
Cheek-Pro-Heart: What Can the Buccal Mucosa Do for Arrhythmogenic Cardiomyopathy?

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Genetics of ACM
3. Proteins ‘Shifting’ in the Heart
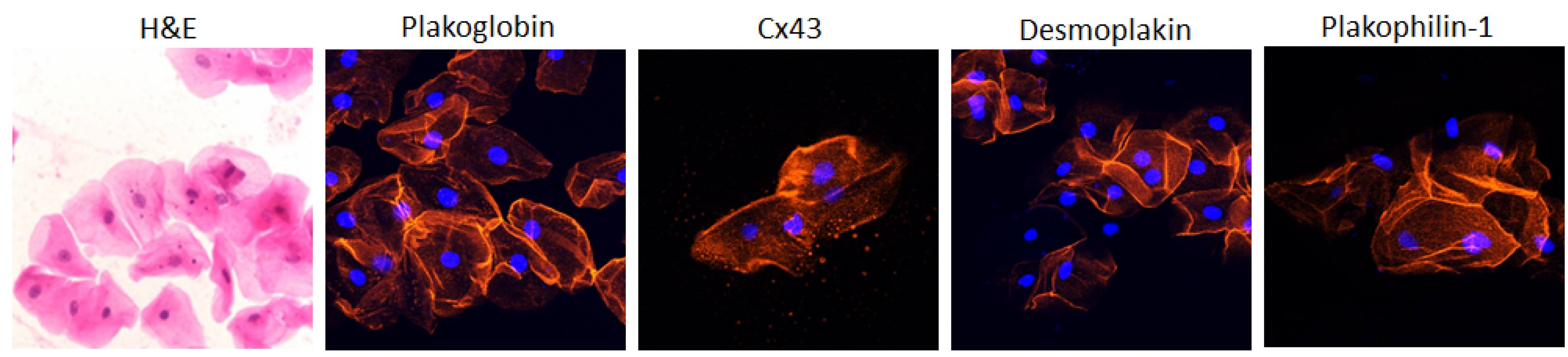
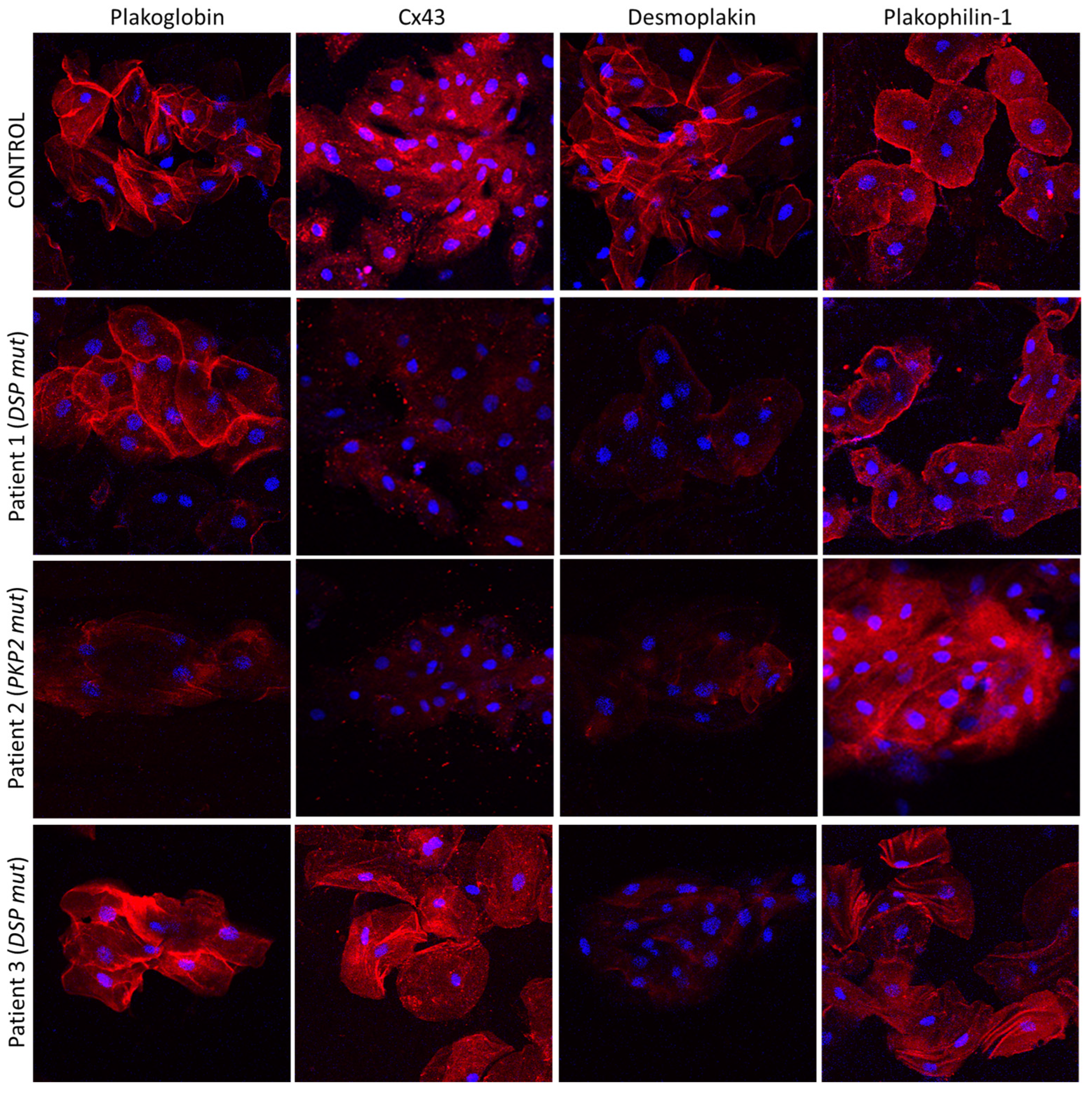
4. The Buccal Mucosa in Adult ACM
5. The Buccal Mucosa in Pediatric ACM
6. Cheeks beyond Diagnosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corrado, D.; Basso, C.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy: Diagnosis, prognosis, and treatment. Heart 2000, 83, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Morgan, R.D.; Chambers, J.C.; McKenna, W.J. Arrhythmogenic Cardiomyopathy: Etiology, Diagnosis, and Treatment. Annu. Rev. Med. 2010, 61, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.; Mejia-Lopez, E.; Manrique, C.; Lucariello, R. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC/D): A Systematic Literature Review. Clin. Med. Insights Cardiol. 2013, 7, 97–114. [Google Scholar] [CrossRef]
- Asimaki, A.; Saffitz, J.E. Remodeling of cell-cell junctions in arrhythmogenic cardiomyopathy. Cell Commun. Adhes. 2014, 21, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Eur. Heart J. 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Saffitz, J.E.; Asimaki, A.; Huang, H. Arrhythmogenic right ventricular cardiomyopathy: New insights into mechanisms of disease. Cardiovasc. Pathol. 2010, 19, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac abnormalities in familial palmoplantar keratosis. Heart 1986, 56, 321–326. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin–intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A novel dominant mutation in plakoglobin causes ar-rhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, C.; Li, Y.; Yu, R. Whole-Exome Sequencing Identified a De Novo Mutation of Junction Plakoglobin (p.R577C) in a Chinese Patient with Arrhythmogenic Right Ventricular Cardiomyopathy. BioMed Res. Int. 2019, 2019, 9103860. [Google Scholar] [CrossRef] [PubMed]
- Castelletti, S.; Vischer, A.S.; Syrris, P.; Crotti, L.; Spazzolini, C.; Ghidoni, A.; Parati, G.; Jenkins, S.; Kotta, M.-C.; McKenna, W.J.; et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int. J. Cardiol. 2017, 249, 268–273. [Google Scholar] [CrossRef]
