
Aging, Physical Exercise, Telomeres, and Sarcopenia: A Narrative Review
 , , , , , and
, , , , , and
Abstract
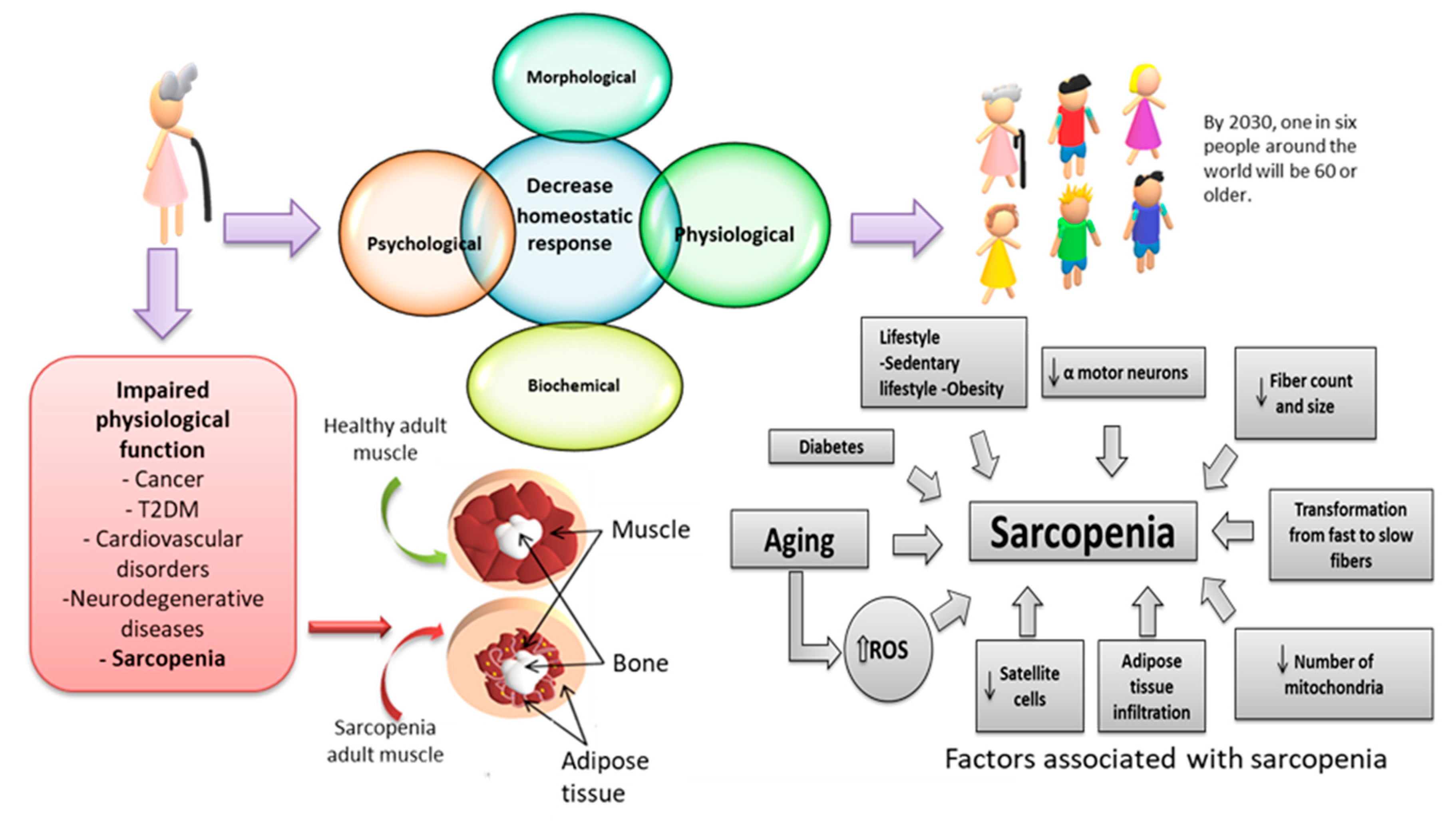
:1. Introduction
2. Human Aging
3. Oxidative Stress
Oxidative Stress and Aging
4. Sarcopenia
Oxidative Stress and Sarcopenia
5. Inflammation
5.1. Inflammation and Sarcopenia
5.2. Sarcopenic Obesity
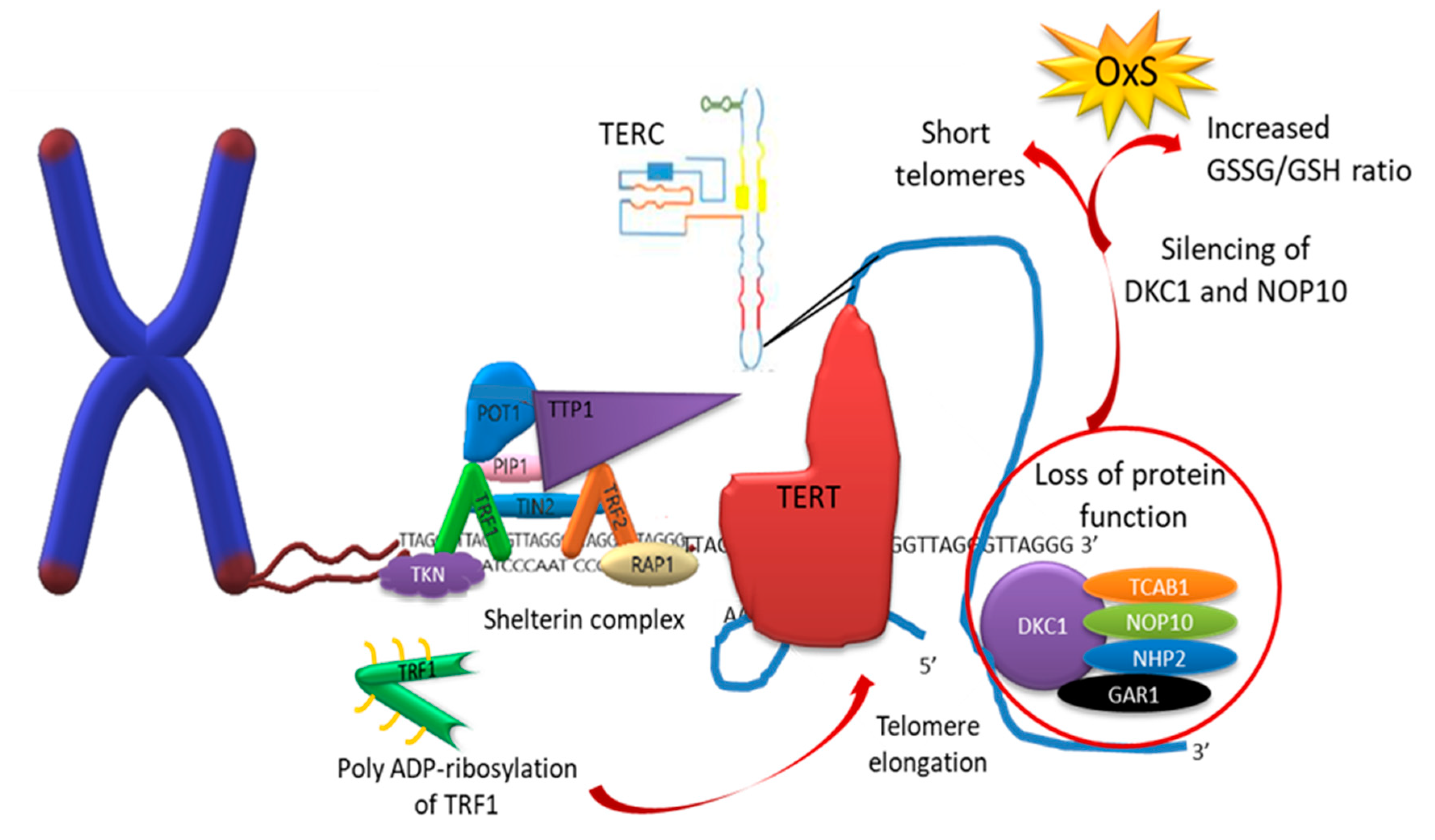
6. Telomeres
6.1. Telomerase
6.2. Sarcopenia and Telomeric Length
7. Base Excision Repair (BER)
7.1. Nucleotide Excision Repair (NER)
7.2. Telomere Repair Mechanisms
8. Sarcopenia Prevention Interventions
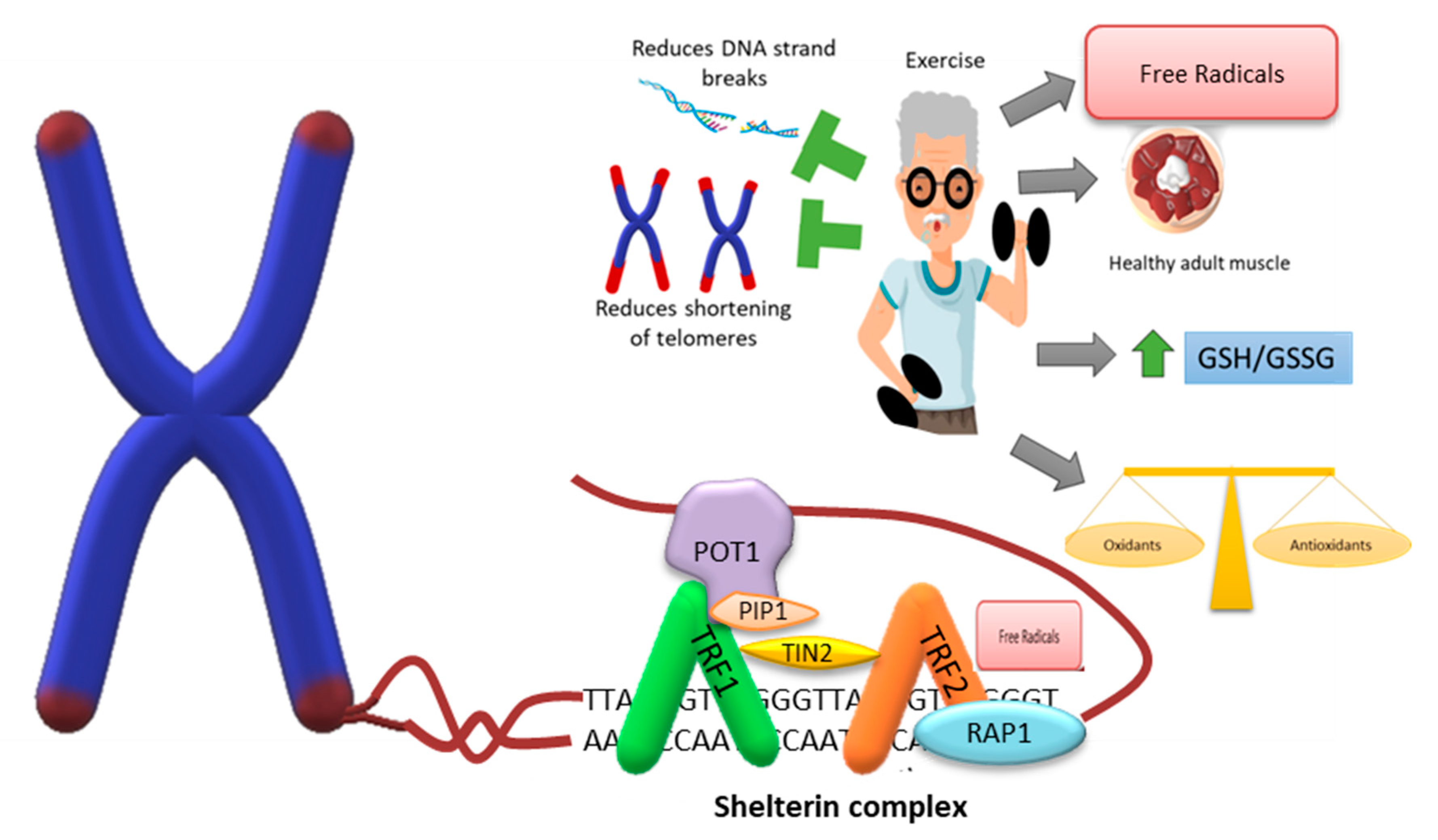
8.1. Physical Exercise
8.2. Exercise and Sarcopenia
8.3. Exercise and Telomere Length
8.4. Exercise and DNA Repair
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mander, T. Longevity and healthy ageing–Will healthcare be drowned by the grey tsunami or sunk by the demographic iceberg? Post. Reprod. Health. 2014, 20, 8–10. [Google Scholar] [CrossRef]
- Barishansky, R.M. The silver tsunami: Are you ready? America’s elderly population is exploding, and EMS services will have to reflect that. EMS. World 2016, 45, 53–56. [Google Scholar]
- World Health Organization. Ageing and Health; WHO: Geneva, Switzerland, 2021.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Arsenis, N.C.; You, T.; Ogawa, E.F.; Tinsley, G.M.; Zuo, L. Physical activity and telomere length: Impact of aging and potential mechanisms of action. Oncotarget 2017, 8, 45008–45019. [Google Scholar] [CrossRef] [Green Version]
- Golubev, A.G. An essay on the nominal vs. real definitions of aging. Biogerontology 2021, 22, 441–457. [Google Scholar] [CrossRef]
- Troen, B.R. The biology of aging. Mt. Sinai. J. Med. 2003, 70, 3–22. [Google Scholar]
- Fulop, T.; Larbi, A.; Khalil, A.; Cohen, A.A.; Witkowski, J.M. Are we ill because we age? Front. Physiol. 2019, 10, 1508. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, M. Defining aging. Biol. Philos. 2020, 35, 1–30. [Google Scholar] [CrossRef]
- Cohen, A.A.; Kennedy, B.K.; Anglas, U.; Bronikowski, A.M.; Deelen, J.; Dufour, F.; Ferbeyre, G.; Ferrucci, L.; Franceschi, C.; Frasca, D.; et al. Lack of consensus on an aging biology paradigm? A global survey reveals an agreement to disagree, and the need for an interdisciplinary framework. Mech. Ageing. Dev. 2020, 191, 111316. [Google Scholar] [CrossRef]
- Mendoza-Núñez, V.M.; Martínez-Maldonado, M.L.; Vivaldo-Martínez, M. What is the onset age of human aging and old age? Int. J. Gerontol. 2016, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Razgonova, M.P.; Zakharenko, A.M.; Golokhvast, K.S.; Thanasoula, M.; Sarandi, E.; Nikolouzakis, K.; Fragkiadaki, P.; Tsoukalas, D.; Spandidos, D.A.; Tsatsakis, A. Telomerase and telomeres in aging theory and chronographic aging theory (Review). Mol. Med. Rep. 2020, 22, 1679–1694. [Google Scholar] [CrossRef]
- Masanés, T.F.; Navarro, L.M.; Sacanella, M.E.; López, S.A. ¿Qué es la sarcopenia? Semin. Fund. Esp. Reumatol. 2010, 11, 14–23. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D. Oxidative stress. In Encyclopedia of Stress, 2nd ed.; Fink, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 45–48. [Google Scholar]
- Cabello-Verrugio, C.; Simon, F.; Trollet, C.; Santibañez, J.F. Oxidative stress in disease and aging: Mechanisms and therapies 2016. Oxid. Med. Cell. Longev. 2017, 2017, 4310469. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Venereo, G.J. Daño oxidante, radicales libres y antioxidantes. Rev. Cub. Med. Mil. 2002, 31, 126–133. [Google Scholar]
- López, D.G.N.; Gutiérrez, R.M.C.; Cortés, B.E.; Zentella, D.A.; Konigsberg, F.M. Daño al DNA y niveles de radicales libres en fibroblastos de ratones jóvenes y viejos. Rev. Cub. Investig. Biomed. 2003, 22, 109–116. [Google Scholar]
- Marnett, L.J.; Riggins, J.N.; West, J.D. Endogenous generation of reactive oxidants and electrophiles and their reactions with and protein. J. Clin. Investig. 2003, 111, 583–593. [Google Scholar] [CrossRef]
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE. 2014, 9, e110963. [Google Scholar] [CrossRef] [PubMed]
- Houben, J.M.; Moonen, H.J.; van Schooten, F.J.; Hageman, G.J. Telomere length assessment: Biomarker of chronic oxidative stress? Free Radic. Biol. Med. 2008, 44, 235–246. [Google Scholar] [CrossRef]
- Kino, K.; Hirao-Suzuki, M.; Morikawa, M.; Sakaga, A.; Miyazawa, H. Generation, repair and replication of guanine oxidation products. Genes Environ. 2017, 39, 21. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Eckl, P.; Ortner, A. Possible mutagens derived from lipids and lipid precursors. Mutat. Res. 1990, 238, 223–233. [Google Scholar] [CrossRef]
- Hwa, Y.B.; Guo, J.; Bellamri, M.; Turesky, R.J. DNA adducts: Formation, biological effects, and new biospecimens for mass spectrometric measurements in humans. Mass Spectrom. Rev. 2020, 39, 55–82. [Google Scholar]
- Díaz-Acosta, A.E.; Membrillo-Hernández, J. Consecuencias fisiológicas de la oxidación de proteínas por carbonilación en diversos sistemas biológicos. TIP Rev. Espec. Cienc. Quim.-Biol. 2006, 9, 34–44. [Google Scholar]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative stress in human pathology and aging: Molecular mechanisms and perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R18–R36. [Google Scholar] [CrossRef]
- Mendoza-Núñez, V.M.; Ruiz-Ramos, M.; Sánchez-Rodríguez, M.A.; Retana-Ugalde, R.; Muñoz-Sánchez, J.L. Aging-related oxidative stress in healthy humans. Tohoku J. Exp. Med. 2007, 213, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Cutler, R.G. Antioxidants and aging. Am. J. Clin. Nutr. 1991, 53, 373S–379S. [Google Scholar] [CrossRef]
- Du, C.; Anderson, A.; Lortie, M.; Parsons, R.; Bodnar, A. Oxidative damage and cellular defense mechanisms in sea urchin models of aging. Free Radic. Biol. Med. 2013, 63, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Campo, R.; López-Torres, M.; Cadenas, S.; Rojas, C.; Barja, G. The rate of free radical production as a determinant of the rate of aging: Evidence from the comparative approach. J. Comp. Physiol. B. 1998, 168, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Starke-Reed, P.E.; Oliver, C.N. Protein oxidation and proteolysis during aging and oxidative stress. Arch. Biochem. Biophys. 1989, 275, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Salmon, A.B.; Richardson, A.; Pérez, V.I. Update on the oxidative stress theory of aging: Does oxidative stress play a role in aging or healthy aging? Free Radic. Biol. Med. 2010, 48, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, C.M.; Ingles, M.; Salvador-Pascual, A.; Cominetti, M.R.; Gomez-Cabrera, M.C.; Viña, J. Sarcopenia, frailty and their prevention by exercise. Free Radic. Biol. Med. 2019, 20, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite cells and skeletal muscle regeneration. Compr. Physiol. 2015, 5, 1027–1109. [Google Scholar]
