
Multifaceted Effects of Kinase Inhibitors on Pancreatic Cancer Cells Reveals Pivotal Entities with Therapeutic Implications


Abstract
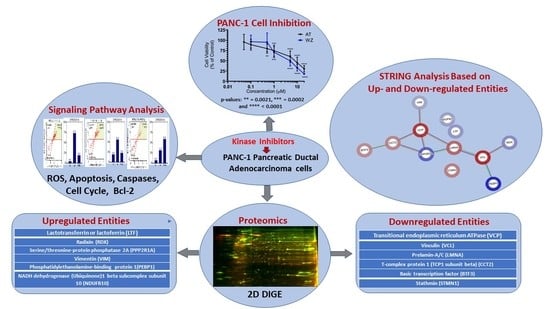
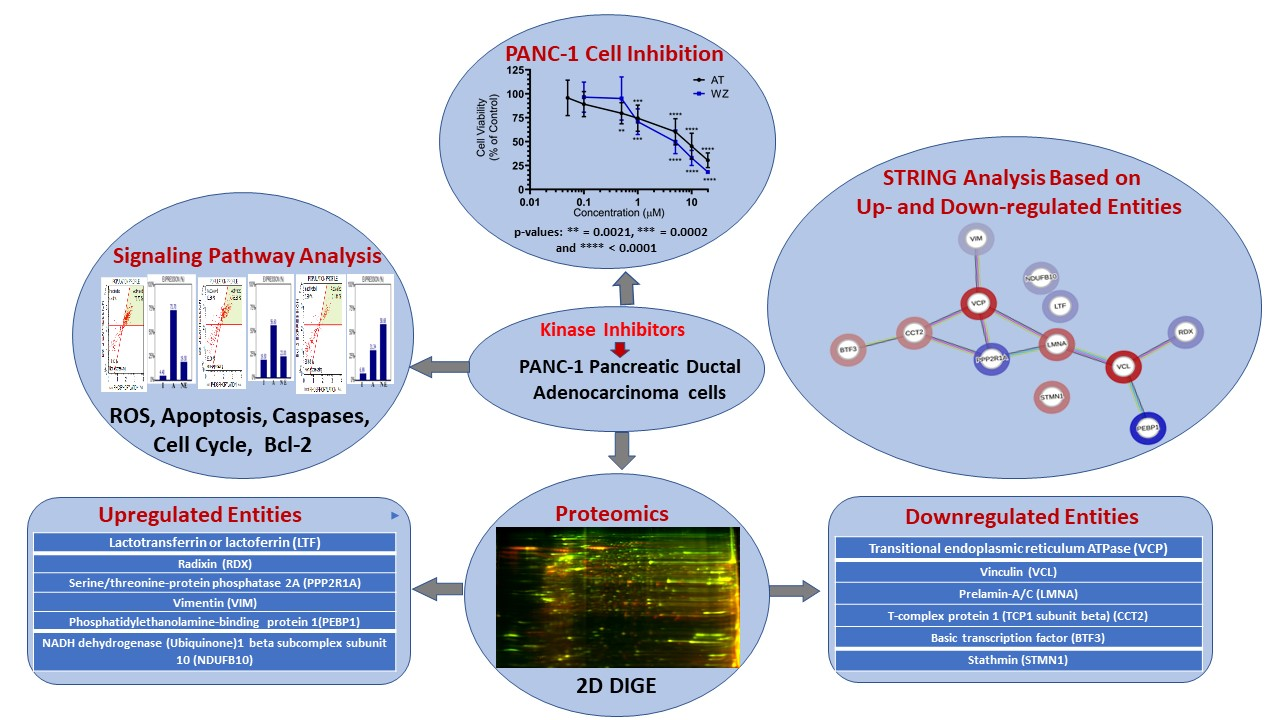
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Materials
2.2. Library Screening and Toxicity Assay
2.3. Flow Cytometry Assays
2.4. Proteomic Analysis
2.5. Statistical Analysis
3. Results
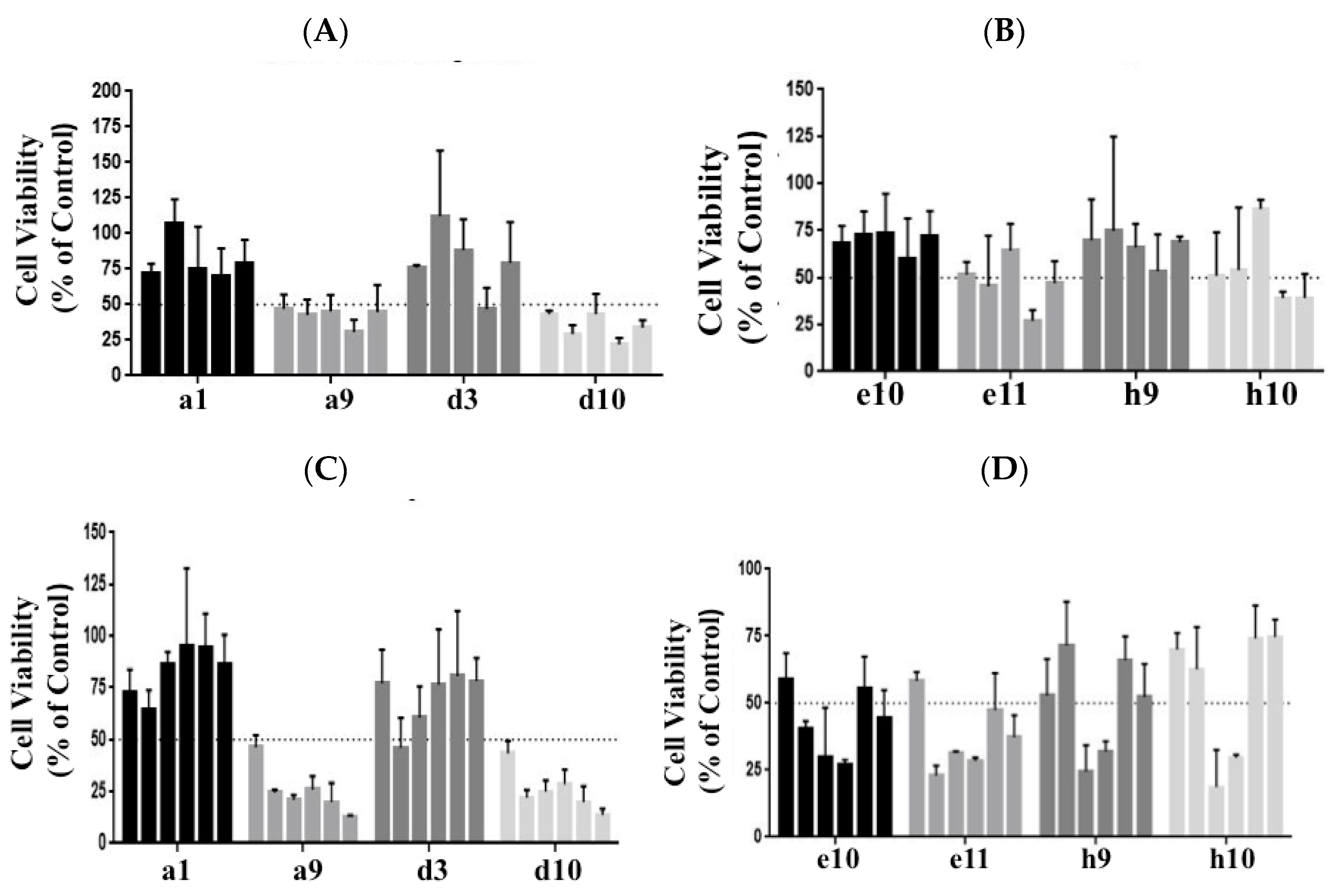
3.1. Identification of Molecules Impacting the Viability of PANC-1 and BxPC-3 Cells by Screening a Library of Kinase Inhibitors
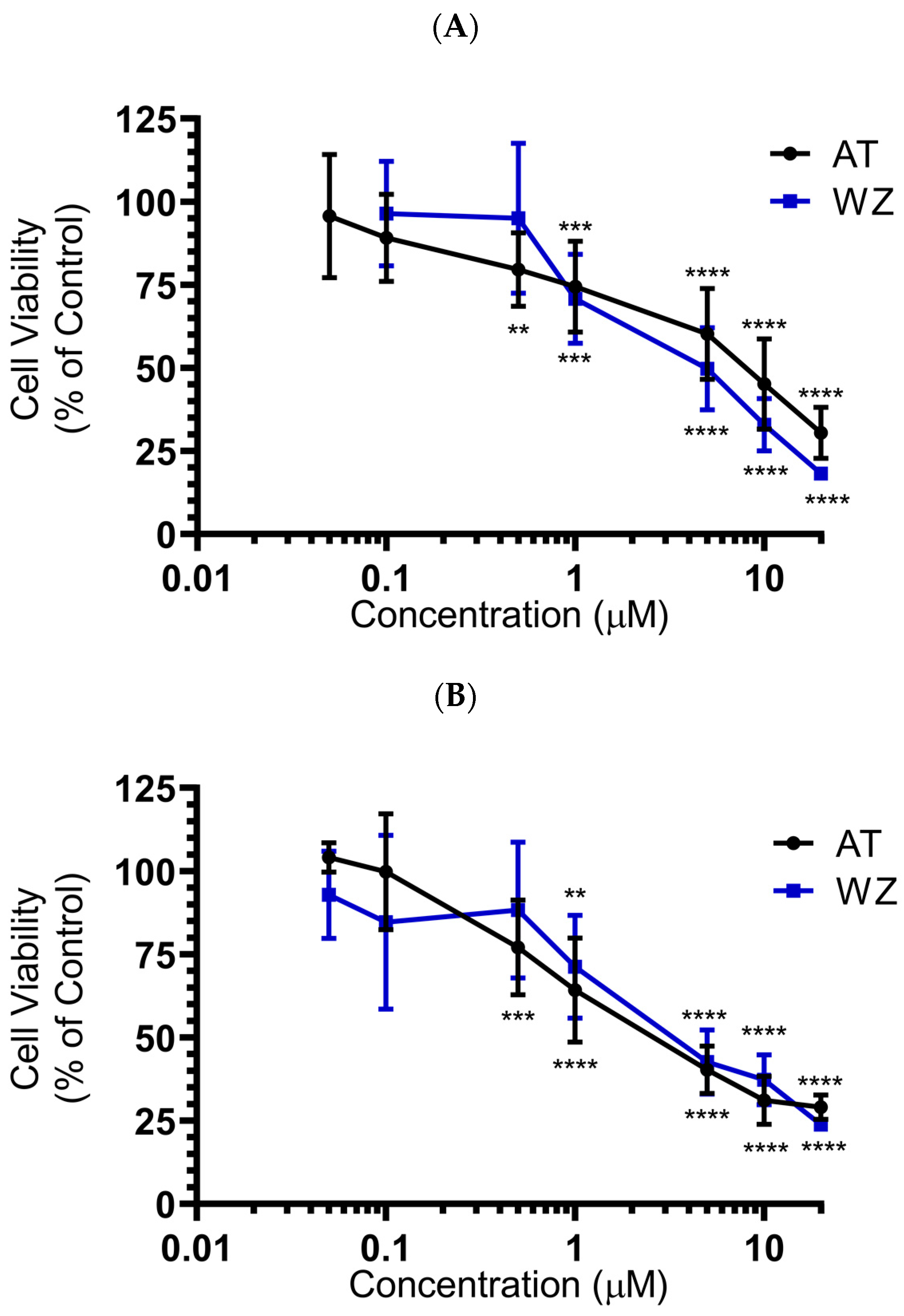
3.2. Cellular Level Analysis Revealed Potent Dose-Dependent Inhibition of Viability of PANC-1 and BxPC-3 Cells by the Kinase Inhibitors AT and WZ
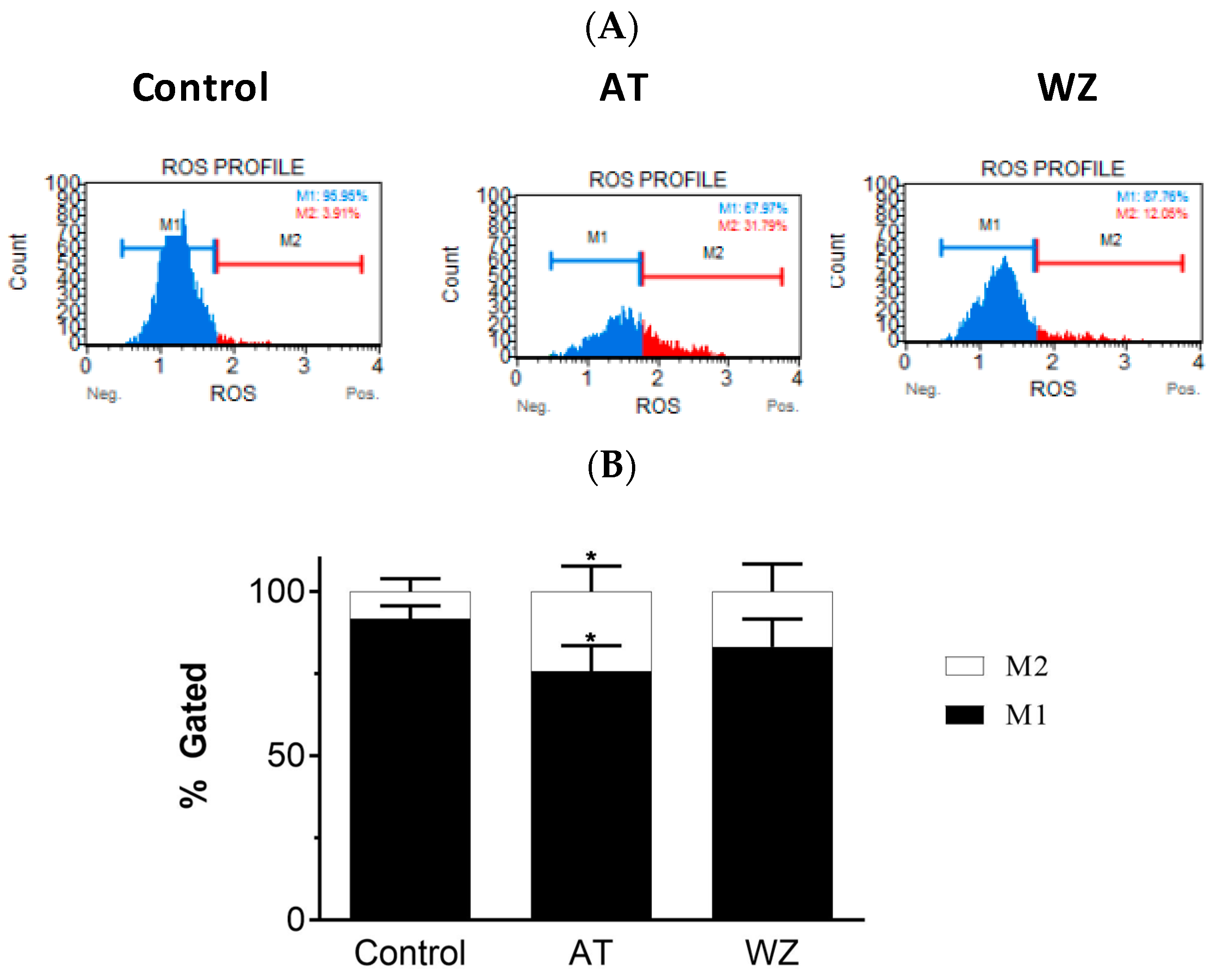
3.3. Effect of Kinase Inhibitors AT and WZ on ROS Levels in PANC-1 Cells
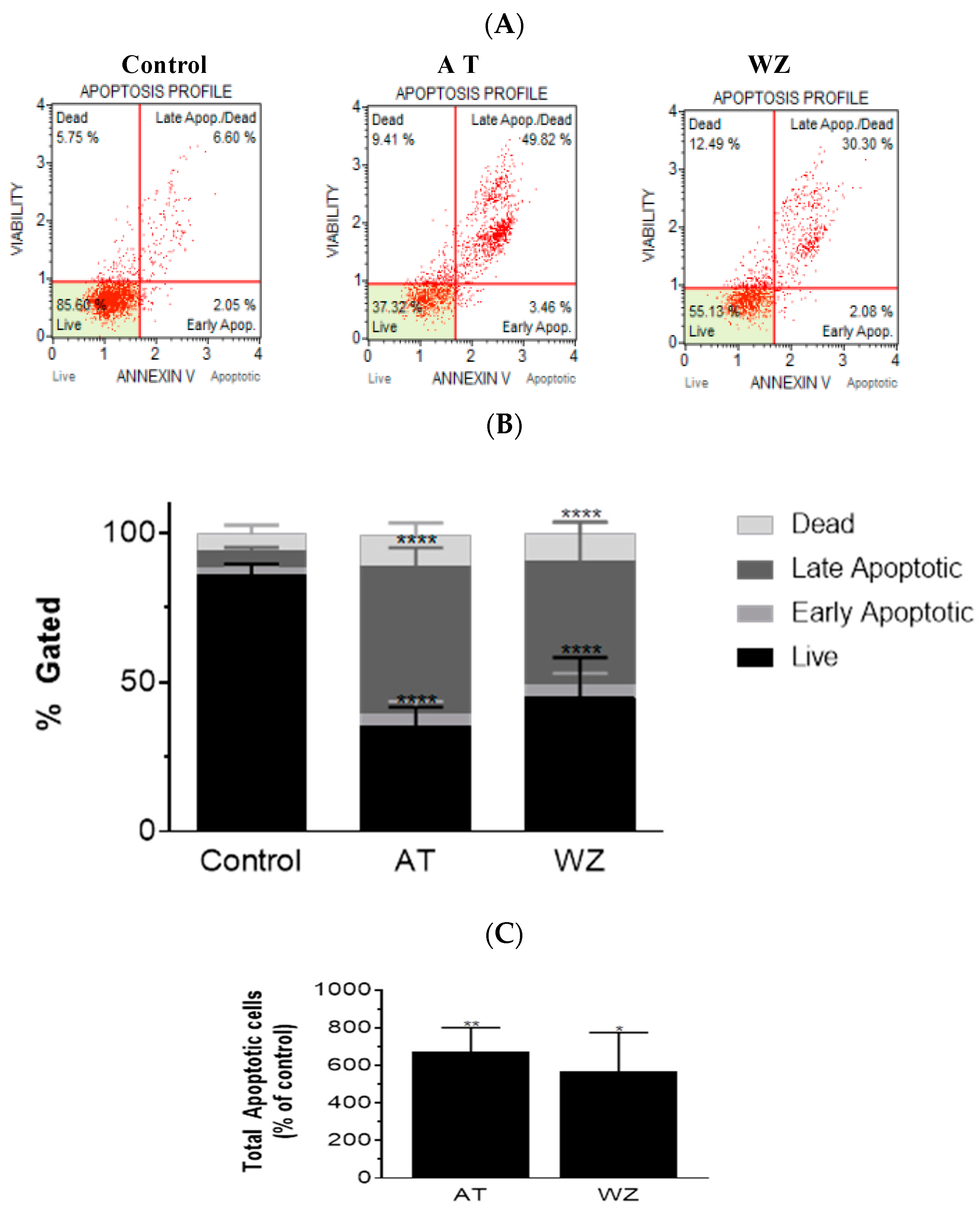
3.4. Kinase Inhibitors AT and WZ Invoked the Programmed Cell Death Pathway in PANC-1 Cells
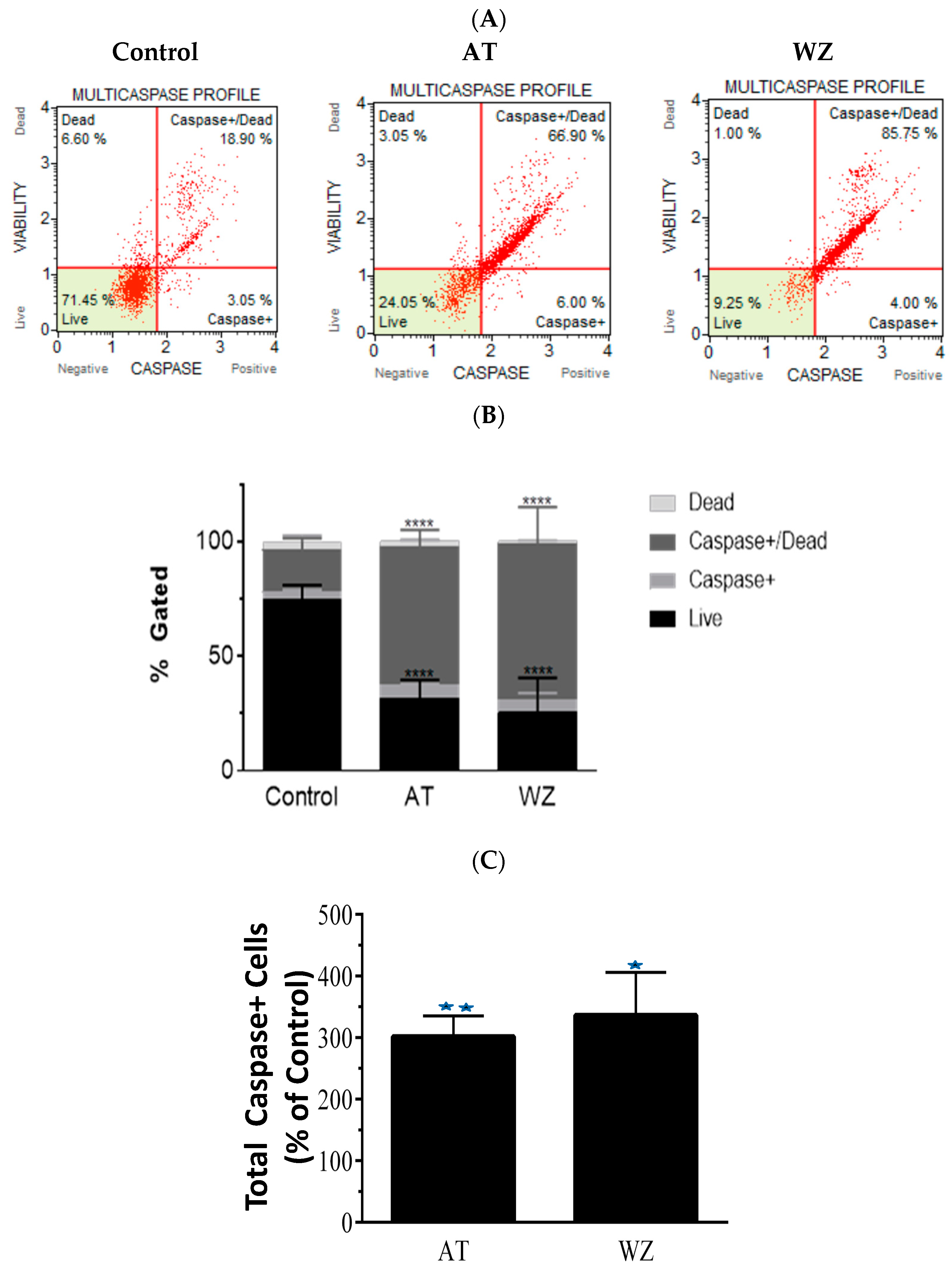
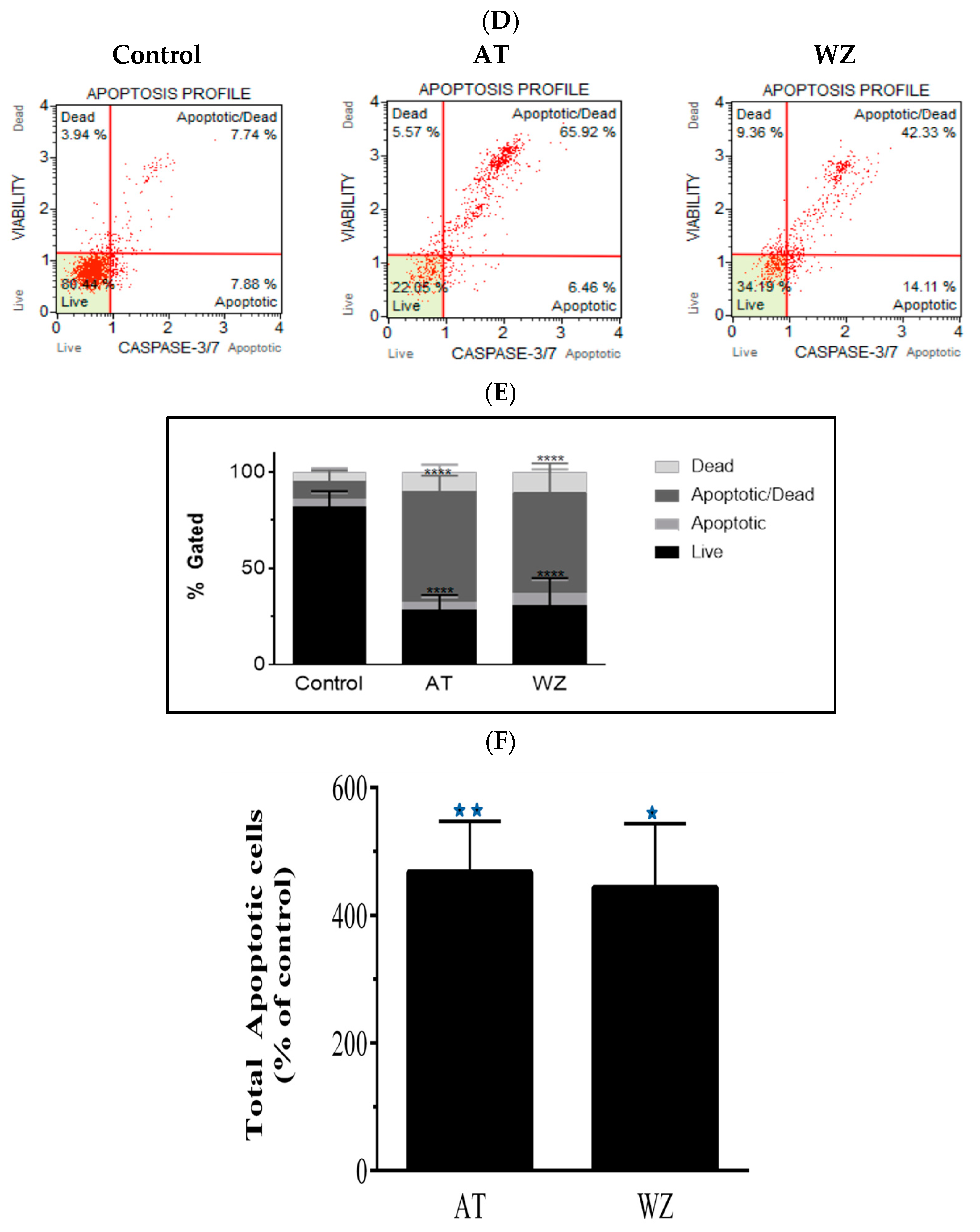
3.5. Multicaspase and Executioner Caspases 3/7 Were Elicited in PANC-1 Cells by AT and WZ
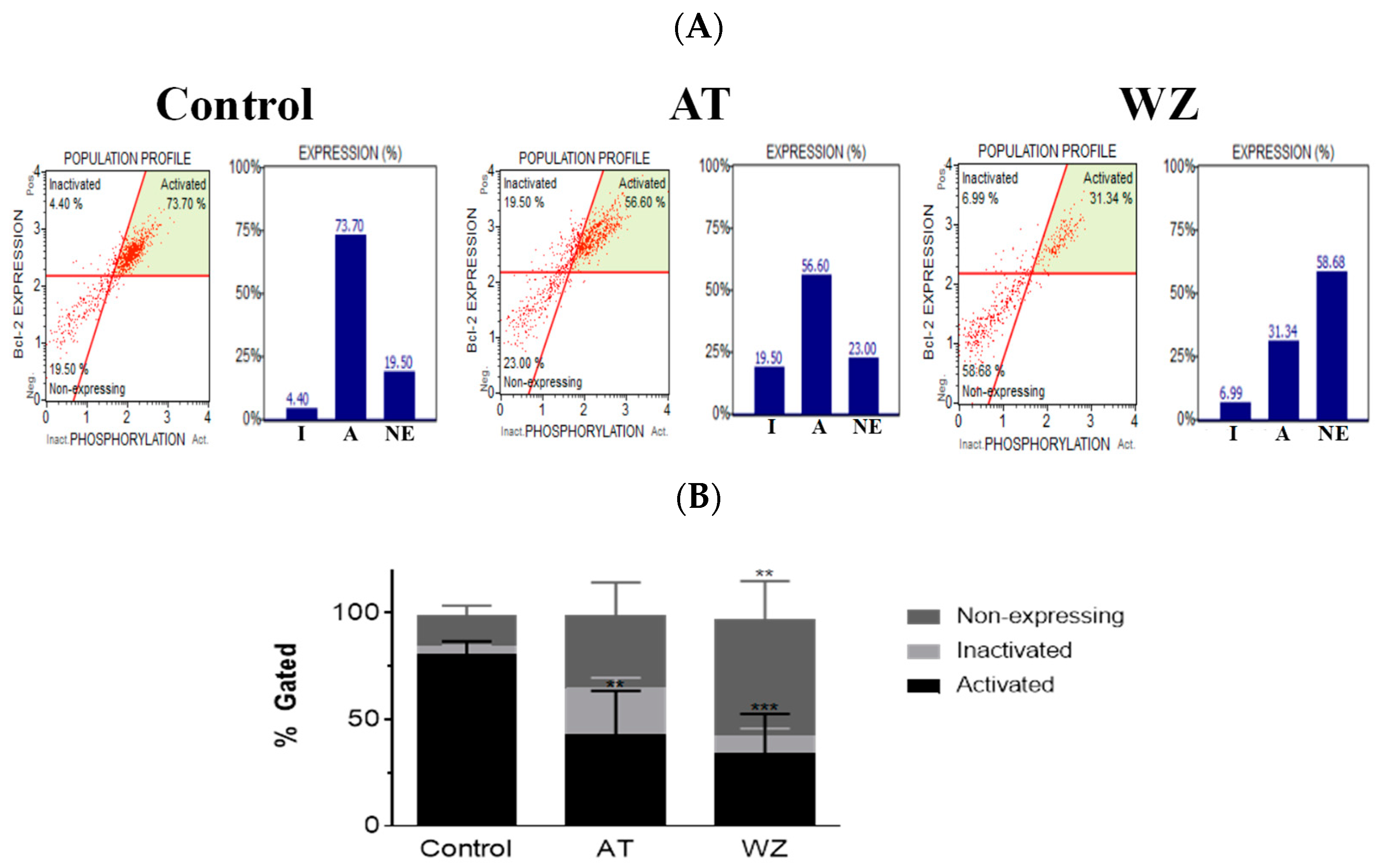
3.6. The Key Cell Survival Molecule Bcl-2 Was Impacted by AT and WZ
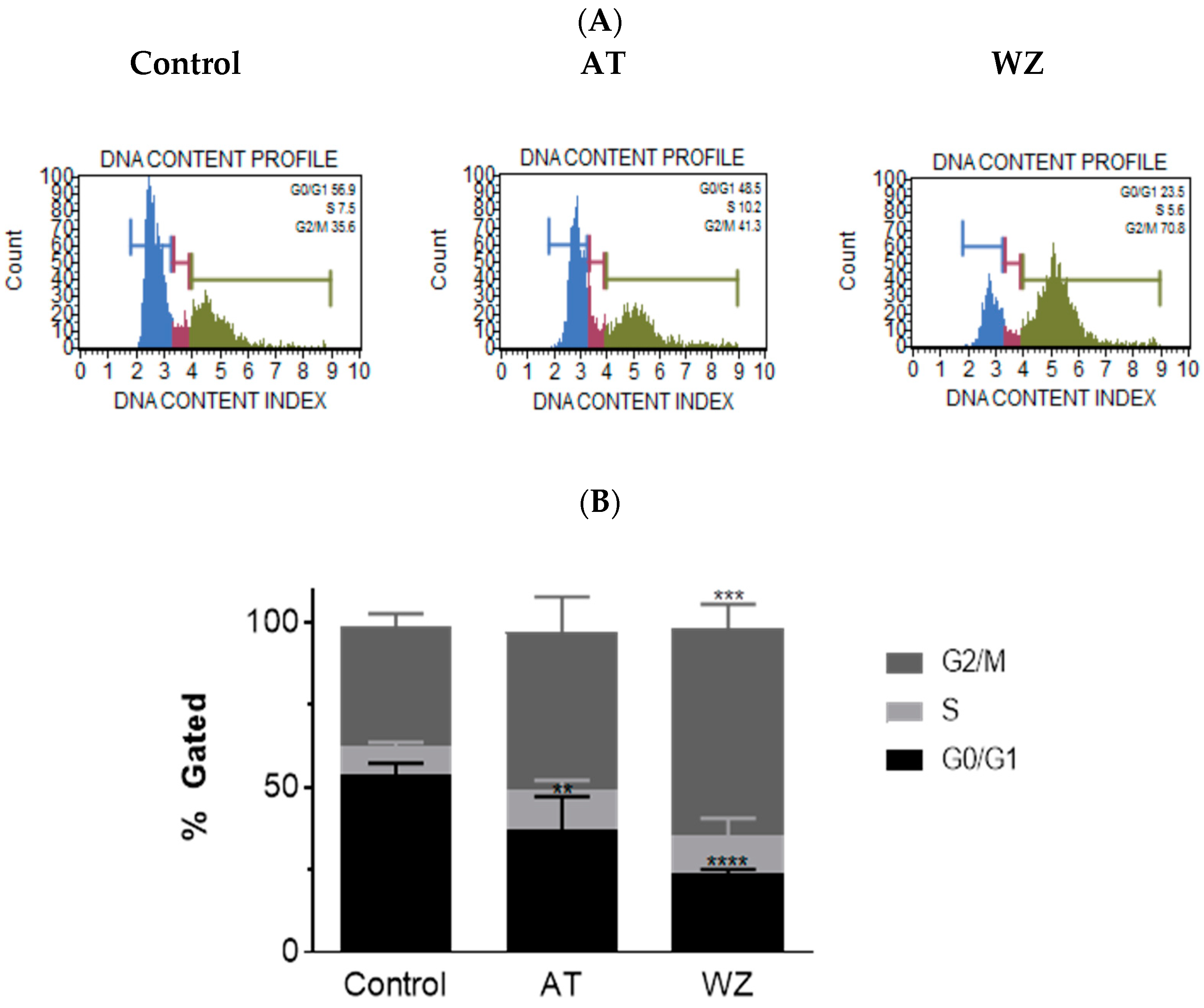
3.7. Cell Cycle of PANC-1 Cells Was Impacted by AT and WZ
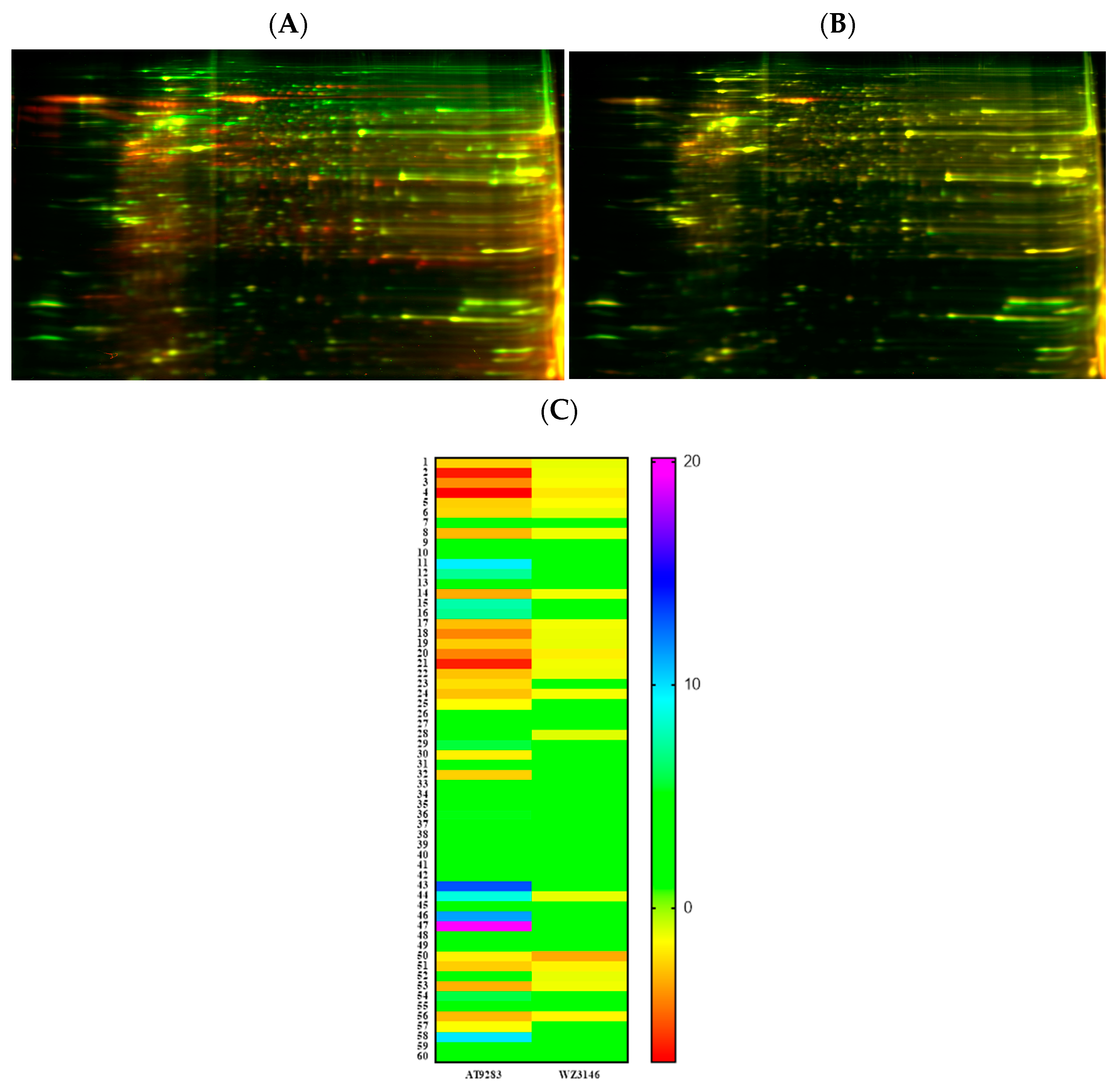
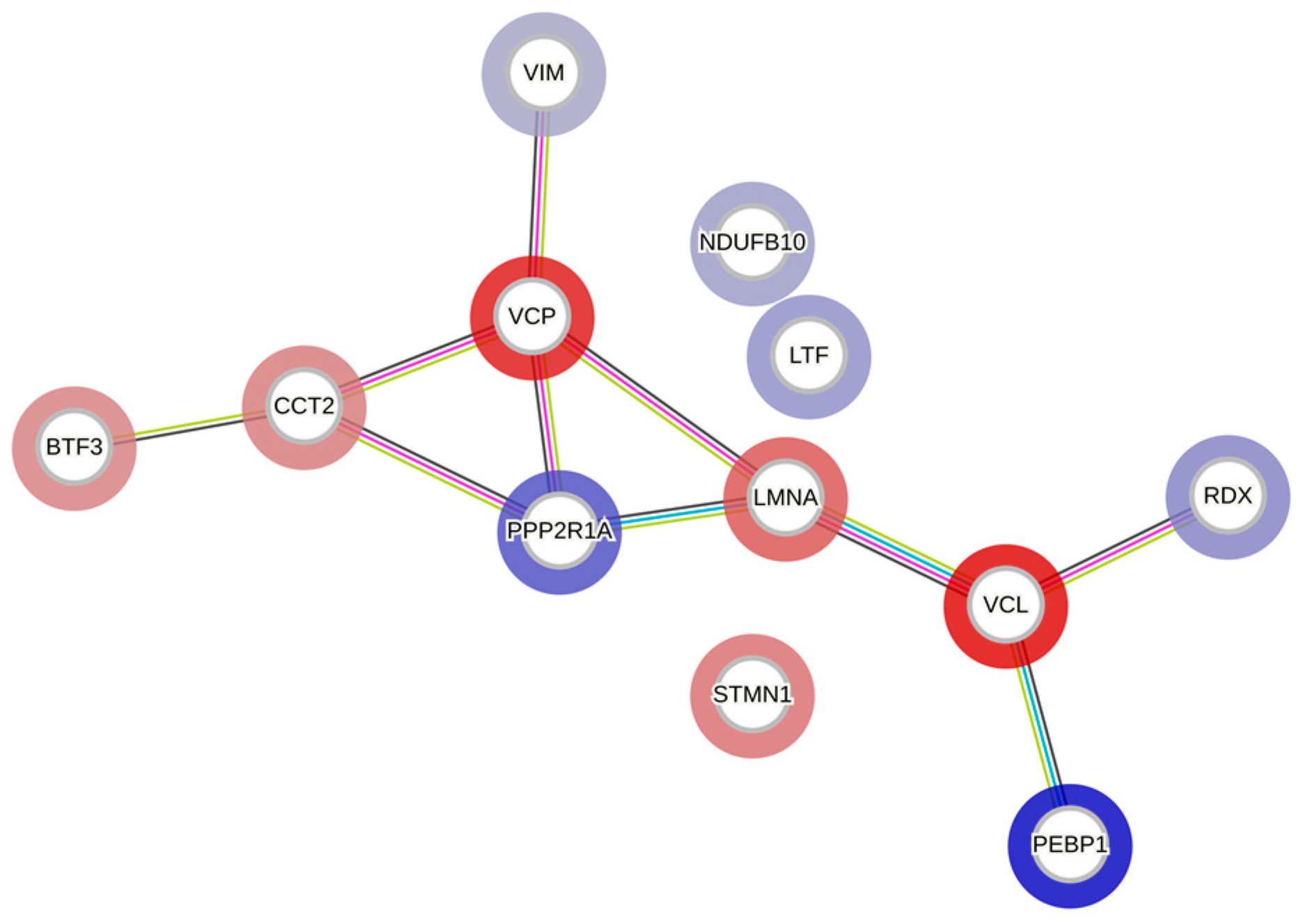
3.8. Proteomic Analysis Reveals Entities of Pivotal Signaling Pathways That Were Differentially Regulated in PANC-1 Cells on Treatment with AT and WZ
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Zijlstra, M.; Bernards, N.; de Hingh, I.H.; van de Wouw, A.J.; Goey, S.H.; Jacobs, E.M.; Lemmens, V.E.; Creemers, G.J. Does long-term survival exist in pancreatic adenocarcinoma? Acta Oncol. 2016, 55, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; van Mansfeld, A.D.; Offerhaus, G.J.; van Weering, D.H.; Allison, D.C.; Goodman, S.N.; Kensler, T.W.; Bose, K.K.; Cameron, J.L.; Bos, J.L. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 1993, 143, 545–554. [Google Scholar] [PubMed]
