
Nonsense-Mediated mRNA Decay Factor Functions in Human Health and Disease


Abstract
:1. Introduction
2. Mammalian NMD Pathways
3. Core NMD Factors
3.1. UPF1

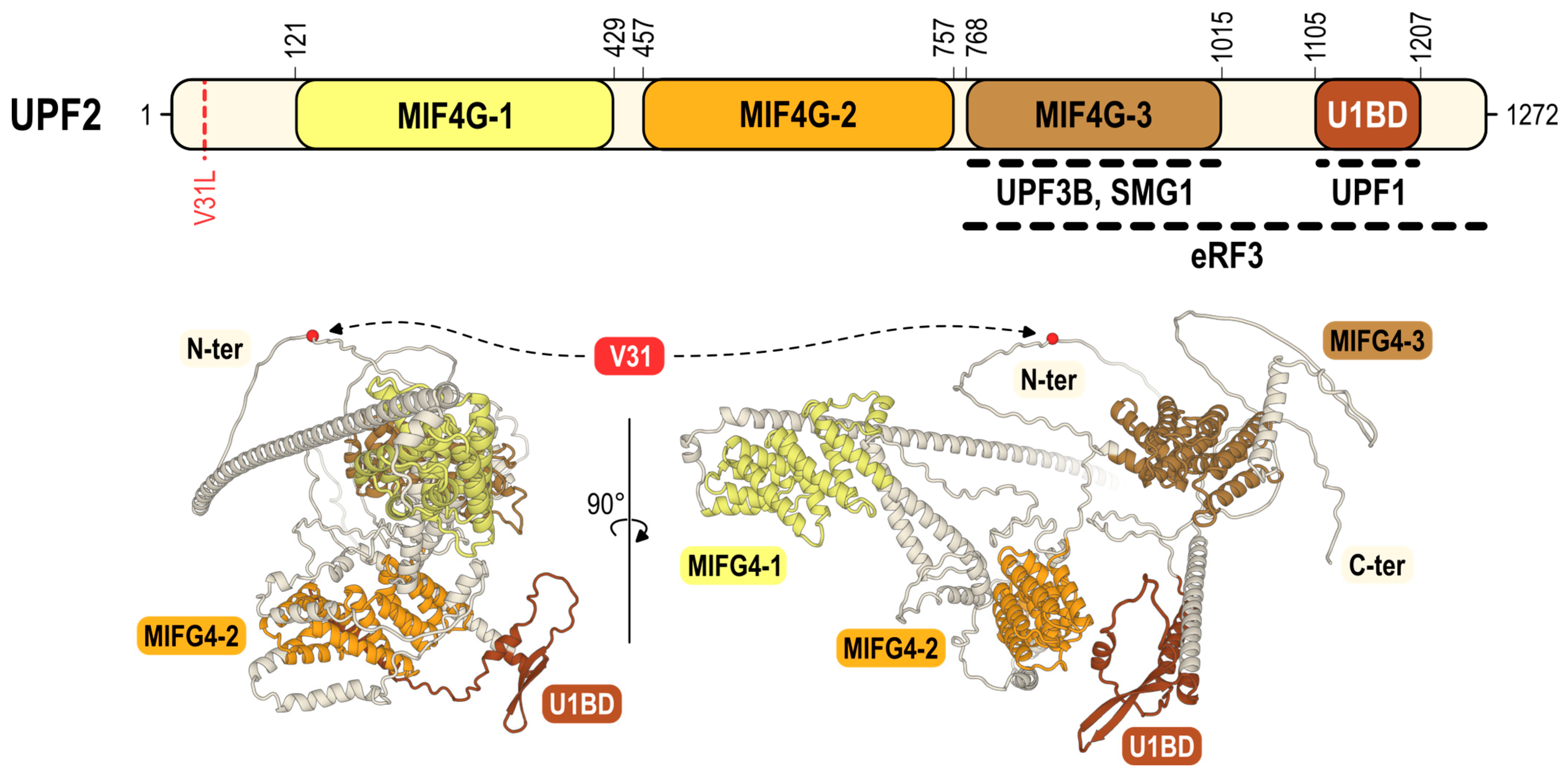
3.2. UPF2
3.3. UPF3A and UPF3B Paralogs
4. Implication of NMD Factors in Human Disease
4.1. Neurodevelopmental Disorders (NDD)
4.2. Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Potential Therapeutic Approach | Ref. | ||||
|---|---|---|---|---|---|---|
| Cancer Type, Cell Line | Characteristics | Strategy | Method | Effect | ||
| NMD inhibition | Small-cell lung cancer, cell line N417 Breast cancer, cell line HDQP-I | PTC mutationsin Tp53 gene | Promoting expression of tumour suppressor proteins | NMDI14 (disruption of UPF1-SMG7 interaction), G418 (PTC read-through) | Restoration of full-length p53 expression, leading to cell death | [141] |
| Microsatellite instability colorectal cancer | Overexpression of UPF1, UPF2, SMG1, SMG6 and SMG7 | Amlexanox or UPF1 siRNA | Decrease in cell proliferation rate | [139] | ||
| Murine colorectal carcinoma, cell line CT26 | n.d. a | Producing tumour-specific neoantigens that can be recognised by the human immune system | PSMA b aptamer-SMG1 siRNA conjugate, 4-1BB aptamer | Suppression of tumour growth by PSMA-Smg1 ± 4-1BB treatment | [142] | |
| B-cell lymphoma | n.d. a | CD40-agonist aptamer SMG1-shRNA chimera | Tumour infiltration by lymphocytes Improved mice survival | [144] | ||
| Murine breast cancer, cell line 4T1E | n.d. a | EpCAM c aptamer- UPF2 siRNA chimera | Improved CD8+ T-cell immunity Inhibition of tumour growth | [143] | ||
| NMD activation | Prostate cancer, cell line PC3 Colon cancer, cell line HCT116 Melanoma, cell line A375 | eIF2α phosphorylation | Downregulating tumorigenesis transcripts | UPF1 overexpression | Inhibition of tumour growth | [146] |
| Human gastric tumour | Inhibition of UPF1 expression | Suppression of cell cycle progression and proliferation Promotion of cell apoptosis Inhibition of EMT d and metastasis | [147] | |||
4.3. Viral Infections
4.3.1. NMD Restriction of Viral Replication
| Virus Classification | Effect of NMD Factors | |||||||
|---|---|---|---|---|---|---|---|---|
| Name a | Realm | Genome b | Family | Genus | Anti-Viral | None | Pro-Viral | Ref. |
| MHV | Riboviria | +ssRNA | Coronaviridae | Betacoronavirus | UPF1 | n.d. c | n.d. c | [157] |
| UPF2 | ||||||||
| SMG5 | ||||||||
| SMG6 | ||||||||
| SFV | Riboviria | +ssRNA | Togaviridae | Alphavirus | UPF1 | SMG6 | n.d. c | [156] |
| SMG5 | ||||||||
| SMG7 | ||||||||
| SINV | Riboviria | +ssRNA | Togaviridae | Alphavirus | UPF1 | n.d. c | n.d. c | [155] |
| ZIKV | Riboviria | +ssRNA | Flaviviridae | Flavivirus | UPF1 | n.d. c | n.d. c | [158] |
| UUKV | Riboviria | -ssRNA | Phenuiviridae | Phlebovirus | n.d. c | UPF1 | n.d. c | [167] |
| HRSV | Riboviria | -ssRNA | Pneumoviridae | Orthopneumovirus | n.d. c | UPF1 | n.d. c | [156] |
| HIV-1 | Riboviria | ssRNA-RT | Retroviridae | Lentivirus | UPF2 | n.d. c | UPF1 d | [159,160,161,162,163] |
| SMG6 | ||||||||
| KSHV | Duplodnaviria | dsDNA | Herpesviridae | Rhadinovirus | UPF1 | n.d. c | n.d. c | [164] |
| UPF3B | ||||||||
4.3.2. Viral NMD Evasion cis-Strategies: NMD Resistance Conferred by Inherent RNA Features
| NMD-Resistant RNA Feature | Virus Classification | |||||
|---|---|---|---|---|---|---|
| Realm | Genome a | Family | Genus | Name b | Ref. | |
| Programmed ribosome readthrough or frameshifting | Riboviria | ssRNA-RT | Retroviridae | Gammaretrovirus | MoMLV | [168] |
| Riboviria | dsRNA | Reoviridae | Coltivirus | CTFV | [169] | |
| Regulated RNA splicing and export | Riboviria | ssRNA-RT | Retroviridae | Lentivirus | HIV-1 | [170,171] |
| 3′-UTR RSE bound by PTBP1 | Riboviria | ssRNA-RT | Retroviridae | Alpharetrovirus | RSV | [95,172] |
4.3.3. Viral NMD Evasion trans-Strategies: NMD Inhibition by Viral Proteins
| NMD Inhibition | Virus Classification | ||||||
|---|---|---|---|---|---|---|---|
| Viral Protein | Mechanism | Realm | Genome a | Family | Genus | Name b | Ref. |
| N (nucleocapsid) | Interacts with UPF1 | Riboviria | +ssRNA | Coronaviridae | Gammacoronavirus | IBV | [179] |
| Riboviria | +ssRNA | Coronaviridae | Betacoronavirus | MHV | [157] | ||
| Riboviria | +ssRNA | Coronaviridae | Betacoronavirus | SARS-CoV-2 | [180] | ||
| C (capsid) | Interacts with UPF1 Interacts with PYM, preventing its interaction with MAGOH and RBM8A | Riboviria | +ssRNA | Togaviridae | Alphavirus | SFV | [181] |
| Riboviria | +ssRNA | Flaviviridae | Hepacivirus | HCV | [182] | ||
| Riboviria | +ssRNA | Flaviviridae | Flavivirus | WNV | [183] | ||
| Riboviria | +ssRNA | Flaviviridae | Flavivirus | DENV | [183] | ||
| Riboviria | +ssRNA | Flaviviridae | Flavivirus | ZIKV | [158] | ||
| Interacts with UPF1, promoting its degradation in the nucleus | Riboviria | +ssRNA | Flaviviridae | Flavivirus | ZIKV | [186] | |
| Rex (regulatory) | Unknown mechanism | Riboviria | ssRNA-RT | Retroviridae | Deltaretrovirus | HTLV-1 | [187] |
| Tax (regulatory) | Interacts with INT6 and UPF1, impeding UPF1 association with RNA and promoting phospho-UPF1 sequestration in P-bodies | Riboviria | ssRNA-RT | Retroviridae | Deltaretrovirus | HTLV-1 | [188,189] |
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Amrani, N.; Ganesan, R.; Kervestin, S.; Mangus, D.A.; Ghosh, S.; Jacobson, A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 2004, 432, 112–118. [Google Scholar] [CrossRef]
- Eberle, A.B.; Stalder, L.; Mathys, H.; Orozco, R.Z.; Mühlemann, O. Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol. 2008, 6, e92. [Google Scholar] [CrossRef]
- Peixeiro, I.; Inácio, Â.; Barbosa, C.; Silva, A.L.; Liebhaber, S.A.; Romão, L. Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res. 2012, 40, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Mikhailova, T.; Eliseev, B.; Yeramala, L.; Sokolova, E.; Susorov, D.; Shuvalov, A.; Schaffitzel, C.; Alkalaeva, E. PABP enhances release factor recruitment and stop codon recognition during translation termination. Nucleic Acids Res. 2016, 44, 7766–7776. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Shu, M.D.; Steitz, J.A. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 2000, 103, 1121–1131. [Google Scholar] [CrossRef]
- Le Hir, H.; Gatfield, D.; Izaurralde, E.; Moore, M.J. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001, 20, 4987–4997. [Google Scholar] [CrossRef]