- Bariani, R.; Cason, M.; Rigato, I.; Cipriani, A.; Celeghin, R.; De Gaspari, M.; Marinas, M.B.; Mattesi, G.; Pergola, V.; Rizzo, S.; et al. Clinical profile and long-term follow-up of a cohort of patients with desmoplakin cardio-myopathy. Heart Rhythm. 2022, 19, 1315–1324. [Google Scholar] [CrossRef]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Green, K.J.; Gaudry, C.A. Are desmosomes more than tethers for intermediate filaments? Nat. Rev. Mol. Cell Biol. 2000, 1, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhu, W.; Wang, C.; Huang, L.; Zhou, Q.; Hu, J.; Cheng, X.; Hong, K. Genotype-phenotype relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 41387. [Google Scholar] [CrossRef]
- Vimalanathan, A.K.; Ehler, E.; Gehmlich, K. Genetics of and pathogenic mechanisms in arrhythmogenic right ventricular cardi-omyopathy. Biophys. Rev. 2018, 10, 973–982. [Google Scholar] [CrossRef]
- Christensen, A.H.; Benn, M.; Bundgaard, H.; Tybjaerg-Hansen, A.; Haunso, S.; Svendsen, J.H. Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2010, 47, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Delmar, M.; McKenna, W.J. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef]
- Ghidoni, A.; Elliott, P.M.; Syrris, P.; Calkins, H.; James, C.A.; Judge, D.P.; Murray, B.; Barc, J.; Probst, V.; Schott, J.J.; et al. Cadherin 2-Related Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2021, 14, e003097. [Google Scholar] [CrossRef] [PubMed]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef] [PubMed]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicatesCDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef] [PubMed]
- van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.A.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, A.; Beffagna, G.; Bauce, B.; De Bortoli, M.; Mura, I.E.L.; Calore, M.; Dazzo, E.; Basso, C.; Nava, A.; Thiene, G.; et al. Desmin Mutations and Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2013, 111, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Brun, F.; Gigli, M.; Graw, S.L.; Judge, D.P.; Merlo, M.; Murray, B.; Calkins, H.; Sinagra, G.; Taylor, M.R.; Mestroni, L.; et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2020, 57, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic Variation in Titin in Arrhythmogenic Right Ventricular Cardiomyopathy–Overlap Syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, F.; Zorio, E.; Jimenez-Jaimez, J.; Salguero-Bodes, R.; Zwart, R.; Gonzalez-Lopez, E.; Molina, P.; Bermúdez-Jiménez, F.; Delgado, J.; Braza-Boïls, A.; et al. Clinical characteristics and determinants of the phenotype in TMEM43 arrhythmogenic right ventricular cardiomyopathy type 5. Heart Rhythm 2020, 17, 945–954. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.-C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- te Riele, A.S.J.M.