- Heinemann, V.; Reni, M.; Ychou, M.; Richel, D.J.; Macarulla, T.; Ducreux, M. Tumour-stroma interactions in pancreatic ductal adenocarcinoma: Rationale and current evidence for new therapeutic strategies. Cancer Treat Rev. 2014, 40, 118–128. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Gupta, S.; El-Rayes, B.F. Small molecule tyrosine kinase inhibitors in pancreatic cancer. Biologics 2008, 2, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Grapa, C.M.; Mocan, T.; Gonciar, D.; Zdrehus, C.; Mosteanu, O.; Pop, T.; Mocan, L. Epidermal Growth Factor Receptor and Its Role in Pancreatic Cancer Treatment Mediated by Nanoparticles. Int. J. Nanomed. 2019, 14, 9693–9706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal growth factor receptor in pancreatic cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef]
- Dong, M.; Nio, Y.; Guo, K.J.; Tamura, K.; Tian, Y.L.; Dong, Y.T. Epidermal growth factor and its receptor as prognostic indicators in Chinese patients with pancreatic cancer. Anticancer Res. 1998, 18, 4613–4619. [Google Scholar]
- Wang, J.P.; Wu, C.Y.; Yeh, Y.C.; Shyr, Y.M.; Wu, Y.Y.; Kuo, C.Y.; Hung, Y.P.; Chen, M.H.; Lee, W.P.; Luo, J.C.; et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: A randomized, open-label, prospective trial. Oncotarget 2015, 6, 18162–18173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhu, J.; Firozi, P.F.; Abbruzzese, J.L.; Evans, D.B.; Cleary, K.; Friess, H.; Sen, S. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin. Cancer Res. 2003, 9, 991–997. [Google Scholar] [PubMed]
- Warner, S.L.; Munoz, R.M.; Bearss, D.J.; Grippo, P.; Han, H.; Von Hoff, D.D. Pdx-1-driven overexpression of aurora a kinase induces mild ductal dysplasia of pancreatic ducts near islets in transgenic mice. Pancreas 2008, 37, e39–e44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Abbruzzese, J.L.; Izzo, J.; Hittelman, W.N.; Li, D. AURKA amplification, chromosome instability, and centrosome abnormality in human pancreatic carcinoma cells. Cancer Genet. Cytogenet. 2005, 159, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kanai, N.; Shiwaku, H.O.; Soga, N.; Uehara, A.; Horii, A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene 2006, 25, 4831–4839. [Google Scholar] [CrossRef] [Green Version]