- Lindeboom, R.G.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, H.; Izaurralde, E.; Maquat, L.E.; Moore, M.J. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000, 19, 6860–6869. [Google Scholar] [CrossRef]
- Gehring, N.H.; Lamprinaki, S.; Kulozik, A.E.; Hentze, M.W. Disassembly of exon junction complexes by PYM. Cell 2009, 137, 536–548. [Google Scholar] [CrossRef]
- Raimondeau, E.; Bufton, J.C.; Schaffitzel, C. New insights into the interplay between the translation machinery and nonsense-mediated mRNA decay factors. Biochem. Soc. Trans. 2018, 46, 503–512. [Google Scholar] [CrossRef]
- Karousis, E.D.; Mühlemann, O. Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb. Perspect. Biol. 2019, 11, a032862. [Google Scholar] [CrossRef] [PubMed]
- Lavysh, D.; Neu-Yilik, G. UPF1-Mediated RNA Decay-Danse Macabre in a Cloud. Biomolecules 2020, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Nasif, S.; Contu, L.; Mühlemann, O. Beyond quality control: The role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin. Cell Dev. Biol. 2018, 75, 78–87. [Google Scholar] [CrossRef]
- Kim, Y.K.; Maquat, L.E. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 2019, 25, 407–422. [Google Scholar] [CrossRef]
- Pawlicka, K.; Kalathiya, U.; Alfaro, J. Nonsense-Mediated mRNA Decay: Pathologies and the Potential for Novel Therapeutics. Cancers 2020, 12, 765. [Google Scholar] [CrossRef]
- Fernandes, R.; Nogueira, G.; Da Costa, P.J.; Pinto, F.; Romão, L. Nonsense-Mediated mRNA Decay in Development, Stress and Cancer. Adv. Exp. Med. Biol. 2019, 1157, 41–83. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Muhlrad, D.; Parker, R. Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 1999, 5, 1299–1307. [Google Scholar] [CrossRef]
- Ruiz-Echevarría, M.J.; Peltz, S.W. The RNA binding protein Pub1 modulates the stability of transcripts containing upstream open reading frames. Cell 2000, 101, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Yepiskoposyan, H.; Aeschimann, F.; Nilsson, D.; Okoniewski, M.; Mühlemann, O. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 2011, 17, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Fujimura, M. Post-transcriptional defects of antioxidant selenoenzymes cause oxidative stress under methylmercury exposure. J. Biol. Chem. 2011, 286, 6641–6649. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, A.A.; Cenik, C.; Chua, H.N.; Roth, F.P.; Moore, M.J. Introns in UTRs: Why we should stop ignoring them. Bioessays 2012, 34, 1025–1034. [Google Scholar] [CrossRef]
- Lykke-Andersen, S.; Chen, Y.; Ardal, B.R.; Lilje, B.; Waage, J.; Sandelin, A.; Jensen, T.H. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 2014, 28, 2498–2517. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Wilusz, J. Nonsense-mediated RNA decay: At the ‘cutting edge’ of regulated snoRNA production. Genes Dev. 2014, 28, 2447–2449. [Google Scholar] [CrossRef]
- Andjus, S.; Morillon, A.; Wery, M. From Yeast to Mammals, the Nonsense-Mediated mRNA Decay as a Master Regulator of Long Non-Coding RNAs Functional Trajectory. Noncoding RNA 2021, 7, 44. [Google Scholar] [CrossRef]
- Yi, Z.; Sanjeev, M.; Singh, G. The Branched Nature of the Nonsense-Mediated mRNA Decay Pathway. Trends Genet. 2021, 37, 143–159. [Google Scholar] [CrossRef]
- Mailliot, J.; Vivoli-Vega, M.; Schaffitzel, C. No-nonsense: Insights into the functional interplay of nonsense-mediated mRNA decay factors. Biochem. J. 2022, 479, 973–993. [Google Scholar] [CrossRef]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Izumi, N.; Kashima, I.; Ohnishi, T.; Saari, B.; Katsuhata, Y.; Muramatsu, R.; Morita, T.; Iwamatsu, A.; Hachiya, T.; et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009, 23, 1091–1105. [Google Scholar] [CrossRef]
- Durand, S.; Franks, T.M.; Lykke-Andersen, J. Hyperphosphorylation amplifies UPF1 activity to resolve stalls in nonsense-mediated mRNA decay. Nat. Commun. 2016, 7, 12434. [Google Scholar] [CrossRef] [PubMed]
- Durand, S.; Cougot, N.; Mahuteau-Betzer, F.; Nguyen, C.H.; Grierson, D.S.; Bertrand, E.; Tazi, J.; Lejeune, F. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J. Cell Biol. 2007, 178, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Serdar, L.D.; Whiteside, D.L.; Nock, S.L.; McGrath, D.; Baker, K.E. Inhibition of post-termination ribosome recycling at premature termination codons in UPF1 ATPase mutants. eLife 2020, 9, e57834. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Mangkalaphiban, K.; Baker, R.E.; He, F.; Jacobson, A. Ribosome-bound Upf1 forms distinct 80S complexes and conducts mRNA surveillance. RNA 2022, 28, 1621–1642. [Google Scholar] [CrossRef]
- Pisarev, A.V.; Hellen, C.U.; Pestova, T.V. Recycling of eukaryotic posttermination ribosomal complexes. Cell 2007, 131, 286–299. [Google Scholar] [CrossRef]
- Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.; Pestova, T.V. Reinitiation and other unconventional posttermination events during eukaryotic translation. Mol. Cell 2013, 51, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Isken, O.; Kim, Y.K.; Hosoda, N.; Mayeur, G.L.; Hershey, J.W.; Maquat, L.E. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 2008, 133, 314–327. [Google Scholar] [CrossRef]
- Okada-Katsuhata, Y.; Yamashita, A.; Kutsuzawa, K.; Izumi, N.; Hirahara, F.; Ohno, S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012, 40, 1251–1266. [Google Scholar] [CrossRef]
- Eberle, A.B.; Lykke-Andersen, S.; Mühlemann, O.; Jensen, T.H. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 2009, 16, 49–55. [Google Scholar] [CrossRef]
- Boehm, V.; Haberman, N.; Ottens, F.; Ule, J.; Gehring, N.H. 3′ UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep. 2014, 9, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Shyu, A.B. Rapid deadenylation triggered by a nonsense codon precedes decay of the RNA body in a mammalian cytoplasmic nonsense-mediated decay pathway. Mol. Cell. Biol. 2003, 23, 4805–4813. [Google Scholar] [CrossRef]
- Lejeune, F.; Li, X.; Maquat, L.E. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol. Cell 2003, 12, 675–687. [Google Scholar] [CrossRef]