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and Digenic Heterozygosity Predicts Lifetime Arrhythmic Outcome and Sudden Cardiac Death in Desmosomal Gene–Related Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, S.R.; Gard, J.J.; Protonotarios, N.; Tsatsopoulou, A.; Spiliopoulou, C.; Anastasakis, A.; Squarcioni, C.P.; McKenna, W.J.; Thiene, G.; Basso, C.; et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventriculazr cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 2004, 1, 3–11. [Google Scholar] [CrossRef]
- Kaplan, S.R.; Gard, J.J.; Carvajal-Huerta, L.; Ruiz-Cabezas, J.C.; Thiene, G.; Saffitz, J.E. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc. Pathol. 2004, 13, 26–32. [Google Scholar] [CrossRef]
- Asimaki, A.; Tandri, H.; Huang, H.; Halushka, M.K.; Gautam, S.; Basso, C.; Thiene, G.; Tsatsopoulou, A.; Protonotarios, N.; McKenna, W.J.; et al. A New Diagnostic Test for Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2009, 360, 1075–1084. [Google Scholar] [CrossRef]
- Saffitz, J.E. Dependence of electrical coupling on mechanical coupling in cardiac myocytes. In Advances in Cardiovascular Medicine; Thiene, G., Dessina, A.C., Eds.; Universitá degli Studi di Padova: Padua, Italy, 2005; pp. 15–28. [Google Scholar]
- Peters, N.S.; Green, C.R.; Poole-Wilson, P.A.; Severs, N.J. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation 1993, 88, 864–875. [Google Scholar] [CrossRef]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.-H. Arrhythmogenic Ion-Channel Remodeling in the Heart: Heart Failure, Myocardial Infarction, and Atrial Fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Polychronopoulou, S.; Tsatsopoulou, A.; Papadhimitriou, S.I.; Panagiotou, J.P.; Anastasakis, A.; Paterakis, G.; Anagnostou, D.; Protonotarious, N.; Haidas, S.A. Myelodysplasia and Naxos disease: A novel pathogenetic association? Leukemia 2002, 16, 2335–2337. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.; Andersen, C.; Tybjaerg-Hansen, A.; Haunso, S.; Svendsen, J. Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2011, 80, 256–264. [Google Scholar] [CrossRef]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell-cell adhesion structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2020, 307, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kawano, H.; Kusumoto, S.; Fukae, S.; Koga, S.; Ikeda, S.; Koide, Y.; Abe, K.; Hayashi, T.; Maemura, K. Relationships between clinical characteristics and decreased plakoglobin and con-nexin 43 expressions in myocardial biopsies from patients with arrhythmogenic right ventricular cardiomyopathy. Int. Heart J. 2015, 56, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Ermakov, S.; Ursell, P.C.; Johnson, C.J.; Meadows, A.; Zhao, S.; Marcus, G.M.; Scheinman, M. Plakoglobin immunolocalization as a diagnostic test for arrhythmogenic right ventricular cardiomyopathy. Pacing Clin. Electrophysiol. 