- Katsha, A.; Belkhiri, A.; Goff, L.; El-Rifai, W. Aurora kinase A in gastrointestinal cancers: Time to target. Mol. Cancer 2015, 14, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes-Filho, S.M.; Dos Santos, E.O.; Bertoldi, E.R.M.; Scalabrini, L.C.; Heidrich, V.; Dazzani, B.; Levantini, E.; Reis, E.M.; Basseres, D.S. Aurora A kinase and its activator TPX2 are potential therapeutic targets in KRAS-induced pancreatic cancer. Cell Oncol. 2020, 43, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Xiao, H.; Shen, P.; Yang, Y.; Xue, J.; Yang, Y.; Shang, Y.; Zhang, L.; Li, X.; Zhang, Y.; et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022, 8, 5. [Google Scholar] [CrossRef]
- Ungefroren, H.; Thurling, I.; Farber, B.; Kowalke, T.; Fischer, T.; De Assis, L.V.M.; Braun, R.; Castven, D.; Oster, H.; Konukiewitz, B.; et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1-A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers 2022, 14, 2057. [Google Scholar] [CrossRef] [PubMed]
- Dhir, H.; Choudhury, M.; Patil, K.; Cheung, C.; Bodlak, A.; Pardo, D.; Adams, A.; Travaglino, S.; Rojas, J.A.; Pai, S.B. Interception of Signaling Circuits of Esophageal Adenocarcinoma Cells by Resveratrol Reveals Molecular and Immunomodulatory Signatures. Cancers 2021, 13, 5811. [Google Scholar] [CrossRef]
- Qu, Y.; Olsen, J.R.; Yuan, X.; Cheng, P.F.; Levesque, M.P.; Brokstad, K.A.; Hoffman, P.S.; Oyan, A.M.; Zhang, W.; Kalland, K.H.; et al. Small molecule promotes beta-catenin citrullination and inhibits Wnt signaling in cancer. Nat. Chem. Biol. 2018, 14, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Tonge, R.; Shaw, J.; Middleton, B.; Rowlinson, R.; Rayner, S.; Young, J.; Pognan, F.; Hawkins, E.; Currie, I.; Davison, M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.L.; Magnus, N.K.C.; Thapar, V.; Morris, R.; Szabolcs, A.; Neyaz, A.; Kulkarni, A.S.; Tai, E.; Chougule, A.; Hillis, A.; et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 