- Ohnishi, T.; Yamashita, A.; Kashima, I.; Schell, T.; Anders, K.R.; Grimson, A.; Hachiya, T.; Hentze, M.W.; Anderson, P.; Ohno, S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell 2003, 12, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Bono, F.; Ebert, J.; Lorentzen, E.; Conti, E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 2006, 126, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.B.; Ballut, L.; Johansen, J.S.; Chamieh, H.; Nielsen, K.H.; Oliveira, C.L.; Pedersen, J.S.; Séraphin, B.; Le Hir, H.; Andersen, G.R. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 2006, 313, 1968–1972. [Google Scholar] [CrossRef]
- Gerbracht, J.V.; Boehm, V.; Britto-Borges, T.; Kallabis, S.; Wiederstein, J.L.; Ciriello, S.; Aschemeier, D.U.; Krüger, M.; Frese, C.K.; Altmüller, J.; et al. CASC3 promotes transcriptome-wide activation of nonsense-mediated decay by the exon junction complex. Nucleic Acids Res. 2020, 48, 8626–8644. [Google Scholar] [CrossRef]
- Gehring, N.H.; Kunz, J.B.; Neu-Yilik, G.; Breit, S.; Viegas, M.H.; Hentze, M.W.; Kulozik, A.E. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol. Cell 2005, 20, 65–75. [Google Scholar] [CrossRef]
- Gehring, N.H.; Lamprinaki, S.; Hentze, M.W.; Kulozik, A.E. The hierarchy of exon-junction complex assembly by the spliceosome explains key features of mammalian nonsense-mediated mRNA decay. PLoS Biol. 2009, 7, e1000120. [Google Scholar] [CrossRef]
- Mabin, J.W.; Woodward, L.A.; Patton, R.D.; Yi, Z.; Jia, M.; Wysocki, V.H.; Bundschuh, R.; Singh, G. The Exon Junction Complex Undergoes a Compositional Switch that Alters mRNP Structure and Nonsense-Mediated mRNA Decay Activity. Cell Rep. 2018, 25, 2431–2446. [Google Scholar] [CrossRef]
- Clerici, M.; Mourão, A.; Gutsche, I.; Gehring, N.H.; Hentze, M.W.; Kulozik, A.; Kadlec, J.; Sattler, M.; Cusack, S. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009, 28, 2293–2306. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Jayachandran, U.; Bonneau, F.; Fiorini, F.; Basquin, C.; Domcke, S.; Le Hir, H.; Conti, E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell 2011, 41, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Maciej, V.D.; Machado de Amorim, A.; Pak, M.; Jayachandran, U.; Chakrabarti, S. Modulation of RNA-binding properties of the RNA helicase UPF1 by its activator UPF2. RNA 2023, 29, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.; Leroy, C.; Salomé-Desnoulez, S.; Werkmeister, E.; Kong, R.; Mongy, M.; Le Hir, H.; Lejeune, F. A role for AKT1 in nonsense-mediated mRNA decay. Nucleic Acids Res. 2021, 49, 11022–11037. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Abshire, E.T.; Popp, M.W.; Pröschel, C.; Schwartz, J.L.; Yeo, G.W.; Maquat, L.E. AKT constitutes a signal-promoted alternative exon-junction complex that regulates nonsense-mediated mRNA decay. Mol. Cell 2022, 82, 2779–2796.e2710. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Shu, M.D.; Steitz, J.A. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science 2001, 293, 1836–1839. [Google Scholar] [CrossRef]
- Zhang, Z.; Krainer, A.R. Involvement of SR proteins in mRNA surveillance. Mol. Cell 2004, 16, 597–607. [Google Scholar] [CrossRef]
- Sato, H.; Hosoda, N.; Maquat, L.E. Efficiency of the pioneer round of translation affects the cellular site of nonsense-mediated mRNA decay. Mol. Cell 2008, 29, 255–262. [Google Scholar] [CrossRef]
- Aznarez, I.; Nomakuchi, T.T.; Tetenbaum-Novatt, J.; Rahman, M.A.; Fregoso, O.; Rees, H.; Krainer, A.R. Mechanism of Nonsense-Mediated mRNA Decay Stimulation by Splicing Factor SRSF1. Cell Rep. 2018, 23, 2186–2198. [Google Scholar] [CrossRef]
- Michlewski, G.; Sanford, J.R.; Cáceres, J.F. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol. Cell 2008, 30, 179–189. [Google Scholar] [CrossRef]
- Chiu, S.Y.; Serin, G.; Ohara, O.; Maquat, L.E. Characterization of human Smg5/7a: A protein with similarities to Caenorhabditis elegans SMG5 and SMG7 that functions in the dephosphorylation of Upf1. RNA 2003, 9, 77–87. [Google Scholar] [CrossRef]
- Anders, K.R.; Grimson, A.; Anderson, P. SMG-5, required for C.elegans nonsense-mediated mRNA decay, associates with SMG-2 and protein phosphatase 2A. EMBO J. 2003, 22, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Ji, S.J.; Porse, B.T.; Jaffrey, S.R. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell 2013, 153, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Longman, D.; Hug, N.; Keith, M.; Anastasaki, C.; Patton, E.E.; Grimes, G.; Cáceres, J.F. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res. 2013, 41, 8319–8331. [Google Scholar] [CrossRef]
- Alrahbeni, T.; Sartor, F.; Anderson, J.; Miedzybrodzka, Z.; McCaig, C.; Müller, B. Full UPF3B function is critical for neuronal differentiation of neural stem cells. Mol. Brain 2015, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, K.; Kaufman, R.J. Interaction between quality control systems for ER protein folding and RNA biogenesis. Worm 2013, 2, e23005. [Google Scholar] [CrossRef]
- Sieber, J.; Hauer, C.; Bhuvanagiri, M.; Leicht, S.; Krijgsveld, J.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E. Proteomic Analysis Reveals Branch-specific Regulation of the Unfolded Protein Response by Nonsense-mediated mRNA Decay. Mol. Cell. Proteomics 2016, 15, 1584–1597. [Google Scholar] [CrossRef]
- Karam, R.; Lou, C.H.; Kroeger, H.; Huang, L.; Lin, J.H.; Wilkinson, M.F. The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 2015, 16, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Longman, D.; Jackson-Jones, K.A.; Maslon, M.M.; Murphy, L.C.; Young, R.S.; Stoddart, J.J.; Hug, N.; Taylor, M.S.; Papadopoulos, D.K.; Cáceres, J.F. Identification of a localized nonsense-mediated decay pathway at the endoplasmic reticulum. Genes Dev. 2020, 34, 1075–1088. [Google Scholar] [CrossRef]
- Karam, R.; Wengrod, J.; Gardner, L.B.; Wilkinson, M.F. Regulation of nonsense-mediated mRNA decay: Implications for physiology and disease. Biochim. Biophys. Acta 2013, 1829, 624–633. [Google Scholar] [CrossRef]
- Kunz, J.B.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E.; Gehring, N.H. Functions of hUpf3a and hUpf3b in nonsense-mediated mRNA decay and translation. RNA 2006, 12, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Bhalla, A.D.; Le Hir, H.; Nguyen, L.S.; Huang, L.; Gécz, J.; Wilkinson, M.F. A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat. Struct. Mol. Biol. 2009, 16, 747–753. [Google Scholar] [CrossRef]