2014, 37, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, J.; Andersen, C.B.; Ottesen, G.L. Plakoglobin: A diagnostic marker of arrhythmogenic right ventricular cardiomyopathy in forensic pathology? Forensic Sci. Med. Pathol. 2015, 11, 47–52. [Google Scholar] [CrossRef]
- Fidler, L.M.; Wilson, G.J.; Liu, F.; Cui, X.; Scherer, S.W.; Taylor, G.P.; Hamilton, R.M. Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J. Cell. Mol. Med. 2009, 13, 4219–4228. [Google Scholar] [CrossRef] [PubMed]
- Noorman, M.; Hakin, S.; Kessler, E.; Groeneweg, J.A.; Cox, M.G.P.J.; Asimaki, A.; Asimaki, H.V.M.; van Stuijvenberg, L.; Chkourko, L.; van der Heyden, M.A.G.; et al. Remodeling of the cardiac sodium channel, connexin43 and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2013, 10, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Musa, H.; Coombs, W.; Guerrero-Serna, G.; Patiño, G.A.; Taffet, S.M.; Isom, L.L.; Delmar, M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velovity in cultured cardiac myocytes. Circ. Res. 2009, 105, 523–526. [Google Scholar] [CrossRef]
- Cerrone, M.; Noorman, M.; Lin, X.; Chkourko, H.; Liang, F.; van der Nagel, R.; Hund, T.; Birchmeier, W.; Mohler, P.; van Veen, T.A.; et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc. Res. 2012, 95, 460–468. [Google Scholar] [CrossRef]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74. [Google Scholar] [CrossRef]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense Mutations in Plakophilin-2 Cause Sodium Current Deficit and Associate with a Brugada Syndrome Phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Milstein, M.L.; Musa, H.; Balbuena, D.P.; Anumonwo, J.M.B.; Auerbach, D.S.; Furspan, P.B.; Hou, L.; Hu, B.; Schumacher, S.M.; Vaidyanathan, R.; et al. Dynamic reciprocity of sedum and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc. Natl. Acad. Sci. USA 2012, 109, E2134–E2143. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; Macrae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. J. Clin. Investig. 2016, 1, e85923. [Google Scholar] [CrossRef]
- Bueno-Beti, C.; Asimaki, A. Histopathological Features and Protein Markers of Arrhythmogenic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 746321. [Google Scholar] [CrossRef] [PubMed]