- Dao, T.; Green, A.E.; Kim, Y.A.; Bae, S.J.; Ha, K.T.; Gariani, K.; Lee, M.R.; Menzies, K.J.; Ryu, D. Sarcopenia and muscle aging: A brief overview. Endocrinol. Metab. 2020, 35, 716–732. [Google Scholar] [CrossRef]
- Fulle, S.; Protasi, F.; Di Tano, G.; Pietrangelo, T.; Beltramin, A.; Boncompagni, S.; Vecchiet, L.; Fanò, G. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp. Gerontol. 2004, 39, 17–24. [Google Scholar] [CrossRef]
- Derbré, F.; Gratas-Delamarche, A.; Gómez-Cabrera, M.C.; Viña, J. Inactivity-induced oxidative stress: A central role in age-related sarcopenia? Eur. J. Sport. Sci. 2014, 14, S98–S108. [Google Scholar] [CrossRef]
- Dhillon, R.J.; Hasni, S. Pathogenesis and management of sarcopenia. Clin. Geriatr. Med. 2017, 33, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, A.; Seino, S.; Abe, T.; Nofuji, Y.; Yokoyama, Y.; Amano, H.; Nishi, M.; Taniguchi, Y.; Narita, M.; Fujiwara, Y.; et al. Sarcopenia: Prevalence, associated factors, and the risk of mortality and disability in Japanese older adults. J. Cachexia Sarcopenia Muscle 2021, 12, 30–38. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Vellas, B.; van Kan, G.A.; Anker, S.D.; Bauer, J.M.; Bernabei, R.; Cesari, M.; Chumlea, W.C.; Doehner, W.; Evans, J.; et al. Frailty consensus: A call to action. J. Am. Med. Dir. Assoc. 2013, 14, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anker, S.D.; Morley, J.E.; von Haehling, S. Welcome to the ICD-10 code for sarcopenia. J. Cachexia Sarcopenia Muscle 2016, 7, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.J. Aging and sarcopenia. J. Appl Physiol. 2003, 95, 1717–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiee, G.; Keshtkar, A.; Soltani, A.; Ahadi, Z.; Larijani, B.; Heshmat, R. Prevalence of sarcopenia in the world: A systematic review and meta- analysis of general population studies. J. Diabetes Metab. Disord. 2017, 16, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Haehling, S.; Morley, J.E.; Anker, S.D. An overview of sarcopenia: Facts and numbers on prevalence and clinical impact. J. Cachexia Sarcopenia Muscle 2010, 1, 129–133. [Google Scholar] [CrossRef]
- González, G. Tipo de fibra muscular y su relación con el abordaje fenoaudiológico en los trastornos de la deglución. Rev. Chil. Fono. 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.Y.; Zhao, Y. Efficacy of Exercise on Muscle Function and Physical Performance in Older Adults with Sarcopenia: An Updated Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 8212. [Google Scholar] [CrossRef]
- Nishikawa, H.; Asai, A.; Fukunishi, S.; Nishiguchi, S.; Higuchi, K. Metabolic syndrome and sarcopenia. Nutrients 2021, 13, 3519. [Google Scholar] [CrossRef]
- Beltran, V.M.R.; Wilkinson, D.J.; Narici, M.V.; Smith, K.; Phillips, B.E.; Caporossi, D.; Atherton, P.J. Protein carbonylation and heat shock proteins in human skeletal muscle: Relationships to age and sarcopenia. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Bellanti, F.; Romano, A.D.; Lo Buglio, A.; Castriotta, V.; Guglielmi, G.; Greco, A.; Serviddio, G.; Vendemiale, G. Oxidative stress is increased in sarcopenia and associated with cardiovascular disease risk in sarcopenic obesity. Maturitas 2018, 109, 6–12. [Google Scholar] [CrossRef]
- Delrieu, L.; Martin, A.; Touillaud, M.; Pérol, O.; Morelle, M.; Febvey-Combres, O.; Freyssenet, D.; Friendenreich, C.; Dufresne, A.; Bachelot, T.; et al. Sarcopenia and serum biomarkers of oxidative stress after a 6-month physical activity intervention in women with metastatic breast cancer: Results from the ABLE feasibility trial. Breast Cancer Res. Treat. 2021, 188, 601–613. [Google Scholar] [CrossRef]
- Song, Y.R.; Kim, J.K.; Lee, H.S.; Kim, S.G.; Choi, E.K. Serum levels of protein carbonyl, a marker of oxidative stress, are associated with overhydration, sarcopenia and mortality in hemodialysis patients. BMC Nephrol. 2020, 21, 281. [Google Scholar] [CrossRef]
- Sánchez-Castellano, C.; Martín-Aragón, S.; Bermejo-Bescós, P.; Vaquero-Pinto, N.; Miret-Corchado, C.; Merello de Miguel, A.; Cruz-Jentoft, A.J. Biomarkers of sarcopenia in very old patients with hip fracture. J. Cachexia Sarcopenia Muscle 2020, 11, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda-Loyola, W.; de Castro, L.A.; Matsumoto, A.K.; Camillo, C.A.; Barbosa, D.S.; Galvan, C.C.R.; Probst, V.S. NOVEL antioxidant and oxidant biomarkers related to sarcopenia in COPD. Heart Lung 2021, 50, 184–191. [Google Scholar] [CrossRef]
- Bernabeu-Wittel, M.; Gómez-Díaz, R.; González-Molina, Á.; Vidal-Serrano, S.; Díez-Manglano, J.; Salgado, F.; Soto-Martín, M.; Ollero-Baturone, M.; on behalf of the PROTEO RESEARCHERS. Oxidative stress, telomere shortening, and apoptosis associated to sarcopenia and frailty in patients with multimorbidity. J. Clin. Med. 2020, 9, 2669. [Google Scholar] [CrossRef] [PubMed]
- Kadoguchi, T.; Shimada, K.; Miyazaki, T.; Kitamura, K.; Kunimoto, M.; Aikawa, T.; Sugita, Y.; Ouchi, S.; Shiozawa, T.; Yokoyama-Nishitani, M.; et al. Promotion of oxidative stress is associated with mitochondrial dysfunction and muscle atrophy in aging mice. Geriatr. Gerontol. Int. 2020, 20, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [Green Version]
- Adhihetty, P.J.; Ljubicic, V.; Menzies, K.J.; Hood, D.A. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am. J. Physiol. Cell Physiol. 2005, 289, C994–C1001. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.J.; Yu, L.J. Oxidative stress, molecular inflammation and sarcopenia. Int. J. Mol. Sci. 2010, 11, 1509–1526. [Google Scholar] [CrossRef] [Green Version]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Johnson, M.L.; Robinson, M.M.; Nair, K.S. Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 2013, 24, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirks, A.; Leeuwenburgh, C. Apoptosis in skeletal muscle with aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R519–R527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, M.; Vecino, E. Vías de señalización intracelular que conducen a la apoptosis de las células de la retina. Arch. Soc. Esp. Oftalmol. 2003, 78, 351–364. [Google Scholar] [CrossRef]