26835–26845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtlein, P.M.; Volz, C.; Hackel, A.; Thurling, I.; Castven, D.; Braun, R.; Wellner, U.F.; Konukiewitz, B.; Riemekasten, G.; Lehnert, H.; et al. Activation of a Ductal-to-Endocrine Transdifferentiation Transcriptional Program in the Pancreatic Cancer Cell Line PANC-1 Is Controlled by RAC1 and RAC1b through Antagonistic Regulation of Stemness Factors. Cancers 2021, 13, 5541. [Google Scholar] [CrossRef]
- Kumari, G.; Ulrich, T.; Krause, M.; Finkernagel, F.; Gaubatz, S. Induction of p21CIP1 protein and cell cycle arrest after inhibition of Aurora B kinase is attributed to aneuploidy and reactive oxygen species. J. Biol. Chem. 2014, 289, 16072–16084. [Google Scholar] [CrossRef] [Green Version]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.C.; Nicolau, A.; Teixeira, J.A.; Rodrigues, L.R. The effect of bovine milk lactoferrin on human breast cancer cell lines. J. Dairy Sci. 2011, 94, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mader, J.S.; Salsman, J.; Conrad, D.M.; Hoskin, D.W. Bovine lactoferricin selectively induces apoptosis in human leukemia and carcinoma cell lines. Mol. Cancer Ther. 2005, 4, 612–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Monitto, C.L.; Minhas, K.M.; Sidransky, D. Lactoferrin down-regulates G1 cyclin-dependent kinases during growth arrest of head and neck cancer cells. Clin. Cancer Res. 2004, 10, 8683–8686. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.S.; Li, G.; Varadhachary, A.; Petrak, K.; Schneyer, M.; Li, D.; Ongkasuwan, J.; Zhang, X.; Taylor, R.J.; Strome, S.E.; et al. Oral lactoferrin results in T cell-dependent tumor inhibition of head and neck squamous cell carcinoma in vivo. Clin. Cancer Res. 2007, 13, 1601–1610. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, P.; Kaczynska, K.; Kleczkowska, P.; Bukowska-Osko, I.; Kramkowski, K.; Sulejczak, D. The Lactoferrin Phenomenon-A Miracle Molecule. Molecules 2022, 27, 2941. [Google Scholar] [CrossRef]