- Bao, J.; Tang, C.; Yuan, S.; Porse, B.T.; Yan, W. UPF2, a nonsense-mediated mRNA decay factor, is required for prepubertal Sertoli cell development and male fertility by ensuring fidelity of the transcriptome. Development 2015, 142, 352–362. [Google Scholar] [CrossRef]
- Shum, E.Y.; Jones, S.H.; Shao, A.; Dumdie, J.; Krause, M.D.; Chan, W.K.; Lou, C.H.; Espinoza, J.L.; Song, H.W.; Phan, M.H.; et al. The Antagonistic Gene Paralogs Upf3a and Upf3b Govern Nonsense-Mediated RNA Decay. Cell 2016, 165, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Wallmeroth, D.; Lackmann, J.W.; Kueckelmann, S.; Altmüller, J.; Dieterich, C.; Boehm, V.; Gehring, N.H. Human UPF3A and UPF3B enable fault-tolerant activation of nonsense-mediated mRNA decay. EMBO J. 2022, 41, e109191. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Arvola, R.M.; Myers, S.; Dilsavor, C.N.; Abu Alhasan, R.; Carter, B.N.; Patton, R.D.; Bundschuh, R.; Singh, G. Mammalian UPF3A and UPF3B can activate nonsense-mediated mRNA decay independently of their exon junction complex binding. EMBO J. 2022, 41, e109202. [Google Scholar] [CrossRef]
- Gowravaram, M.; Bonneau, F.; Kanaan, J.; Maciej, V.D.; Fiorini, F.; Raj, S.; Croquette, V.; Le Hir, H.; Chakrabarti, S. A conserved structural element in the RNA helicase UPF1 regulates its catalytic activity in an isoform-specific manner. Nucleic Acids Res. 2018, 46, 2648–2659. [Google Scholar] [CrossRef]
- Fritz, S.E.; Ranganathan, S.; Wang, C.D.; Hogg, J.R. An alternative UPF1 isoform drives conditional remodeling of nonsense-mediated mRNA decay. EMBO J. 2022, 41, e108898. [Google Scholar] [CrossRef]
- Kadlec, J.; Guilligay, D.; Ravelli, R.B.; Cusack, S. Crystal structure of the UPF2-interacting domain of nonsense-mediated mRNA decay factor UPF1. RNA 2006, 12, 1817–1824. [Google Scholar] [CrossRef]
- Cheng, Z.; Muhlrad, D.; Lim, M.K.; Parker, R.; Song, H. Structural and functional insights into the human Upf1 helicase core. EMBO J. 2007, 26, 253–264. [Google Scholar] [CrossRef]
- Weng, Y.; Czaplinski, K.; Peltz, S.W. ATP is a cofactor of the Upf1 protein that modulates its translation termination and RNA binding activities. RNA 1998, 4, 205–214. [Google Scholar]
- Bhattacharya, A.; Czaplinski, K.; Trifillis, P.; He, F.; Jacobson, A.; Peltz, S.W. Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA 2000, 6, 1226–1235. [Google Scholar] [CrossRef]
- Franks, T.M.; Singh, G.; Lykke-Andersen, J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense-mediated mRNA decay. Cell 2010, 143, 938–950. [Google Scholar] [CrossRef]
- Chapman, J.H.; Craig, J.M.; Wang, C.D.; Gundlach, J.H.; Neuman, K.C.; Hogg, J.R. UPF1 mutants with intact ATPase but deficient helicase activities promote efficient nonsense-mediated mRNA decay. Nucleic Acids Res. 2022, 50, 11876–11894. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, F.; Bagchi, D.; Le Hir, H.; Croquette, V. Human Upf1 is a highly processive RNA helicase and translocase with RNP remodelling activities. Nat. Commun. 2015, 6, 7581. [Google Scholar] [CrossRef] [PubMed]
- Serdar, L.D.; Whiteside, D.L.; Baker, K.E. ATP hydrolysis by UPF1 is required for efficient translation termination at premature stop codons. Nat. Commun. 2016, 7, 14021. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.E.; Ranganathan, S.; Wang, C.D.; Hogg, J.R. The RNA-binding protein PTBP1 promotes ATPase-dependent dissociation of the RNA helicase UPF1 to protect transcripts from nonsense-mediated mRNA decay. J. Biol. Chem. 2020, 295, 11613–11625. [Google Scholar] [CrossRef]
- Kurosaki, T.; Li, W.; Hoque, M.; Popp, M.W.; Ermolenko, D.N.; Tian, B.; Maquat, L.E. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev. 2014, 28, 1900–1916. [Google Scholar] [CrossRef]
- Lee, S.R.; Pratt, G.A.; Martinez, F.J.; Yeo, G.W.; Lykke-Andersen, J. Target Discrimination in Nonsense-Mediated mRNA Decay Requires Upf1 ATPase Activity. Mol. Cell 2015, 59, 413–425. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar (accessed on 13 January 2023).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Chamieh, H.; Ballut, L.; Bonneau, F.; Le Hir, H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008, 15, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Melero, R.; Buchwald, G.; Castaño, R.; Raabe, M.; Gil, D.; Lázaro, M.; Urlaub, H.; Conti, E.; Llorca, O. The cryo-EM structure of the UPF-EJC complex shows UPF1 poised toward the RNA 3′ end. Nat. Struct. Mol. Biol. 2012, 19, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Padariya, M.; Fahraeus, R.; Hupp, T.; Kalathiya, U. Molecular Determinants and Specificity of mRNA with Alternatively-Spliced UPF1 Isoforms, Influenced by an Insertion in the ‘Regulatory Loop’. Int. J. Mol. Sci. 2021, 22, 2744. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Quek, B.L.; Beemon, K.L.; Hogg, J.R. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. eLife 2016, 5, e11155. [Google Scholar] [CrossRef] [PubMed]
- Kishor, A.; Ge, Z.; Hogg, J.R. hnRNP L-dependent protection of normal mRNAs from NMD subverts quality control in B cell lymphoma. EMBO J. 2019, 38, e99128. [Google Scholar] [CrossRef]
- Kishor, A.; Fritz, S.E.; Haque, N.; Ge, Z.; Tunc, I.; Yang, W.; Zhu, J.; Hogg, J.R. Activation and inhibition of nonsense-mediated mRNA decay control the abundance of alternative polyadenylation products. Nucleic Acids Res. 2020, 48, 7468–7482. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; Karuppasamy, M.; Bock, T.; Masiulis, S.; Huard, K.; Garzoni, F.; Kerschgens, K.; Hentze, M.W.; Kulozik, A.E.; Beck, M.; et al. A network of SMG-8, SMG-9 and SMG-1 C-terminal insertion domain regulates UPF1 substrate recruitment and phosphorylation. Nucleic Acids Res. 2015, 43, 7600–7611. [Google Scholar] [CrossRef]
- Brumbaugh, K.M.; Otterness, D.M.; Geisen, C.; Oliveira, V.; Brognard, J.; Li, X.; Lejeune, F.; Tibbetts, R.S.; Maquat, L.E.; Abraham, R.T. The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol. Cell 2004, 14, 585–598. [Google Scholar] [CrossRef]
- Langer, L.M.; Gat, Y.; Bonneau, F.; Conti, E. Structure of substrate-bound SMG1-8-9 kinase complex reveals molecular basis for phosphorylation specificity. eLife 2020, 9, e57127. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Bonneau, F.; Schüssler, S.; Eppinger, E.; Conti, E. Phospho-dependent and phospho-independent interactions of the helicase UPF1 with the NMD factors SMG5-SMG7 and SMG6. Nucleic Acids Res. 2014, 42, 9447–9460. [Google Scholar] [CrossRef]