- Stankov, S.V. Definition of inflammation, causes of inflammation and possible anti-inflammatory strategies. Open. Inflamm. J. 2012, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [Green Version]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Kuprash, D.V.; Nedospasov, S.A. Molecular and cellular mechanisms of inflammation. Biochemistry 2016, 81, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Scrivo, R.; Vasile, M.; Bartosiewicz, I.; Valesini, G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun. Rev. 2011, 10, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Soysal, P.; Arik, F.; Smith, L.; Jackson, S.E.; Isik, A.T. Inflammation, frailty and cardiovascular disease. Adv. Exp. Med. Biol. 2020, 1216, 55–64. [Google Scholar] [PubMed] [Green Version]
- Opal, S.M.; DePalo, V.A. Anti-inflammatory cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Role of pro- and anti-inflammatory cytokines during inflammation: Experimental and clinical findings. J. Biol. Regul. Homeost. 1997, 11, 91–103. [Google Scholar]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Zembroń-Łacny, A.; Dziubek, W.; Rogowski, Ł.; Skorupka, E.; Dąbrowska, G. Sarcopenia: Monitoring, molecular mechanisms, and physical intervention. Physiol. Res. 2014, 63, 683–691. [Google Scholar] [CrossRef]
- Serra, R.J. Consecuencias Clínicas de la sarcopenia. Nutr. Hosp. 2006, 21, 46–50. [Google Scholar]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The Role of inflammation in age-related sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef] [Green Version]
- Burd, N.A.; Gorissen, S.H.; van Loon, L.J.C. Anabolic resistance of muscle protein synthesis with aging. Exerc. Sport Sci. Rev. 2013, 41, 169–173. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and oxidative stress in human diseases: From molecular mechanisms to novel treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [Green Version]
- Can, B.; Kara, O.; Kizilarslanoglu, M.C.; Arik, G.; Aycicek, G.S.; Sumer, F.; Civelek, R.; Demirtas, C.; Ulger, Z. Serum markers of inflammation and oxidative stress in sarcopenia. Aging Clin. Exp. Res. 2017, 29, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Bano, G.; Trevisan, C.; Carraro, S.; Solmi, M.; Luchini, C.; Stubbs, B.; Manzar, E.; Sergi, G.; Veronese, N. Inflammation and sarcopenia: A systematic review and meta-analysis. Maturitas 2017, 96, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Jensen, G.L. Inflammation: Roles in aging and sarcopenia. J. Parenter. Enteral Nutr. 2008, 32, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Bian, A.L.; Hu, H.Y.; Rong, Y.D.; Wang, J.; Wang, J.X.; Zhou, X.Z. A study on relationship between elderly sarcopenia and inflammatory factors IL-6 and TNF-α. Eur. J. Med. Res. 2017, 25, 2017. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.D.; Bian, A.L.; Hu, H.Y.; Ma, Y.; Zhuo, X.Z. Study on relationship between elderly sarcopenia and inflammatory cytokine IL-6, anti-inflammatory cytokine IL-10. BMC Geriatr. 2018, 18, 2018. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, F.; Prattichizzo, F.; Lattanzio, F.; Bonfigli, A.R.; Spazzafumo, L. Antifragility and antiinflammaging: Can they play a role for a healthy longevity? Ageing Res. Rev. 2023, 84, 101836. [Google Scholar] [CrossRef]
- Donini, L.M.; Busetto, L.; Bischoff, S.C.; Cederholm, T.; Ballesteros-Pomar, M.D.; Batsis, J.A.; Bauer, J.M.; Boirie, Y.; Cruz-Jentoft, A.J.; Dicker, D.; et al. Definition and diagnostic criteria for κ: ESPEN and EASO consensus statement. Obes. Facts 2022, 15, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Stenholm, S.; Harris, T.B.; Rantanen, T.; Visser, M.; Kritchevsky, S.B.; Ferrucci, L. Sarcopenic obesity: Definition, cause and consequences. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 693–700. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Simon, F.; Achiardi, O.; Vilos, C.; Cabrera, D.; Cabello-Verrugio, C. The critical role of oxidative stress in sarcopenic obesity. Oxid. Med. Cell. Longev. 2021, 2021, 4493817. [Google Scholar] [CrossRef]
- Schrager, M.A.; Metter, E.J.; Simonsick, E.; Ble, A.; Bandinelli, S.; Lauretani, F.; Ferrucci, L. Sarcopenic obesity and inflammation in the InCHIANTI study. J. Appl. Physio. 2007, 102, 919–925. [Google Scholar] [CrossRef]
- Batsis, J.A.; Mackenzie, T.A.; Jones, J.D.; Lopez-Jimenez, F.; Bartels, S.J. Sarcopenia, sarcopenic obesity and inflammation: Results from the 1999–2004 National Health and Nutrition Examination Survey. Clin. Nutr. 2016, 35, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Zoico, E.; Roubenoff, R. The role of cytokines in regulating protein metabolism and muscle function. Nutr. Rev. 2002, 60, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M. Sarcopenia and sarcopenic obesity. Endocrinol Metab. 2013, 28, 86–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, C.H.; Frost, R.A.; Nairn, A.C.; MacLean, D.A.; Vary, T.C. TNF-alpha impairs heart and skeletal muscle protein synthesis by altering translation initiation. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E336–E347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belizário, J.E.; Fontes-Oliveira, C.C.; Borges, J.P.; Kashiabara, J.A.; Vannier, E. Skeletal muscle wasting and renewal: A pivotal role of myokine IL-6. Springerplus 2016, 13, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wåhlin-Larsson, B.; Carnac, G.; Kadi, F. The influence of systemic inflammation on skeletal muscle in physically active elderly women. Age 2014, 36, 9718. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moylan, J.S.; Chambers, M.A.; Smith, J.; Reid, M.B. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell. Physiol. 2009, 297, C706–C714. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, C.S.L.; Thang, L.A.N.; Maier, A.B. Markers of inflammation and their association with muscle strength and mass: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 64, 101185. [Google Scholar] [CrossRef]
- de Lange, T. Protection of mammalian telomeres. Oncogene 2002, 21, 532–540. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [Green Version]
- Diotti, R.; Loayza, D. Shelterin complex and associated factors at human telomeres. Nucleus 2011, 2, 119–135. [Google Scholar] [CrossRef] [Green Version]
- Fairall, L.; Chapman, L.; Moss, H.; de Lange, T.; Rhodes, D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol. Cell 2001, 8, 351–361. [Google Scholar] [CrossRef]