- Bukowska-Osko, I.; Sulejczak, D.; Kaczynska, K.; Kleczkowska, P.; Kramkowski, K.; Popiel, M.; Wietrak, E.; Kowalczyk, P. Lactoferrin as a Human Genome “Guardian”—An Overall Point of View. Int. J. Mol. Sci. 2022, 23, 5248. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.W.; Pyo, C.W.; Yoo, N.K.; Kim, J.; Choi, S.Y. Requirement of the JNK-associated Bcl-2 pathway for human lactoferrin-induced apoptosis in the Jurkat leukemia T cell line. Biochimie 2009, 91, 102–108. [Google Scholar] [CrossRef]
- Fujita, K.; Matsuda, E.; Sekine, K.; Iigo, M.; Tsuda, H. Lactoferrin enhances Fas expression and apoptosis in the colon mucosa of azoxymethane-treated rats. Carcinogenesis 2004, 25, 1961–1966. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Debbabi, H.; Dubarry, M.; Boyaka, P.; Tome, D. Regulation of physiological and pathological Th1 and Th2 responses by lactoferrin. Biochem. Cell Biol. 2006, 84, 303–311. [Google Scholar] [CrossRef]
- Kuhara, T.; Iigo, M.; Itoh, T.; Ushida, Y.; Sekine, K.; Terada, N.; Okamura, H.; Tsuda, H. Orally administered lactoferrin exerts an antimetastatic effect and enhances production of IL-18 in the intestinal epithelium. Nutr. Cancer 2000, 38, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Massodi, I.; Thomas, E.; Raucher, D. Application of thermally responsive elastin-like polypeptide fused to a lactoferrin-derived peptide for treatment of pancreatic cancer. Molecules 2009, 14, 1999–2015. [Google Scholar] [CrossRef] [Green Version]
- Roseanu, A.; Florian, P.E.; Moisei, M.; Sima, L.E.; Evans, R.W.; Trif, M. Liposomalization of lactoferrin enhanced its anti-tumoral effects on melanoma cells. Biometals 2010, 23, 485–492. [Google Scholar] [CrossRef]
- Tang, T.; Huang, X.; Zhang, G.; Lu, M.; Hong, Z.; Wang, M.; Huang, J.; Zhi, X.; Liang, T. Oncolytic peptide LTX-315 induces anti-pancreatic cancer immunity by targeting the ATP11B-PD-L1 axis. J. Immunother. Cancer 2022, 10, e004129. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Wennerberg, E.; Hensler, M.; Buque, A.; Kraynak, J.; Fucikova, J.; Zhou, X.K.; Sveinbjornsson, B.; Rekdal, O.; Demaria, S.; et al. LTX-315-enabled, radiotherapy-boosted immunotherapeutic control of breast cancer by NK cells. Oncoimmunology 2021, 10, 1962592. [Google Scholar] [CrossRef]
