and is required for the release of mRNAs from gene loci. eLife 2019, 8, e41444. [Google Scholar] [CrossRef] [PubMed]
- Taiana, M.; Govoni, A.; Salani, S.; Kleinschmidt, N.; Galli, N.; Saladini, M.; Ghezzi, S.B.; Melzi, V.; Bersani, M.; Del Bo, R.; et al. Molecular analysis of SMARD1 patient-derived cells demonstrates that nonsense-mediated mRNA decay is impaired. J. Neurol. Neurosurg. Psychiatry 2022, 93, 908–910. [Google Scholar] [CrossRef]
- Krieger, F.; Elflein, N.; Ruiz, R.; Guerra, J.; Serrano, A.L.; Asan, E.; Tabares, L.; Jablonka, S. Fast motor axon loss in SMARD1 does not correspond to morphological and functional alterations of the NMJ. Neurobiol. Dis. 2013, 54, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Villalon, E.; Shababi, M.; Kline, R.; Lorson, Z.C.; Florea, K.M.; Lorson, C.L. Selective vulnerability in neuronal populations in nmd/SMARD1 mice. Hum. Mol. Genet. 2018, 27, 679–690. [Google Scholar] [CrossRef]
- Maddatu, T.P.; Garvey, S.M.; Schroeder, D.G.; Hampton, T.G.; Cox, G.A. Transgenic rescue of neurogenic atrophy in the nmd mouse reveals a role for Ighmbp2 in dilated cardiomyopathy. Hum. Mol. Genet. 2004, 13, 1105–1115. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, B.J.; Cox, G.A.; Maddatu, T.P.; Judex, S.; Rubin, C.T. Devastation of bone tissue in the appendicular skeleton parallels the progression of neuromuscular disease. J. Musculoskelet Neuronal Interact. 2009, 9, 215–224. [Google Scholar] [PubMed]
- Shababi, M.; Feng, Z.; Villalon, E.; Sibigtroth, C.M.; Osman, E.Y.; Miller, M.R.; Williams-Simon, P.A.; Lombardi, A.; Sass, T.H.; Atkinson, A.K.; et al. Rescue of a Mouse Model of Spinal Muscular Atrophy With Respiratory Distress Type 1 by AAV9-IGHMBP2 Is Dose Dependent. Mol. Ther. 2016, 24, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Shababi, M.; Smith, C.E.; Kacher, M.; Alrawi, Z.; Villalon, E.; Davis, D.; Bryda, E.C.; Lorson, C.L. Development of a novel severe mouse model of spinal muscular atrophy with respiratory distress type 1: FVB-nmd. Biochem. Biophys. Res. Commun. 2019, 520, 341–346. [Google Scholar] [CrossRef]
- Smith, C.E.; Lorson, M.A.; Ricardez Hernandez, S.M.; Al Rawi, Z.; Mao, J.; Marquez, J.; Villalon, E.; Keilholz, A.N.; Smith, C.L.; Garro-Kacher, M.O.; et al. The Ighmbp2D564N mouse model is the first SMARD1 model to demonstrate respiratory defects. Hum. Mol. Genet. 2022, 31, 1293–1307. [Google Scholar] [CrossRef]
- Martin, P.B.; Holbrook, S.E.; Hicks, A.N.; Hines, T.J.; Bogdanik, L.P.; Burgess, R.W.; Cox, G.A. Clinically relevant mouse models of Charcot-Marie-Tooth type 2S. Hum. Mol. Genet. 2023, 32, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Krieger, F.; Metzger, F.; Jablonka, S. Differentiation defects in primary motoneurons from a SMARD1 mouse model that are insensitive to treatment with low dose PEGylated IGF1. Rare. Dis. 2014, 2, e29415. [Google Scholar] [CrossRef][Green Version]
- Krieger, F.; Elflein, N.; Saenger, S.; Wirthgen, E.; Rak, K.; Frantz, S.; Hoeflich, A.; Toyka, K.V.; Metzger, F.; Jablonka, S. Polyethylene glycol-coupled IGF1 delays motor function defects in a mouse model of spinal muscular atrophy with respiratory distress type 1. Brain 2014, 137, 1374–1393. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, R.; Lin, J.; Forgie, A.; Foletti, D.; Shelton, D.; Rosenthal, A.; Tabares, L. Treatment with trkC agonist antibodies delays disease progression in neuromuscular degeneration (nmd) mice. Hum. Mol. Genet. 2005, 14, 1825–1837. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.; Nizzardo, M.; Rizzo, F.; Ruggieri, M.; Riboldi, G.; Salani, S.; Bucchia, M.; Bresolin, N.; Comi, G.P.; Corti, S. iPSC-Derived neural stem cells act via kinase inhibition to exert neuroprotective effects in spinal muscular atrophy with respiratory distress type 1. Stem Cell Rep. 2014, 3, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Locatelli, F.; Papadimitriou, D.; Donadoni, C.; Del, B.R.; Crimi, M.; Bordoni, A.; Fortunato, F.; Strazzer, S.; Menozzi, G.; et al. Transplanted ALDHhiSSClo neural stem cells generate motor neurons and delay disease progression of nmd mice, an animal model of SMARD1. Hum. Mol. Genet. 2006, 15, 167–187. [Google Scholar] [CrossRef][Green Version]