- Nicholson, P.; Josi, C.; Kurosawa, H.; Yamashita, A.; Mühlemann, O. A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res. 2014, 42, 9217–9235. [Google Scholar] [CrossRef]
- Takahashi, S.; Araki, Y.; Ohya, Y.; Sakuno, T.; Hoshino, S.; Kontani, K.; Nishina, H.; Katada, T. Upf1 potentially serves as a RING-related E3 ubiquitin ligase via its association with Upf3 in yeast. RNA 2008, 14, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Jagannathan, S.; Bradley, R.K. The RNA Surveillance Factor UPF1 Represses Myogenesis via Its E3 Ubiquitin Ligase Activity. Mol. Cell 2017, 67, 239–251.e236. [Google Scholar] [CrossRef]
- Chu, V.; Feng, Q.; Lim, Y.; Shao, S. Selective destabilization of polypeptides synthesized from NMD-targeted transcripts. Mol. Biol. Cell 2021, 32, ar38. [Google Scholar] [CrossRef]
- Inglis, A.J.; Guna, A.; Merchán, Á.G.; Pal, A.; Esantsi, T.K.; Keys, H.R.; Frenkel, E.M.; Oania, R.; Weissman, J.S.; Voorhees, R.M. Coupled protein quality control during nonsense mediated mRNA decay. bioRxiv 2022. [Google Scholar] [CrossRef]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef]
- Powers, K.T.; Szeto, J.A.; Schaffitzel, C. New insights into no-go, non-stop and nonsense-mediated mRNA decay complexes. Curr. Opin. Struct. Biol. 2020, 65, 110–118. [Google Scholar] [CrossRef]
- Tatsuno, T.; Nakamura, Y.; Ma, S.; Tomosugi, N.; Ishigaki, Y. Nonsense-mediated mRNA decay factor Upf2 exists in both the nucleoplasm and the cytoplasm. Mol. Med. Rep. 2016, 14, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Deniaud, A.; Boehm, V.; Gehring, N.H.; Schaffitzel, C.; Cusack, S. Structural and functional analysis of the three MIF4G domains of nonsense-mediated decay factor UPF2. Nucleic Acids Res. 2014, 42, 2673–2686. [Google Scholar] [CrossRef]
- López-Perrote, A.; Castaño, R.; Melero, R.; Zamarro, T.; Kurosawa, H.; Ohnishi, T.; Uchiyama, A.; Aoyagi, K.; Buchwald, G.; Kataoka, N.; et al. Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res. 2016, 44, 1909–1923. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, J.; Izaurralde, E.; Cusack, S. The structural basis for the interaction between nonsense-mediated mRNA decay factors UPF2 and UPF3. Nat. Struct. Mol. Biol. 2004, 11, 330–337. [Google Scholar] [CrossRef]
- Neu-Yilik, G.; Raimondeau, E.; Eliseev, B.; Yeramala, L.; Amthor, B.; Deniaud, A.; Huard, K.; Kerschgens, K.; Hentze, M.W.; Schaffitzel, C.; et al. Dual function of UPF3B in early and late translation termination. EMBO J. 2017, 36, 2968–2986. [Google Scholar] [CrossRef]
- Bufton, J.C.; Powers, K.T.; Szeto, J.A.; Toelzer, C.; Berger, I.; Schaffitzel, C. Structures of nonsense-mediated mRNA decay factors UPF3B and UPF3A in complex with UPF2 reveal molecular basis for competitive binding and for neurodevelopmental disorder-causing mutation. Nucleic Acids Res. 2022, 50, 5934–5947. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Leoyklang, P.; Suphapeetiporn, K.; Srichomthong, C.; Tongkobpetch, S.; Fietze, S.; Dorward, H.; Cullinane, A.R.; Gahl, W.A.; Huizing, M.; Shotelersuk, V. Disorders with similar clinical phenotypes reveal underlying genetic interaction: SATB2 acts as an activator of the UPF3B gene. Hum. Genet. 2013, 132, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.S.; Jolly, L.; Shoubridge, C.; Chan, W.K.; Huang, L.; Laumonnier, F.; Raynaud, M.; Hackett, A.; Field, M.; Rodriguez, J.; et al. Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol. Psychiatry 2012, 17, 1103–1115. [Google Scholar] [CrossRef]
- Buchwald, G.; Ebert, J.; Basquin, C.; Sauliere, J.; Jayachandran, U.; Bono, F.; Le Hir, H.; Conti, E. Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc. Natl. Acad. Sci. USA 2010, 107, 10050–10055. [Google Scholar] [CrossRef]
- Gehring, N.H.; Neu-Yilik, G.; Schell, T.; Hentze, M.W.; Kulozik, A.E. Y14 and hUpf3b form an NMD-activating complex. Mol. Cell 2003, 11, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shum, E.Y.; Jones, S.H.; Lou, C.H.; Chousal, J.; Kim, H.; Roberts, A.J.; Jolly, L.A.; Espinoza, J.L.; Skarbrevik, D.M.; et al. A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol. Psychiatry 2018, 23, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Domingo, D.; Nawaz, U.; Corbett, M.; Espinoza, J.L.; Tatton-Brown, K.; Coman, D.; Wilkinson, M.F.; Gecz, J.; Jolly, L.A. A synonymous UPF3B variant causing a speech disorder implicates NMD as a regulator of neurodevelopmental disorder gene networks. Hum. Mol. Genet. 2020, 29, 2568–2578. [Google Scholar] [CrossRef]
- Bruno, I.G.; Karam, R.; Huang, L.; Bhardwaj, A.; Lou, C.H.; Shum, E.Y.; Song, H.W.; Corbett, M.A.; Gifford, W.D.; Gecz, J.; et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol. Cell 2011, 42, 500–510. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Kim, H.G.; Rosenfeld, J.A.; Shen, Y.; Gusella, J.F.; Lacassie, Y.; Layman, L.C.; Shaffer, L.G.; Gécz, J. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet. 2013, 22, 1816–1825. [Google Scholar] [CrossRef]
- Johnson, J.L.; Stoica, L.; Liu, Y.; Zhu, P.J.; Bhattacharya, A.; Buffington, S.A.; Huq, R.; Eissa, N.T.; Larsson, O.; Porse, B.T.; et al. Inhibition of Upf2-Dependent Nonsense-Mediated Decay Leads to Behavioral and Neurophysiological Abnormalities by Activating the Immune Response. Neuron 2019, 104, 665–679.e668. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, P.S.; Raymond, F.L.; Nguyen, L.S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C.; Edkins, S.; Stevens, C.; et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007, 39, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Shoubridge, C.; Antar, C.; Nguyen, L.S.; Van Esch, H.; Kleefstra, T.; Briault, S.; Fryns, J.P.; Hamel, B.; Chelly, J.; et al. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry 2010, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Addington, A.M.; Gauthier, J.; Piton, A.; Hamdan, F.F.; Raymond, A.; Gogtay, N.; Miller, R.; Tossell, J.; Bakalar, J.; Inoff-Germain, G.; et al. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol. Psychiatry 2011, 16, 238–239. [Google Scholar] [CrossRef]
- Szyszka, P.; Sharp, S.I.; Dedman, A.; Gurling, H.M.; McQuillin, A. A nonconservative amino acid change in the UPF3B gene in a patient with schizophrenia. Psychiatr. Genet. 2012, 22, 150–151. [Google Scholar] [CrossRef]