- Kobori, T.; Tanaka, C.; Tameishi, M.; Urashima, Y.; Ito, T.; Obata, T. Role of Ezrin/Radixin/Moesin in the Surface Localization of Programmed Cell Death Ligand-1 in Human Colon Adenocarcinoma LS180 Cells. Pharmaceuticals 2021, 14, 864. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Iizuka, A.; Maeda, C.; Tanaka, E.; Kondou, R.; Miyata, H.; Sugino, T.; Kawata, T.; Deguchi, S.; Mitsuya, K.; et al. Impact of combination therapy with anti-PD-1 blockade and a STAT3 inhibitor on the tumor-infiltrating lymphocyte status. Immunol. Lett. 2019, 216, 43–50. [Google Scholar] [CrossRef]
- Kobori, T.; Doukuni, R.; Ishikawa, H.; Ito, Y.; Okada, R.; Tanaka, C.; Tameishi, M.; Urashima, Y.; Ito, T.; Obata, T. Ezrin and Radixin Differentially Modulate Cell Surface Expression of Programmed Death Ligand-1 in Human Pancreatic Ductal Adenocarcinoma KP-2. Immunol. Lett. 2022, 2, 68–84. [Google Scholar] [CrossRef]
- Chen, S.D.; Song, M.M.; Zhong, Z.Q.; Li, N.; Wang, P.L.; Cheng, S.; Bai, R.X.; Yuan, H.S. Knockdown of radixin by RNA interference suppresses the growth of human pancreatic cancer cells in vitro and in vivo. Asian Pac. J. Cancer Prev. 2012, 13, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yogesha, S.D.; Mayfield, J.E.; Gill, G.N.; Zhang, Y. Viewing serine/threonine protein phosphatases through the eyes of drug designers. FEBS J. 2013, 280, 4739–4760. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.J.; Vellody, K.; Patel, S.; Bergstrom, R.; Meisinger, J.; Jackson, J.; Wright, M.A.; Young, M.R. Vitamin D3 and ceramide reduce the invasion of tumor cells through extracellular matrix components by elevating protein phosphatase-2A. Invasion Metastasis 1996, 16, 280–290. [Google Scholar]