- Kim, S.H.; Kaminker, P.; Campisi, J. TIN2, a new regulator of telomere length in human cells. Nat. Genet. 1999, 23, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Beausejour, C.; Davalos, A.R.; Kaminker, P.; Heo, S.J.; Campisi, J. TIN2 mediates functions of TRF2 at human telomeres. J. Biol. Chem. 2004, 279, 43799–43804. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Podell, E.R.; Cech, T.R. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol. 2004, 11, 1223–1239. [Google Scholar] [CrossRef]
- Ye, J.Z.; Hockemeyer, D.; Krutchinsky, A.N.; Loayza, D.; Hooper, S.M.; Chait, B.T.; de Lange, T. POT1-interacting protein PIP1: A telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004, 18, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.; Taxman, D.J. Short hairpin RNA (shRNA): Design, delivery, and assessment of gene knockdown. Methods Mol. Biol. 2010, 629, 141–158. [Google Scholar] [PubMed] [Green Version]
- Li, B.; Oestreich, S.; de Lange, T. Identification of human Rap1: Implications for telomere evolution. Cell 2000, 101, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Kandula, V.; Kosuru, R.; Ye, X.; Irwin, M.G.; Xia, Z. Decoding telomere protein Rap1: Its telomeric and nontelomeric functions and potential implications in diabetic cardiomyopathy. Cell Cycle 2017, 16, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Zhang, Y.; Liu, D.; Songyang, Z.; Wan, M. Telomeres-structure, function, and regulation. Exp. Cell. Res. 2013, 319, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, M.J.; Baribault, M.E.; Israel, J.N.; Bae, N.S. Telomere protein RAP1 levels are affected by cellular aging and oxidative stress. Biomed. Rep. 2016, 5, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Teo, H.; Ghosh, S.; Luesch, H.; Ghosh, A.; Wong, E.T.; Malik, N.; Orth, A.; de Jesus, P.; Perry, A.S.; Oliver, J.D.; et al. Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat. Cell Biol. 2010, 12, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, E.; Leone, S.; Sgura, A. Oxidative stress induces telomere dysfunction and senescence by replication fork arrest. Cells 2019, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef]
- Smith, S.; Giriat, I.; Schmitt, A.; de Lange, T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998, 282, 1484–1487. [Google Scholar] [CrossRef] [Green Version]
- La Torre, D.; Conti, A.; Aguennouz, M.H.; De Pasquale, M.G.; Romeo, S.; Angileri, F.F.; Cardali, S.; Tomasello, C.; Alafaci, C.; Germanò, A. Telomere length modulation in human astroglial brain tumors. PLoS ONE 2013, 8, e64296. [Google Scholar] [CrossRef] [Green Version]
- d’Adda di Fagagna, F.; Hande, M.P.; Tong, W.M.; Lansdorp, P.M.; Wang, Z.Q.; Jackson, S.P. Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat. Genet. 1999, 23, 76–80. [Google Scholar] [CrossRef]
- Wang, Y.; Sušac, L.; Feigon, J. Structural biology of telomerase. Cold Spring Harb. Perspect. Biol. 2019, 11, a032383. [Google Scholar] [CrossRef]
- Collins, K.; Gandhi, L. The reverse transcriptase component of the Tetrahymena telomerase ribonucleoprotein complex. Proc. Natl. Acad. Sci. USA 1998, 95, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Trahan, C.; Martel, C.; Dragon, F. Effects of dyskeratosis congenita mutations in dyskerin, NHP2 and NOP10 on assembly of H/ACA pre-RNPs. Hum. Mol. Genet. 2010, 19, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A.; Niewisch, M.R. Dyskeratosis congenita and related telomere biology disorders. In GeneReviews; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2009; pp. 1993–2022. [Google Scholar]
- Ibáñez-Cabellos, J.S.; Pérez-Machado, G.; Seco-Cervera, M.; Berenguer-Pascual, E.; García-Giménez, J.L.; Pallardó, F.V. Acute telomerase components depletion triggers oxidative stress as an early event previous to telomeric shortening. Redox Biol. 2018, 14, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Centeno, J.; Perona, R.; Sastre, L. Dyskerin mutations present in dyskeratosis congenita patients increase oxidative stress and dna damage signalling in dictyostelium discoideum. Cells 2019, 8, 1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2012, 2, 135. [Google Scholar] [CrossRef] [Green Version]
- Koliada, A.K.; Krasnenkov, D.S.; Vaiserman, A.M. Telomeric aging: Mitotic clock or stress indicator? Front. Genet. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Monaghan, P.; Haussmann, M.F. Do telomere dynamics link lifestyle and lifespan? Trends Ecol. Evol. 2006, 21, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, K.; Inoue, E.; Nagao, N.; Hiyama, E.; Miwa, N. Age-dependent telomere shortening is slowed down by enrichment of intracellular vitamin C via suppression of oxidative stress. Life Sci. 1998, 63, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Lorenzi, M.; Antocicco, M.; Bonassi, S.; Celi, M.; Mastropaolo, S.; Settanni, S.; Valdiglesias, V.; Landi, F.; Bernabei, R.; et al. Shorter telomeres in peripheral blood mononuclear cells from older persons with sarcopenia: Results from an exploratory study. Front. Aging Neurosci. 2014, 6, 233. [Google Scholar] [CrossRef] [PubMed]
- Babizhayev, M.A.; Kasus-Jacobi, A.; Vishnyakova, K.S.; Yegorov, Y.E. Novel neuroendocrine and metabolic mechanism provides the patented platform for important rejuvenation therapies: Targeted therapy of telomere attrition and lifestyle changes of telomerase activity with the timing of neuron-specific imidazole-containing dipeptide-dominant pharmaconutrition provision. Recent Pat. Endocr. Metab. Immune Drug Discov. 2014, 8, 153–179. [Google Scholar] [PubMed]
- Woo, J.; Yu, R.; Tang, N.; Leung, J. Telomere length is associated with decline in grip strength in older persons aged 65 years and over. Age 2014, 36, 9711. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 143. [Google Scholar] [CrossRef] [Green Version]
- Kirk, B.; Kuo, C.L.; Xiang, M.; Duque, G. Associations between leukocyte telomere length and osteosarcopenia in 20,400 adults aged 60 years and over: Data from the UK Biobank. Bone 2022, 161, 116425. [Google Scholar] [CrossRef]
- Goddard, T.; Tsintzas, K.; Stephan, B.C.M.; Prado, C.M.; Mazidi, M.; Siervo, M. Sarcopenic obesity is associated with telomere shortening: Findings from the NHANES 1999–2002. Int. J. Obes. 2022, 46, 437–440. [Google Scholar] [CrossRef]