- Lynch, S.A.; Nguyen, L.S.; Ng, L.Y.; Waldron, M.; McDonald, D.; Gecz, J. Broadening the phenotype associated with mutations in UPF3B: Two further cases with renal dysplasia and variable developmental delay. Eur. J. Med. Genet. 2012, 55, 476–479. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; Tong, P.; Xun, G.; Su, W.; Xiong, Z.; Zhu, T.; Zheng, Y.; Luo, S.; Pan, Y.; et al. Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin. Genet. 2013, 83, 560–564. [Google Scholar] [CrossRef]
- Tejada, M.I.; Villate, O.; Ibarluzea, N.; De la Hoz, A.B.; Martínez-Bouzas, C.; Beristain, E.; Martínez, F.; Friez, M.J.; Sobrino, B.; Barros, F. Molecular and Clinical Characterization of a Novel Nonsense Variant in Exon 1 of the UPF3B Gene Found in a Large Spanish Basque Family (MRX82). Front. Genet. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Guo, H.; Duyzend, M.H.; Coe, B.P.; Baker, C.; Hoekzema, K.; Gerdts, J.; Turner, T.N.; Zody, M.C.; Beighley, J.S.; Murali, S.C.; et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet. Med. 2019, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Tejada, M.I.; Gecz, J.; Shaw, M.; Corbett, M.; Haan, E.; Thompson, E.; Friend, K.; et al. Targeted Next-Generation Sequencing Analysis of 1,000 Individuals with Intellectual Disability. Hum. Mutat. 2015, 36, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Medghalchi, S.M.; Frischmeyer, P.A.; Mendell, J.T.; Kelly, A.G.; Lawler, A.M.; Dietz, H.C. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet. 2001, 10, 99–105. [Google Scholar] [CrossRef]
- Alzahrani, F.; Kuwahara, H.; Long, Y.; Al-Owain, M.; Tohary, M.; AlSayed, M.; Mahnashi, M.; Fathi, L.; Alnemer, M.; Al-Hamed, M.H.; et al. Recessive, Deleterious Variants in SMG8 Expand the Role of Nonsense-Mediated Decay in Developmental Disorders in Humans. Am. J. Hum. Genet. 2020, 107, 1178–1185. [Google Scholar] [CrossRef]
- Abdel-Salam, G.M.H.; Duan, R.; Abdel-Hamid, M.S.; Sayed, I.S.M.; Jhangiani, S.N.; Khan, Z.; Du, H.; Gibbs, R.A.; Posey, J.E.; Marafi, D.; et al. Expanding the phenotypic and allelic spectrum of SMG8: Clinical observations reveal overlap with SMG9-associated disease trait. Am. J. Med. Genet. A 2022, 188, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Imamachi, N.; Pröschel, C.; Mitsutomi, S.; Nagao, R.; Akimitsu, N.; Maquat, L.E. Loss of the fragile X syndrome protein FMRP results in misregulation of nonsense-mediated mRNA decay. Nat. Cell Biol. 2021, 23, 40–48. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Bokhari, A.; Jonchere, V.; Lagrange, A.; Bertrand, R.; Svrcek, M.; Marisa, L.; Buhard, O.; Greene, M.; Demidova, A.; Jia, J.; et al. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis 2018, 7, 70. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef]
- Martin, L.; Grigoryan, A.; Wang, D.; Wang, J.; Breda, L.; Rivella, S.; Cardozo, T.; Gardner, L.B. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014, 74, 3104–3113. [Google Scholar] [CrossRef]
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature 2010, 465, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, X.; Yeganeh, P.N.; Lee, D.J.; Valle-Garcia, D.; Meza-Sosa, K.F.; Junqueira, C.; Su, J.; Luo, H.R.; Hide, W.; et al. Immunotherapy for breast cancer using EpCAM aptamer tumor-targeted gene knockdown. Proc. Natl. Acad. Sci. USA 2021, 118, e2022830118. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; Villanueva, H.; Bendandi, M.; Inoges, S.; López-Díaz de Cerio, A.; Pastor, F. 2-fluoro-RNA oligonucleotide CD40 targeted aptamers for the control of B lymphoma and bone-marrow aplasia. Biomaterials 2015, 67, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Cheruiyot, A.; Li, S.; Nonavinkere Srivatsan, S.; Ahmed, T.; Chen, Y.; Lemacon, D.S.; Li, Y.; Yang, Z.; Wadugu, B.A.; Warner, W.A.; et al. Nonsense-Mediated RNA Decay Is a Unique Vulnerability of Cancer Cells Harboring SF3B1 or U2AF1 Mutations. Cancer Res. 2021, 81, 4499–4513. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zavadil, J.; Martin, L.; Parisi, F.; Friedman, E.; Levy, D.; Harding, H.; Ron, D.; Gardner, L.B. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell. Biol. 2011, 31, 3670–3680. [Google Scholar] [CrossRef]
- Li, L.; Geng, Y.; Feng, R.; Zhu, Q.; Miao, B.; Cao, J.; Fei, S. The Human RNA Surveillance Factor UPF1 Modulates Gastric Cancer Progression by Targeting Long Non-Coding RNA MALAT1. Cell. Physiol. Biochem. 2017, 42, 2194–2206. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, S.; Qin, W.; Wang, Y.; Li, L.; Li, Q.; Yuan, X. A Novel RNA Binding Protein-Related Prognostic Signature for Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 580513. [Google Scholar] [CrossRef]
- Man, Z.; Chen, Y.; Gao, L.; Xei, G.; Li, Q.; Lu, Q.; Yan, J. A Prognostic Model Based on RNA Binding Protein Predicts Clinical Outcomes in Hepatocellular Carcinoma Patients. Front. Oncol. 2020, 10, 613102. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Wang, Q.; Zhang, M.; Wang, B.; Jiang, K.; Ye, Y.; Wang, S.; Shen, Z. Molecular Characterization and Clinical Relevance of RNA Binding Proteins in Colorectal Cancer. Front. Genet. 2020, 11, 580149. [Google Scholar] [CrossRef]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef]
- Popp, M.W.; Cho, H.; Maquat, L.E. Viral subversion of nonsense-mediated mRNA decay. RNA 2020, 26, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Contu, L.; Steiner, S.; Thiel, V.; Mühlemann, O. The Role of Stress Granules and the Nonsense-mediated mRNA Decay Pathway in Antiviral Defence. Chimia 2019, 73, 374–379. [Google Scholar] [CrossRef]
- Leon, K.; Ott, M. An ‘Arms Race’ between the Nonsense-mediated mRNA Decay Pathway and Viral Infections. Semin. Cell Dev. Biol. 2021, 111, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Wernet, M.F.; Klovstad, M.; Clandinin, T.R. Generation of infectious virus particles from inducible transgenic genomes. Curr. Biol. 2014, 24, R107–R108. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zünd, D.; McInerney, G.; Merits, A.; Mühlemann, O.; Azzalin, C.; Helenius, A. The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe 2014, 16, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Lokugamage, K.G.; Nakagawa, K.; Narayanan, K.; Makino, S. Interplay between coronavirus, a cytoplasmic RNA virus, and nonsense-mediated mRNA decay pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10157–E10166. [Google Scholar] [CrossRef]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J.; et al. The Cellular NMD Pathway Restricts Zika Virus Infection and Is Targeted by the Viral Capsid Protein. mBio 2018, 9, e02126-18. [Google Scholar] [CrossRef] [PubMed]