- Nagahara, Y.; Matsuoka, Y.; Saito, K.; Ikekita, M.; Higuchi, S.; Shinomiya, T. Coordinate involvement of cell cycle arrest and apoptosis strengthen the effect of FTY720. Jpn. J. Cancer Res. 2001, 92, 680–687. [Google Scholar] [CrossRef]
- Handra-Luca, A.; Hong, S.M.; Walter, K.; Wolfgang, C.; Hruban, R.; Goggins, M. Tumour epithelial vimentin expression and outcome of pancreatic ductal adenocarcinomas. Br. J. Cancer 2011, 104, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.H.; Misek, D.E.; Wang, H.; Puravs, E.; Hinderer, R.; Giordano, T.J.; Greenson, J.K.; Brenner, D.E.; Simeone, D.M.; Logsdon, C.D.; et al. Identification of a Specific Vimentin Isoform That Induces an Antibody Response in Pancreatic Cancer. Biomark Insights 2006, 1, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Lamiman, K.; Keller, J.M.; Mizokami, A.; Zhang, J.; Keller, E.T. Survey of Raf kinase inhibitor protein (RKIP) in multiple cancer types. Crit. Rev. Oncog. 2014, 19, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Song, S.P.; Zhang, S.B.; Li, Z.H.; Zhou, Y.S.; Li, B.; Bian, Z.W.; Liao, Q.D.; Zhang, Y.D. Reduced expression of Raf kinase inhibitor protein correlates with poor prognosis in pancreatic cancer. Clin. Transl. Oncol. 2012, 14, 848–852. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Zlobec, I.; Gloor, B.; Kondi-Pafiti, A.; Lugli, A.; Perren, A. Loss of Raf-1 kinase inhibitor protein (RKIP) is strongly associated with high-grade tumor budding and correlates with an aggressive phenotype in pancreatic ductal adenocarcinoma (PDAC). J. Transl. Med. 2013, 11, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Kim, G.Y.; Lim, S.J.; Kim, Y.W. Loss of Raf-1 kinase inhibitory protein in pancreatic ductal adenocarcinoma. Pathology 2010, 42, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Quinonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The challenge of drug resistance in pancreatic ductal adenocarcinoma: A current overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Lowery, M.A.; Capanu, M.; Stadler, Z.K.; Moore, M.J.; Dhani, N.; Kindler, H.L.; Estrella, H.; Maynard, H.; et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: Germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer 2018, 124, 1374–1382. [Google Scholar] [CrossRef] [Green Version]