- Chang, K.V.; Chen, Y.C.; Wu, W.T.; Shen, H.J.; Huang, K.C.; Chu, H.P.; Han, D.S. Expression of telomeric repeat-containing RNA decreases in sarcopenia and increases after exercise and nutrition intervention. Nutrients 2020, 12, 3766. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Chowhan, R.K.; Rahaman, H.; Singh, L.R. Structural basis of peroxidase catalytic cycle of human Prdx6. Sci. Rep. 2020, 10, 17416. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, F.; Arriga, R.; Sorice, G.P.; Capuani, B.; Scioli, M.G.; Pastore, D.; Donadel, G.; Bellia, A.; Caratelli, S.; Coppola, A.; et al. Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes 2014, 63, 3210–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacifici, F.; Della-Morte, D.; Piermarini, F.; Arriga, R.; Scioli, M.G.; Capuani, B.; Pastore, D.; Coppola, A.; Rea, S.; Donadel, G.; et al. Prdx6 plays a main role in the crosstalk between aging and metabolic sarcopenia. Antioxidants 2020, 9, 329. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the mitochondria. Mol. Cells. 2016, 39, 87–95. [Google Scholar]
- Kordinas, V.; Ioannidis, A.; Chatzipanagiotou, S. The telomere/telomerase system in chronic inflammatory diseases. cause or effect? Genes 2016, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.; Bourdat, A.G.; D’Ham, C.; Duarte, V.; Gasparutto, D.; Romieu, A.; Ravanat, J.L. Oxidative base damage to DNA: Specificity of base excision repair enzymes. Mutat. Res. 2000, 462, 121–128. [Google Scholar] [CrossRef]
- van Loon, B.; Markkanen, E.; Hübscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–616. [Google Scholar] [CrossRef]
- Dianov, G.L.; Hübscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and persistent 8-Oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol. Cell 2019, 75, 117–130. [Google Scholar] [CrossRef]
- Peterson, L.A. Formation, repair, and genotoxic properties of bulky DNA adducts formed from tobacco-specific nitrosamines. J. Nucleic Acids 2010, 2010, 284935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, N.; Bazar, G.; Kovacs, Z.; Kunisada, M.; Morita, H.; Kizaki, S.; Sugiyama, H.; Tsenkova, R.; Nishigori, C. Detection of UV-induced cyclobutane pyrimidine dimers by near-infrared spectroscopy and aquaphotomics. Sci. Rep. 2015, 5, 11808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.; Mu, H.; Zhao, H.; Ouerfelli, O.; Jeffrey, P.D.; Broyde, S.; Min, J.H. Structure and mechanism of pyrimidine-pyrimidone (6-4) photoproduct recognition by the Rad4/XPC nucleotide excision repair complex. Nucleic Acids Res. 2019, 47, 6015–6028. [Google Scholar] [CrossRef] [Green Version]
- McCullough, A.K.; Lloyd, R.S. Mechanisms underlying aflatoxin-associated mutagenesis—Implications in carcinogenesis. DNA Repair 2019, 77, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; Chasin, L.A.; Tang, M.S. Effects of genomic context and chromatin structure on transcription-coupled and global genomic repair in mammalian cells. Nucleic Acids Res. 2003, 31, 5897–5906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shell, S.M.; Zou, Y. Other proteins interacting with XP proteins. Adv. Exp. Med. Biol. 2008, 637, 103–112. [Google Scholar]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar] [CrossRef]
- Rimel, J.K.; Taatjes, D.J. The essential and multifunctional TFIIH complex. Protein Sci. 2018, 27, 1018–1037. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Tan, R.; Xu, J.; LaFace, J.; Gao, Y.; Xiao, Y.; Attar, M.; Neumann, C.; Li, G.M.; Su, B.; et al. Targeted DNA damage at individual telomeres disrupts their integrity and triggers cell death. Nucleic Acids Res. 2015, 43, 6334–6347. [Google Scholar] [CrossRef]
- Zhou, J.; Chan, J.; Lambelé, M.; Yusufzai, T.; Stumpff, J.; Opresko, P.L.; Thali, M.; Wallace, S.S. NEIL3 repairs telomere damage during s phase to secure chromosome segregation at mitosis. Cell Rep. 2017, 20, 2044–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Liu, M.; Fleming, A.M.; Burrows, C.J.; Wallace, S.S. Neil3 and Neil1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J.Biol. Chem. 2013, 288, 27263–27272. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Mitchell, T.R.; Zhu, X.D.; Human, X.P.F. Controls TRF2 and telomere length maintenance through distinctive mechanisms. Mech. Ageing Dev. 2008, 129, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Blanco, R.; Flores, J.M.; Blasco, M.A. XPF nuclease-dependent telomere loss and increased DNA damage in mice overexpressing TRF2 result in premature aging and cancer. Nat. Genet. 2005, 37, 1063–1071. [Google Scholar] [CrossRef]
- Fukagawa, N.K. Aging: Is oxidative stress a marker or is it causal? Proc. Soc. Exp. Biol. Med. 1999, 222, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.R.; Ryan, M.J.; Alway, S.E. Long-term supplementation with resveratrol alleviates oxidative stress but does not attenuate sarcopenia in aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Brioche, T.; Kireev, R.A.; Cuesta, S.; Gratas-Delamarche, A.; Tresguerres, J.A.; Gomez-Cabrera, M.C.; Viña, J. Growth hormone replacement therapy prevents sarcopenia by a dual mechanism: Improvement of protein balance and of antioxidant defenses. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1186–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, E.C.; Marqués-Jiménez, D.; Casas-Agustench, P.; Bach-Faig, A. Efecto de la suplementación con leucina sola, junto con otro nutriente o con ejercicio físico en personas adultas mayores con sarcopenia: Una revisión sistemática. Endocrinología, Diabetes y Nutrición 2022, 69, 601–613. [Google Scholar] [CrossRef]
- Cereda, E.; Pisati, R.; Rondanelli, M.; Caccialanza, R. Whey protein, leucine- and vitamin-d-enriched oral nutritional supplementation for the treatment of sarcopenia. Nutrients 2022, 14, 1524. [Google Scholar] [CrossRef]
- Bauer, J.M.; Verlaan, S.; Bautmans, I.; Brandt, K.; Donini, L.M.; Maggio, M.; McMurdo, M.E.; Mets, T.; Seal, C.; Wijers, S.L.; et al. Effects of a vitamin D and leucine-enriched whey protein nutritional supplement on measures of sarcopenia in older adults, the PROVIDE study: A randomized, double-blind, placebo-controlled trial. J Am Med Dir Assoc. 2015, 16, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Verlaan, S.; Maier, A.B.; Bauer, J.M.; Bautmans, I.; Brandt, K.; Donini, L.M.; Maggio, M.; McMurdo, M.E.T.; Mets, T.; Seal, C.; et al. Sufficient levels of 25-hydroxyvitamin D and protein intake required to increase muscle mass in sarcopenic older adults—The PROVIDE study. Clin. Nutr. 2018, 37, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Bo, Y.; Liu, C.; Ji, Z.; Yang, R.; An, Q.; Zhang, X.; You, J.; Duan, D.; Sun, Y.; Zhu, Y.; et al. A high whey protein, vitamin D and E supplement preserves muscle mass, strength, and quality of life in sarcopenic older adults: A double-blind randomized controlled trial. Clin. Nutr. 2019, 38, 159–164. [Google Scholar] [CrossRef]