- Vahidnezhad, H.; Youssefian, L.; Faghankhani, M.; Mozafari, N.; Saeidian, A.H.; Niaziorimi, F.; Abdollahimajd, F.; Sotoudeh, S.; Rajabi, F.; Mirsafaei, L.; et al. Arrhythmogenic right ventricular cardiomyopathy in patients with bi-allelic JUP-associated skin fragility. Sci. Rep. 2020, 10, 21622. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Ward, D.; Guereta, L.G.; Saffitz, J.E.; McKenna, W.J. Unique epidermolytic bullous dermatosis with associated lethal cardiomyopathy related to novel desmoplakin mutations. J. Cutan. Pathol. 2009, 36, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Leifert, W.R.; François, M.; Thomas, P.; Luther, E.; Holden, E.; Fenech, M. Automation of the buccal micronucleus cytome assay using laser scanning cytometry. Methods Cell Biol. 2011, 102, 321–339. [Google Scholar]
- Asimaki, A.; Protonotarios, A.; James, C.A.; Chelko, S.P.; Tichnell, C.; Murray, B.; Tsatsopoulou, A.; Anastasakis, A.; Riele, A.T.; Kléber, A.G.; et al. Characterizing the Molecular Pathology of Arrhythmogenic Cardiomyopathy in Patient Buccal Mucosa Cells. Circ. Arrhythmia Electrophysiol. 2016, 9, e003688. [Google Scholar] [CrossRef] [PubMed]
- Driessen, H.E.; van der Voorn, S.M.; Bourfiss, M.; van Lint, F.H.M.; Mirzad, F.; El Onsri, L.; Vos, M.A.; van Veen, T.A.B. Buccal mucosa cells as a potential diagnostic tool to study onset and progression of arrhythmogenic cardiomyopathy. Int. J. Mol. Sci. 2021, 23, 57. [Google Scholar] [CrossRef] [PubMed]
- Bueno-Beti, C.; Field, E.; Tsatsopoulou, A.; Perry, G.; Sheppard, M.N.; Behr, E.R.; Saffitz, J.E.; Kaski, J.P.; Asimaki, A. Analysis of buccal mucosa as a prognostic tool in children with arrhythmogenic cardiomyopathy. Prog. Pediatr. Cardiol. 2022, 64, 101458. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueno-Beti, C.; Asimaki, A. Cheek-Pro-Heart: What Can the Buccal Mucosa Do for Arrhythmogenic Cardiomyopathy? Biomedicines 2023, 11, 1207. https://doi.org/10.3390/biomedicines11041207
Bueno-Beti C, Asimaki A. Cheek-Pro-Heart: What Can the Buccal Mucosa Do for Arrhythmogenic Cardiomyopathy? Biomedicines. 2023; 11(4):1207. https://doi.org/10.3390/biomedicines11041207
Chicago/Turabian StyleBueno-Beti, Carlos, and Angeliki Asimaki. 2023. "Cheek-Pro-Heart: What Can the Buccal Mucosa Do for Arrhythmogenic Cardiomyopathy?" Biomedicines 11, no. 4: 1207. https://doi.org/10.3390/biomedicines11041207
APA StyleBueno-Beti, C., & Asimaki, A. (2023). Cheek-Pro-Heart: What Can the Buccal Mucosa Do for Arrhythmogenic Cardiomyopathy? Biomedicines, 11(4), 1207. https://doi.org/10.3390/biomedicines11041207