- Ajamian, L.; Abrahamyan, L.; Milev, M.; Ivanov, P.V.; Kulozik, A.E.; Gehring, N.H.; Mouland, A.J. Unexpected roles for UPF1 in HIV-1 RNA metabolism and translation. RNA 2008, 14, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Ajamian, L.; Abel, K.; Rao, S.; Vyboh, K.; García-de-Gracia, F.; Soto-Rifo, R.; Kulozik, A.E.; Gehring, N.H.; Mouland, A.J. HIV-1 Recruits UPF1 but Excludes UPF2 to Promote Nucleocytoplasmic Export of the Genomic RNA. Biomolecules 2015, 5, 2808–2839. [Google Scholar] [CrossRef]
- Rao, S.; Amorim, R.; Niu, M.; Temzi, A.; Mouland, A.J. The RNA surveillance proteins UPF1, UPF2 and SMG6 affect HIV-1 reactivation at a post-transcriptional level. Retrovirology 2018, 15, 42. [Google Scholar] [CrossRef]
- Rao, S.; Amorim, R.; Niu, M.; Breton, Y.; Tremblay, M.J.; Mouland, A.J. Host mRNA decay proteins influence HIV-1 replication and viral gene expression in primary monocyte-derived macrophages. Retrovirology 2019, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Serquiña, A.K.; Das, S.R.; Popova, E.; Ojelabi, O.A.; Roy, C.K.; Göttlinger, H.G. UPF1 is crucial for the infectivity of human immunodeficiency virus type 1 progeny virions. J. Virol. 2013, 87, 8853–8861. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ye, X.; Shehata, M.; Dunker, W.; Xie, Z.; Karijolich, J. The RNA quality control pathway nonsense-mediated mRNA decay targets cellular and viral RNAs to restrict KSHV. Nat. Commun. 2020, 11, 3345. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Tsao, E.H.; Webb, B.L.; Ye, H.; Dalton-Griffin, L.; Tsantoulas, C.; Gale, C.V.; Du, M.Q.; Whitehouse, A.; Kellam, P. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 2007, 81, 13578–13586. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Feng, J.; Harada, J.N.; Chanda, S.K.; Kenney, S.C.; Sun, R. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi’s sarcoma-associated herpesvirus. FEBS Lett. 2007, 581, 3485–3488. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Franceschini, A.; Horvath, P.; Tetard, M.; Mancini, R.; von Mering, C.; Helenius, A.; Lozach, P.Y. Genome-wide small interfering RNA screens reveal VAMP3 as a novel host factor required for Uukuniemi virus late penetration. J. Virol. 2014, 88, 8565–8578. [Google Scholar] [CrossRef]
- Tang, X.; Zhu, Y.; Baker, S.L.; Bowler, M.W.; Chen, B.J.; Chen, C.; Hogg, J.R.; Goff, S.P.; Song, H. Structural basis of suppression of host translation termination by Moloney Murine Leukemia Virus. Nat. Commun. 2016, 7, 12070. [Google Scholar] [CrossRef]
- Baker, S.L.; Hogg, J.R. A system for coordinated analysis of translational readthrough and nonsense-mediated mRNA decay. PLoS ONE 2017, 12, e0173980. [Google Scholar] [CrossRef]
- Fischer, U.; Huber, J.; Boelens, W.C.; Mattaj, I.W.; Lührmann, R. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 1995, 82, 475–483. [Google Scholar] [CrossRef]
- Bohne, J.; Wodrich, H.; Kräusslich, H.G. Splicing of human immunodeficiency virus RNA is position-dependent suggesting sequential removal of introns from the 5′ end. Nucleic Acids Res. 2005, 33, 825–837. [Google Scholar] [CrossRef]
- Weil, J.E.; Beemon, K.L. A 3′ UTR sequence stabilizes termination codons in the unspliced RNA of Rous sarcoma virus. RNA 2006, 12, 102–110. [Google Scholar] [CrossRef]
- Beemon, K.L. Retroviral RNA Processing. Viruses 2022, 14, 1113. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Felber, B.K.; Benko, D.M.; Fenyö, E.M.; Pavlakis, G.N. Cloning and functional analysis of multiply spliced mRNA species of human immunodeficiency virus type 1. J. Virol. 1990, 64, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Purcell, D.F.; Martin, M.A. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 1993, 67, 6365–6378. [Google Scholar] [CrossRef]
- Daly, T.J.; Cook, K.S.; Gray, G.S.; Maione, T.E.; Rusche, J.R. Specific binding of HIV-1 recombinant Rev protein to the Rev-responsive element in vitro. Nature 1989, 342, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Weil, J.E.; Hadjithomas, M.; Beemon, K.L. Structural characterization of the Rous sarcoma virus RNA stability element. J. Virol. 2009, 83, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Withers, J.B.; Beemon, K.L. Structural features in the Rous sarcoma virus RNA stability element are necessary for sensing the correct termination codon. Retrovirology 2010, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Munday, D.; Bickerton, E.; Britton, P.; Rodgers, M.A.; Whitehouse, A.; Zhou, E.M.; Hiscox, J.A. The cellular interactome of the coronavirus infectious bronchitis virus nucleocapsid protein and functional implications for virus biology. J. Virol. 2013, 87, 9486–9500. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Contu, L.; Balistreri, G.; Domanski, M.; Uldry, A.C.; Mühlemann, O. Characterisation of the Semliki Forest Virus-host cell interactome reveals the viral capsid protein as an inhibitor of nonsense-mediated mRNA decay. PLoS Pathog. 2021, 17, e1009603. [Google Scholar] [CrossRef]
- Ramage, H.R.; Kumar, G.R.; Verschueren, E.; Johnson, J.R.; Von Dollen, J.; Johnson, T.; Newton, B.; Shah, P.; Horner, J.; Krogan, N.J.; et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 2015, 57, 329–340. [Google Scholar] [CrossRef]
- Li, M.; Johnson, J.R.; Truong, B.; Kim, G.; Weinbren, N.; Dittmar, M.; Shah, P.S.; Von Dollen, J.; Newton, B.W.; Jang, G.M.; et al. Identification of antiviral roles for the exon-junction complex and nonsense-mediated decay in flaviviral infection. Nat. Microbiol. 2019, 4, 985–995. [Google Scholar] [CrossRef]
- Bono, F.; Ebert, J.; Unterholzner, L.; Güttler, T.; Izaurralde, E.; Conti, E. Molecular insights into the interaction of PYM with the Mago-Y14 core of the exon junction complex. EMBO Rep. 2004, 5, 304–310. [Google Scholar] [CrossRef]
- Diem, M.D.; Chan, C.C.; Younis, I.; Dreyfuss, G. PYM binds the cytoplasmic exon-junction complex and ribosomes to enhance translation of spliced mRNAs. Nat. Struct. Mol. Biol. 2007, 14, 1173–1179. [Google Scholar] [CrossRef]
- Leon, K.E.; Khalid, M.M.; Flynn, R.A.; Fontaine, K.A.; Nguyen, T.T.; Kumar, G.R.; Simoneau, C.R.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; et al. Nuclear accumulation of host transcripts during Zika Virus Infection. PLoS Pathog. 2023, 19, e1011070. [Google Scholar] [CrossRef]