- Camajani, E.; Persichetti, A.; Watanabe, M.; Contini, S.; Vari, M.; Di Bernardo, S.; Faro, M.; Lubrano, C.; Gnessi, L.; Caprio, M.; et al. Whey protein, L-Leucine and vitamin D supplementation for preserving lean mass during a low-calorie diet in sarcopenic obese women. Nutrients 2022, 14, 1884. [Google Scholar] [CrossRef]
- Ronis, M.J.J.; Pedersen, K.B.; Watt, J. Adverse Effects of Nutraceuticals and Dietary Supplements. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 583–601. [Google Scholar] [CrossRef]
- Razzaque, M.S. Can adverse effects of excessive vitamin D supplementation occur without developing hypervitaminosis D? J. Steroid Biochem. Mol. Biol. 2018, 180, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Zdzieblik, D.; Oesser, S.; Baumstark, M.W.; Gollhofer, A.; König, D. Collagen peptide supplementation in combination with resistance training improves body composition and increases muscle strength in elderly sarcopenic men: A randomised controlled trial. Br. J. Nutr. 2015, 114, 1237–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutz, N.E.; Pereira, S.L.; Hays, N.P.; Oliver, J.S.; Edens, N.K.; Evans, C.M.; Wolfe, R.R. Effect of β-hydroxy-β-methylbutyrate (HMB) on lean body mass during 10 days of bed rest in older adults. Clin. Nutr. 2013, 32, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Courel-Ibáñez, J.; Vetrovsky, T.; Dadova, K.; Pallarés, J.G.; Steffl, M. Health benefits of β-Hydroxy-β-Methylbutyrate (HMB) supplementation in addition to physical exercise in older adults: A systematic review with meta-analysis. Nutrients 2019, 11, 2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, J.Y.; Kwon, K.S. Pharmacological interventions for treatment of sarcopenia: Current status of drug development for sarcopenia. Ann. Geriatr. Med. Res. 2019, 23, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Rooks, D.; Swan, T.; Goswami, B.; Filosa, L.A.; Bunte, O.; Panchaud, N.; Coleman, L.A.; Miller, R.R.; Garcia, G.E.; Praestgaard, J.; et al. Bimagrumab vs. optimized standard of care for treatment of sarcopenia in community-dwelling older adults: A randomized clinical trial. JAMA Netw. Open 2020, 3, e2020836. [Google Scholar] [CrossRef]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F.; et al. Myostatin antibody (LY2495655) in older weak fallers: A proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, D.A.; Ather, S.N.; Zhu, H.; Zhou, Y.; Lutkiewicz, J.; Scott, B.B.; Chandler, J. A phase IIA randomized, placebo-controlled clinical trial to study the efficacy and safety of the selective androgen receptor modulator (SARM), MK-0773 in female participants with sarcopenia. J. Nutr. Health Aging 2013, 17, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Vinel, C.; Lukjanenko, L.; Batut, A.; Deleruyelle, S.; Pradère, J.P.; Le Gonidec, S.; Dortignac, A.; Geoffre, N.; Pereira, O.; Karaz, S.; et al. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018, 24, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, C.; Chim, Y.N.; Yao, H.; Shi, L.; Xu, J.; Wang, J.; Wong, R.M.Y.; Leung, K.S.; Chow, S.K.; et al. Vibration and β-hydroxy-β-methylbutyrate treatment suppresses intramuscular fat infiltration and adipogenic differentiation in sarcopenic mice. J. Cachexia Sarcopenia Muscle 2020, 11, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef]
- Liu, P.; Li, Y.; Ma, L. Caloric restriction may help delay the onset of frailty and support frailty management. Front. Nutr. 2021, 8, 731356. [Google Scholar] [CrossRef]
- Yang, L.; Licastro, D.; Cava, E.; Veronese, N.; Spelta, F.; Rizza, W.; Bertozzi, B.; Villareal, D.T.; Hotamisligil, G.S.; Holloszy, J.O.; et al. Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 2016, 14, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.Q.; Xiao, W.F.; Tang, K.; Wu, Y.X.; Hu, P.W.; Li, Y.S.; Duan, Y.; Lv, S. Caloric restriction: Implications for sarcopenia and potential mechanisms. Aging 2020, 12, 24441–24452. [Google Scholar] [CrossRef]
- Marzetti, E.; Carter, C.S.; Wohlgemuth, S.E.; Lees, H.A.; Giovannini, S.; Anderson, B.; Quinn, L.S.; Leeuwenburgh, C. Changes in IL-15 expression and death-receptor apoptotic signaling in rat gastrocnemius muscle with aging and life-long calorie restriction. Mech. Ageing Dev. 2009, 130, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E.; CALERIE Pennington Team. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76. [Google Scholar] [CrossRef] [Green Version]
- Mizunoe, Y.; Kobayashi, M.; Saito, H.; Goto, A.; Migitaka, R.; Miura, K.; Okita, N.; Sudo, Y.; Tagawa, R.; Yoshida, M.; et al. Prolonged caloric restriction ameliorates age-related atrophy in slow and fast muscle fibers of rat soleus muscle. Exp. Gerontol. 2021, 154, 111519. [Google Scholar] [CrossRef]
- Caspersen, C.J.; Powell, K.E.; Christenson, G.M. Physical activity, exercise, and physical fitness: Definitions and distinctions for health-related research. Public Health Rep. 1985, 100, 126–131. [Google Scholar] [PubMed]
- Garber, C.E.; Blissmer, B.; Deschenes, M.R.; Franklin, B.A.; Lamonte, M.J.; Lee, I.M.; Nieman, D.C.; Swain, D.P. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: Guidance for prescribing exercise. Med. Sci. Sports Exerc. 2011, 43, 1334–1359. [Google Scholar] [CrossRef] [PubMed]
- Phu, S.; Boersma, D.; Duque, G. Exercise and sarcopenia. J. Clin. Densitom. 2015, 18, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Schöne, D.; Freiberger, E.; Sieber, C.C. Influence of skeletal muscles on the risk of falling in old age. Internist 2017, 58, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Volkert, D. The role of nutrition in the prevention of sarcopenia. Wien. Med. Wochenschr. 2011, 161, 409–415. [Google Scholar] [CrossRef]
- Sindi, S.; Solomon, A.; Kåreholt, I.; Hovatta, I.; Antikainen, R.; Hänninen, T.; Levälahti, E.; Laatikainen, T.; Lehtisalo, J.; Lindström, J.; et al. Telomere length change in a multidomain lifestyle intervention to prevent cognitive decline: A randomized clinical trial. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 491–498. [Google Scholar] [CrossRef]
- Paltoglou, G.; Raftopoulou, C.; Nicolaides, N.C.; Genitsaridi, S.M.; Karampatsou, S.I.; Papadopoulou, M.; Kassari, P.; Charmandari, E. A comprehensive, multidisciplinary, personalized, lifestyle intervention program is associated with increased leukocyte telomere length in children and adolescents with overweight and obesity. Nutrients 2021, 13, 2682. [Google Scholar] [CrossRef]
- Puterman, E.; Weiss, J.; Lin, J.; Schilf, S.; Slusher, A.L.; Johansen, K.L.; Epel, E.S. Aerobic exercise lengthens telomeres and reduces stress in family caregivers: A randomized controlled trial—curt richter award paper 2018. Psychoneuroendocrinology 2018, 98, 245–252. [Google Scholar] [CrossRef]