- Simone, C.; Ramirez, A.; Bucchia, M.; Rinchetti, P.; Rideout, H.; Papadimitriou, D.; Re, D.B.; Corti, S. Is spinal muscular atrophy a disease of the motor neurons only: Pathogenesis and therapeutic implications? Cell Mol. Life Sci. 2016, 73, 1003–1020. [Google Scholar] [CrossRef] [PubMed]
- Wichterle, H.; Lieberam, I.; Porter, J.A.; Jessell, T.M. Directed differentiation of embryonic stem cells into motor neurons. Cell 2002, 110, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.M.; Krishnan, C.; Darman, J.S.; Deshpande, D.M.; Peck, S.; Shats, I.; Backovic, S.; Rothstein, J.D.; Kerr, D.A. Axonal growth of embryonic stem cell-derived motoneurons in vitro and in motoneuron-injured adult rats. Proc. Natl. Acad. Sci. USA 2004, 101, 7123–7128. [Google Scholar] [CrossRef]
- Li, X.J.; Du, Z.W.; Zarnowska, E.D.; Pankratz, M.; Hansen, L.O.; Pearce, R.A.; Zhang, S.C. Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 2005, 23, 215–221. [Google Scholar] [CrossRef]
- Corti, S.; Nizzardo, M.; Nardini, M.; Donadoni, C.; Salani, S.; Del Bo, R.; Papadimitriou, D.; Locatelli, F.; Mezzina, N.; Gianni, F.; et al. Motoneuron transplantation rescues the phenotype of SMARD1 (spinal muscular atrophy with respiratory distress type 1). J. Neurosci. 2009, 29, 11761–11771. [Google Scholar] [CrossRef][Green Version]
- Perego, M.G.L.; Galli, N.; Nizzardo, M.; Govoni, A.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1). Cell Mol. Life Sci. 2020, 77, 3351–3367. [Google Scholar] [CrossRef]
- Sierra-Delgado, J.A.; Sinha-Ray, S.; Kaleem, A.; Ganjibakhsh, M.; Parvate, M.; Powers, S.; Zhang, X.; Likhite, S.; Meyer, K. In Vitro Modeling as a Tool for Testing Therapeutics for Spinal Muscular Atrophy and IGHMBP2-Related Disorders. Biology 2023, 12, 867. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Simone, C.; Rizzo, F.; Salani, S.; Dametti, S.; Rinchetti, P.; Del Bo, R.; Foust, K.; Kaspar, B.K.; Bresolin, N.; et al. Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model. Sci. Adv. 2015, 1, e1500078. [Google Scholar] [CrossRef] [PubMed]
- Shababi, M.; Villalon, E.; Kaifer, K.A.; DeMarco, V.; Lorson, C.L. A Direct Comparison of IV and ICV Delivery Methods for Gene Replacement Therapy in a Mouse Model of SMARD1. Mol. Ther. Methods Clin. Dev. 2018, 10, 348–360. [Google Scholar] [CrossRef]
- Shababi, M.; Smith, C.E.; Ricardez Hernandez, S.M.; Marquez, J.; Al Rawi, Z.; Villalon, E.; Farris, K.D.; Garro-Kacher, M.O.; Lorson, C.L. Defining the optimal dose and therapeutic window in SMA with respiratory distress type I model mice, FVB/NJ-Ighmpb2 (nmd-2J). Mol. Ther. Methods Clin. Dev. 2021, 23, 23–32. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jablonka, S.; Yildirim, E. Disease Mechanisms and Therapeutic Approaches in SMARD1—Insights from Animal Models and Cell Models. Biomedicines 2024, 12, 845. https://doi.org/10.3390/biomedicines12040845
Jablonka S, Yildirim E. Disease Mechanisms and Therapeutic Approaches in SMARD1—Insights from Animal Models and Cell Models. Biomedicines. 2024; 12(4):845. https://doi.org/10.3390/biomedicines12040845
Chicago/Turabian StyleJablonka, Sibylle, and Ezgi Yildirim. 2024. "Disease Mechanisms and Therapeutic Approaches in SMARD1—Insights from Animal Models and Cell Models" Biomedicines 12, no. 4: 845. https://doi.org/10.3390/biomedicines12040845
APA StyleJablonka, S., & Yildirim, E. (2024). Disease Mechanisms and Therapeutic Approaches in SMARD1—Insights from Animal Models and Cell Models. Biomedicines, 12(4), 845. https://doi.org/10.3390/biomedicines12040845