- Nakano, K.; Ando, T.; Yamagishi, M.; Yokoyama, K.; Ishida, T.; Ohsugi, T.; Tanaka, Y.; Brighty, D.W.; Watanabe, T. Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: Implications for retroviral replication. Microbes Infect. 2013, 15, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Mocquet, V.; Neusiedler, J.; Rende, F.; Cluet, D.; Robin, J.P.; Terme, J.M.; Duc Dodon, M.; Wittmann, J.; Morris, C.; Le Hir, H.; et al. The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J. Virol. 2012, 86, 7530–7543. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, F.; Robin, J.P.; Kanaan, J.; Borowiak, M.; Croquette, V.; Le Hir, H.; Jalinot, P.; Mocquet, V. HTLV-1 Tax plugs and freezes UPF1 helicase leading to nonsense-mediated mRNA decay inhibition. Nat. Commun. 2018, 9, 431. [Google Scholar] [CrossRef]
- Ferraris, P.; Cochet, M.; Hamel, R.; Gladwyn-Ng, I.; Alfano, C.; Diop, F.; Garcia, D.; Talignani, L.; Montero-Menei, C.N.; Nougairède, A.; et al. Zika virus differentially infects human neural progenitor cells according to their state of differentiation and dysregulates neurogenesis through the Notch pathway. Emerg. Microbes Infect. 2019, 8, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Watanabe, T. Tuning Rex rules HTLV-1 pathogenesis. Front. Immunol. 2022, 13, 959962. [Google Scholar] [CrossRef]
- Morris, C.; Wittmann, J.; Jäck, H.M.; Jalinot, P. Human INT6/eIF3e is required for nonsense-mediated mRNA decay. EMBO Rep. 2007, 8, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Lehner, B.; Lindeboom, R.G.H. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2021, 37, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Yang, S.; Sun, Y.; Guo, J.U. Regulation of nonsense-mediated mRNA decay in neural development and disease. J. Mol. Cell Biol. 2021, 13, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Stupack, D.G.; Wilkinson, M.F. Nonsense-mediated RNA decay: An emerging modulator of malignancy. Nat. Rev. Cancer 2022, 22, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wei, Y.; Wang, H.; Wang, F.; Ju, Z.; Li, T. Nonsense-mediated mRNA decay: A ‘nonsense’ pathway makes sense in stem cell biology. Nucleic Acids Res. 2018, 46, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Lingner, J. The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr. Biol. 2006, 16, 433–439. [Google Scholar] [CrossRef]
- Choe, J.; Ahn, S.H.; Kim, Y.K. The mRNP remodeling mediated by UPF1 promotes rapid degradation of replication-dependent histone mRNA. Nucleic Acids Res. 2014, 42, 9334–9349. [Google Scholar] [CrossRef]
- Ngo, G.H.P.; Grimstead, J.W.; Baird, D.M. UPF1 promotes the formation of R loops to stimulate DNA double-strand break repair. Nat. Commun. 2021, 12, 3849. [Google Scholar] [CrossRef]


| Protein | Domain | Gene Variation | Isoform 1 | Isoform 2 | Related Disease | ClinVar Accession [90] | Ref. |
|---|---|---|---|---|---|---|---|
| UPF1 | Helicase | c.2381C > T | T805M | T794M | Autism, developmental delay, joint hypermobility, hypotonia | VCV000996716.1 | - |
| c.2489A > G | Q841R | Q830R | Intellectual disability | VCV000930204.1 | - | ||
| UPF2 | N-terminus | c.91G > T | V31L | Autism spectrum disorder | VCV000996716.1 | [132] | |
| UPF3B | N-terminus | c.52A > C | T18P | T18P | Syndromic X-linked intellectual disability 14 | VCV000287042.5 | - |
| c.53C > T | T18I | T18I | Syndromic X-linked intellectual disability 14 | VCV000698418.4 | - | ||
| RRM-L | c.277A > G | M93V | M93V | Syndromic X-linked intellectual disability 14 | VCV001012886.1 | - | |
| NOPS-L | c.470A > G | D157G | D157G | Syndromic X-linked intellectual disability 14 | VCV001485374.3 | - | |
| c.478T > G | Y160D | Y160D | Syndromic X-linked intellectual disability 14 | VCV000011401.3 | [125] | ||
| Interdomain | c.520A > G | M174V | M174V | Syndromic X-linked intellectual disability 14 | VCV001718760.1 | - | |
| CCL-1 | c.667A > G | I223V | I223V | Syndromic X-linked intellectual disability 14 | VCV000985848.4 | [133] | |
| c.672A > C | E224D | E224D | Intellectual disability | VCV000981400.1 | - | ||
| c.758T > C | I253T | I253T | Cataract, Microcephaly, Severe global developmental delay | VCV000284394.21 | - | ||
| c.763A > G | R255G | R255G | Syndromic X-linked intellectual disability 14 | VCV001029871.1 | - | ||
| c.764G > A | R255K | R255K | Syndromic X-linked intellectual disability 14 | VCV000536851.9 | - | ||
| Interdomain | c.962T > C | L321S | L308S | Syndromic X-linked intellectual disability 14 | VCV000212548.9 | - | |
| CCL-2 | c.1049G > A | R350Q | R337Q | Syndromic X-linked intellectual disability 14 | VCV001469888.3 | - | |
| c.1072C > T | R358C | R345C | Syndromic X-linked intellectual disability 14 | VCV000700934.5 | - | ||
| c.1101G > C | K367N | K354N | Syndromic X-linked intellectual disability 14 | VCV000804079.4 | - | ||
| c.1102C > T | R368W | R355W | Syndromic X-linked intellectual disability 14 | VCV001041099.5 | - | ||
| c.1103G > A | R368Q | R355Q | Borderline intellectual disability with autism | - | [126] | ||
| c.1117C > T | R373C | R360C | Syndromic X-linked intellectual disability 14 | VCV001560636.4 | - | ||
| c.1118G > A | R373H | R360H | Syndromic X-linked intellectual disability 14 | VCV000547850.2 | - | ||
| c.1121G > A | R374H | R361H | Syndromic X-linked intellectual disability 14 | VCV000212546.17 | - | ||
| c.1136G > A | R379H | R366H | Syndromic X-linked intellectual disability 14 | VCV001597732.4 | [126] | ||
| c.1201C > T | R401W | R388W | Syndromic X-linked intellectual disability 14 | VCV001601042.3 | - | ||
| c.1202G > A | R401Q | R388Q | Syndromic X-linked intellectual disability 14 | VCV000581972.4 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Mailliot, J.; Schaffitzel, C. Nonsense-Mediated mRNA Decay Factor Functions in Human Health and Disease. Biomedicines 2023, 11, 722. https://doi.org/10.3390/biomedicines11030722
Sun L, Mailliot J, Schaffitzel C. Nonsense-Mediated mRNA Decay Factor Functions in Human Health and Disease. Biomedicines. 2023; 11(3):722. https://doi.org/10.3390/biomedicines11030722
Chicago/Turabian StyleSun, Lingling, Justine Mailliot, and Christiane Schaffitzel. 2023. "Nonsense-Mediated mRNA Decay Factor Functions in Human Health and Disease" Biomedicines 11, no. 3: 722. https://doi.org/10.3390/biomedicines11030722