- Gravina, S.; Sedivy, J.M.; Vijg, J. The dark side of circulating nucleic acids. Aging Cell 2016, 15, 398–399. [Google Scholar] [CrossRef] [Green Version]
- Neuberger, E.W.I.; Sontag, S.; Brahmer, A.; Philippi, K.F.A.; Radsak, M.P.; Wagner, W.; Simon, P. Physical activity specifically evokes release of cell-free DNA from granulocytes thereby affecting liquid biopsy. Clin. Epigenetics 2022, 14, 29. [Google Scholar] [CrossRef]
- Breitbach, S.; Tug, S.; Simon, P. Circulating cell-free DNA: An up-coming molecular marker in exercise physiology. Sports Med. 2012, 42, 565–586. [Google Scholar] [CrossRef] [PubMed]
- Tosevska, A.; Franzke, B.; Hofmann, M.; Vierheilig, I.; Schober-Halper, B.; Oesen, S.; Neubauer, O.; Wessner, B.; Wagner, K.H. Circulating cell-free DNA, telomere length and bilirubin in the Vienna Active Ageing Study: Exploratory analysis of a randomized, controlled trial. Sci. Rep. 2016, 6, 38084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkas, L.F.; Hunkin, J.L.; Kato, B.S.; Richards, J.B.; Gardner, J.P.; Surdulescu, G.L.; Kimura, M.; Lu, X.; Spector, T.D.; Aviv, A. The association between physical activity in leisure time and leukocyte telomere length. Arch. Intern. Med. 2008, 168, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Friedenreich, C.M.; Wang, Q.; Ting, N.S.; Brenner, D.R.; Conroy, S.M.; McIntyre, J.B.; Mickle, A.; Courneya, K.S.; Beattie, T. Effect of a 12-month exercise intervention on leukocyte telomere length: Results from the ALPHA Trial. Cancer Epidemiol. 2018, 56, 67–74. [Google Scholar] [CrossRef]
- Mason, C.; Risques, R.A.; Xiao, L.; Duggan, C.R.; Imayama, I.; Campbell, K.L.; Kong, A.; Foster-Schubert, K.E.; Wang, C.Y.; Alfano, C.M.; et al. Independent and combined effects of dietary weight loss and exercise on leukocyte telomere length in postmenopausal women. Obesity 2013, 21, E549–E554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, L.L.; Dickman, J.R.; Kang, C.; Koenig, R. Exercise-induced hormesis may help healthy aging. Dose Response 2010, 8, 73–79. [Google Scholar] [CrossRef]
- Lippi, G.; Buonocore, R.; Tarperi, C.; Montagnana, M.; Festa, L.; Danese, E.; Benati, M.; Salvagno, G.L.; Bonaguri, C.; Roggenbuck, D.; et al. DNA injury is acutely enhanced in response to increasing bulks of aerobic physical exercise. Clin. Chim. Acta 2016, 460, 146–151. [Google Scholar] [CrossRef]
- Beltran, V.M.R.; Dimauro, I.; Brunelli, A.; Tranchita, E.; Ciminelli, E.; Caserotti, P.; Duranti, G.; Sabatini, S.; Parisi, P.; Parisi, A.; et al. Explosive type of moderate-resistance training induces functional, cardiovascular, and molecular adaptations in the elderly. Age 2014, 36, 759–772. [Google Scholar] [CrossRef]
- Soares, J.P.; Silva, A.M.; Oliveira, M.M.; Peixoto, F.; Gaivão, I.; Mota, M.P. Effects of combined physical exercise training on DNA damage and repair capacity: Role of oxidative stress changes. Age 2015, 37, 9799. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Villanueva, M.; Kramer, A.; Hammes, T.; Venegas-Carro, M.; Thumm, P.; Bürkle, A.; Gruber, M. Influence of acute exercise on dna repair and parp activity before and after irradiation in lymphocytes from trained and untrained individuals. Int. J. Mol. Sci. 2019, 20, 2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, J.P.; Silva, A.I.; Silva, A.M.; Almeida, V.; Teixeira, J.P.; Matos, M.; Gaivão, I.; Mota, M.P. Effects of physical exercise training in DNA damage and repair activity in humans with different genetic polymorphisms of hOGG1 (Ser326Cys). Cell Biochem. Funct. 2015, 33, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Hughes, C.M.; Burke, G.; Davison, G.W. A combined γ-H2AX and 53BP1 approach to determine the DNA damage-repair response to exercise in hypoxia. Free Radic. Biol. Med. 2020, 154, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.G.; Vasudev, M.M.; Mavathur, R. Role of yoga and its plausible mechanism in the mitigation of DNA damage in type-2 diabetes: A randomized clinical trial. Ann. Behav. Med. 2022, 56, 235–244. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Population Sarcopenia | Determinations | Objective | Findings | Ref. |
|---|---|---|---|---|
| 142 persons aged ≥65 years. | The presence of sarcopenia was established according to the EWGSOP. Whole-body fat-free mass was measured by BIA. The frailty status of participants was assessed according to both Fried’s criteria and the elative telomere length of qRT-PCR. | Determine whether PBMC telomeres obtained from sarcopenic older persons were shorter relative to non-sarcopenic peers. | PBMC telomere length, expressed as T/S values, is shorter in older outpatients with sarcopenia. The cross-sectional assessment of PBMC telomere length is not sufficient for capturing the complex, multidimensional syndrome of frailty. | [135] |
| The stratified sample includes a total of 976 males and 1030 females, in order that approximately 33% would each be aged 65–69, 70–74, and 75 years and older. | Diagnosis of sarcopenia. The T/S ratio was assessed by qRT-PCR. | To examine the association between telomeric length and diagnosis of sarcopenia based on an appendicular skeletal mass index (ASMI), grip strength, walking speed, and chair sit-to-stand in a 5-year prospective study. | Longer telomere length was associated with a slower decline in grip strength in Chinese older persons. | [137] |
| 36 sarcopenic people and 36 healthy people. Older adults (age ≥ 65 years). | Anthropometric measurement. Grip strength. Measurement of telomere length analysis was performed by qPCR. RNA isolation and quantification of TERRA. | To explore the impact of sarcopenia on telomere length and TERRA expression, and changes following exercise and nutrition intervention in the sarcopenic population. | No significant difference in telomere length between control subjects and participants with sarcopenia. | [138] |
| Included 444 patients with an average age of 77.3 ± 8.4 years. | Determination of sarcopenia. Determination of frailty by meeting three or more of Fried’s criteria. OxS markers. Telomere length. DNA fragmentation. | To explore the main markers of OxS, telomere length, and apoptosis parameters in a multicenter cohort of patients with multimorbidity in a hospital. | OxS markers and telomere length were enhanced and shortened, respectively, in blood samples of poly-pathological patients with sarcopenia and/or frailty. Both were associated with decreased survival. | [57] |
| 20,400 older adults (average age: 67.79 ± 4.9 years, 53% male). | Baseline leukocyte telomere length was measured using a multiplex qPCR technique and expressed as a T/S ratio. | Examined the association between leukocyte telomere length and osteosarcopenia. | Telomere length was not associated with osteosarcopenia; however, a slow walking pace was associated. | [139] |
| 5397 individuals; (average age: 44.7 years, 51.3% male). | Body composition evaluation using dual-energy X-ray absorptiometry (DXA). Evaluation of whole blood telomeric length by qPCR. Average telomere length is expressed as the ratio T/S. | Examine the relationship between sarcopenic obesity (SO) and telomere length (TL) in a representative adult population. | Sarcopenia and obesity may act synergistically to shorten telomeres. | [140] |
| Causing Damage | |||
|---|---|---|---|
| ROS, alkylating agents, and UV | UV and ROS | ||
| Injuries | |||
| 8-OHdG, 8-oxodG, alkylated bases, or mismatch bases | Pyrimidine dimers bulky lesions | ||
| BER | NER | ||
| Recognition of the damage | Recognition of the damage | ||
| Short patch pathway | Long patch pathway | GG | TC |
| Bifunctional glycosylases OGG1, NTH, NEIL1, NEIL2, and NEIL3 | Monofunctional glycosylases UNG, MUTY, MBD4, MPG, and SMUG | Complex Cul4-DDB (XPE): RBX1, Cul4, DDB1, DDB2 Complex XPC: XPC, HR23B, CETN2 | Complex Cul4-CSA: RBX1, Cul4, DDB1, CSA, CSB |
| Chain excision sugar removal | Relaxed DNA | ||
| APE1 and APE2 | APE1 and APE2 | Complex TFIIH: CDK7, XPB, TFIIH1, MNAT1, XPD, TFIIH2, CCNH, TTDA, TFIIH3, TFIIH4, | XPG, XPA, and RPA |
| Synthesis | Incision, excision, and synthesis of DNA Incision: XPF and ERCC1 Excision: PCNA, RFC Synthesis: Polδ and Polε | ||
| Dpol, XRCC1, Lig3 | Dpol, PCNA, Polδ, Polβ, Polε, Fen1 | ||
| Ligation Lig1 | |||
| Population | Determinations | Objective | Finding | Ref. |
|---|---|---|---|---|
| Fifty-seven healthy males (40 to 74 years) | Strength tests. Power tests. DNA damage. Assessment of repair capacity. Lipid peroxidation. TAC. | This study aimed to determine the effects of a 16-week combined physical training program on DNA damage and DNA repair of human lymphocytes, taking into account the improvement of physical fitness. To investigate the role of OxS in these changes. | Improvement in general physical performance in the experimental group. Decrease in DNA chain breaks and sites, sensitive to formamide-pyrimidine glycosylase, with a concomitant increase in antioxidant activity and a decrease in lipid peroxidation levels after physical training. There are no significant changes in the enzymatic activity of DNA glycosylase and 8-oxoguanine. | [213] |
| Endurance-trained and young healthy males (age 20 to 36 years) | Simple DNA single break detection. Poly detection (ADP-ribose) and phosphorylation of the H2AX histone (γh2ax). | Determine the general effect of acute exhaustive exercise and physical aptitude (aerobic capacity) on DNA damage, radiosensitivity, and PLP1 activity induced by radiation in immune cells isolated from trained and non-healthy trained volunteers. | Acute exercise induces DNA strand breaks in lymphocytes in untrained individuals. During acute exercise, trained subjects repaired radiation-induced DNA strand breaks more rapidly than untrained subjects. Trained subjects maintained higher levels of radiation-induced PARP1 activity after acute stress. | [214] |
| Thirty-two healthy Caucasian males (40 to 74 years) | Assessment of strand break DNA (SB) and oxidative damage to DNA. Evaluation of sites sensitive to FPG. Assessment of repair capacity with the comet assay. The activity of OGG1. TAC. Determination of the hOGG1 (Ser326Cys) polymorphism. | To investigate the possible influence of genetic polymorphisms of hOGG1 on DNA damage and repair activity OGG1 enzyme in response to 16 weeks of combined physical training. | At baseline, there were no differences in DNA damage and OGG1 activity between the groups. With 16 weeks of physical exercise, there was a decrease in DNA strand breaks in both groups, as well as a decrease in FPG-sensitive sites and an increase in TAC in WTG. | [215] |
| Fourteen (apparently healthy recreationally active males (age 22 ± 2 years, stature 178 ± 6 cm, mass 83 ± 8 kg, BMI 26.2 ± 2) | DNA single-strand breaks and FPG-sensitive sites. Detection of double-strand breaks via histone γ-H2AX and 53BP1. Lipid hydroperoxides. Soluble antioxidants. EPR. | Characterization of the interplay of exercise and hypoxia about DNA damage repair. Quantification of the effects of exercise in hypoxia on single- and double-strand DNA damage using the comet assay in conjunction with γ-H2AX and colocalized repair protein 53BP1. | Increase in γ-H2AX and 53BP1 foci after high-intensity exercise, with markers, increased in hypoxia. Although normoxia resulted in a marked increase in foci detection, hypoxia challenge resulted in a 2.5- and 3.5-fold increase in γ-H2AX and 53BP1 foci, respectively, after exercise. | [216] |
| Sixty-one T2DM subjects, aged (mean ± SD: 50.3 ± 4.2) | Glycemic status. DNA damage (Comet assay). Oxidative DNA damage. OGG1 protein expression. TAC. | Elucidation of the mechanism of action of yoga on T2DM-related DNA damage in terms of its effect on oxidative DNA damage and DNA repair markers. | The yoga group showed a significant reduction in DNA damage, oxidative DNA damage marker, and fasting blood sugar compared to the control. The beneficial effect of yoga on DNA damage in T2DM subjects was found to be mediated by the mitigation of oxidative DNA damage and enhancement of DNA repair. | [217] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Álvarez, D.; Rosado-Pérez, J.; Gavia-García, G.; Arista-Ugalde, T.L.; Aguiñiga-Sánchez, I.; Santiago-Osorio, E.; Mendoza-Núñez, V.M. Aging, Physical Exercise, Telomeres, and Sarcopenia: A Narrative Review. Biomedicines 2023, 11, 598. https://doi.org/10.3390/biomedicines11020598
Hernández-Álvarez D, Rosado-Pérez J, Gavia-García G, Arista-Ugalde TL, Aguiñiga-Sánchez I, Santiago-Osorio E, Mendoza-Núñez VM. Aging, Physical Exercise, Telomeres, and Sarcopenia: A Narrative Review. Biomedicines. 2023; 11(2):598. https://doi.org/10.3390/biomedicines11020598
Chicago/Turabian StyleHernández-Álvarez, David, Juana Rosado-Pérez, Graciela Gavia-García, Taide Laurita Arista-Ugalde, Itzen Aguiñiga-Sánchez, Edelmiro Santiago-Osorio, and Víctor Manuel Mendoza-Núñez. 2023. "Aging, Physical Exercise, Telomeres, and Sarcopenia: A Narrative Review" Biomedicines 11, no. 2: 598. https://doi.org/10.3390/biomedicines11020598