- Skripova, V.; Vlasenkova, R.; Zhou, Y.; Astsaturov, I.; Kiyamova, R. Identification of New Regulators of Pancreatic Cancer Cell Sensitivity to Oxaliplatin and Cisplatin. Molecules 2022, 27, 1289. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Nagano, H.; Dono, K.; Umeshita, K.; Sakon, M.; Ishikawa, O.; Ohigashi, H.; Nakamori, S.; et al. Increased expression of valosin-containing protein (p97) is associated with lymph node metastasis and prognosis of pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2004, 11, 165–172. [Google Scholar] [CrossRef]
- Islam, S.; Kitagawa, T.; Azuma, T.; Kuramitsu, Y. The Expression Levels of Vinculin in Pancreatic Cancer Tissues Significantly Correlates With Patient Survival. Anticancer Res. 2021, 41, 4979–4984. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Nyberg, K.D.; Scott, M.B.; Welsh, A.M.; Nguyen, A.H.; Wu, N.; Hohlbauch, S.V.; Geisse, N.A.; Gibb, E.A.; Robertson, A.G.; et al. Stiffness of pancreatic cancer cells is associated with increased invasive potential. Integr. Biol. 2016, 8, 1232–1245. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.X.; Lin, Y.F.; Chen, C.L.; Huang, M.S.; Hsiao, M.; Liang, P.H. Chaperonin-Containing TCP-1 Promotes Cancer Chemoresistance and Metastasis through the AKT-GSK3beta-beta-Catenin and XIAP-Survivin Pathways. Cancers 2020, 12, 3865. [Google Scholar] [CrossRef]
- Kusumawidjaja, G.; Kayed, H.; Giese, N.; Bauer, A.; Erkan, M.; Giese, T.; Hoheise, J.D.; Friess, H.; Kleeff, J. Basic transcription factor 3 (BTF3) regulates transcription of tumor-associated genes in pancreatic cancer cells. Cancer Biol. Ther. 2007, 6, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, C.; Cheng, H.; Xu, Y.; Jiang, J.; Xu, J.; Long, J.; Liu, L.; Yu, X. Stathmin, interacting with Nf-kappaB, promotes tumor growth and predicts poor prognosis of pancreatic cancer. Curr. Mol. Med. 2014, 14, 328–339. [Google Scholar] [CrossRef] [PubMed]










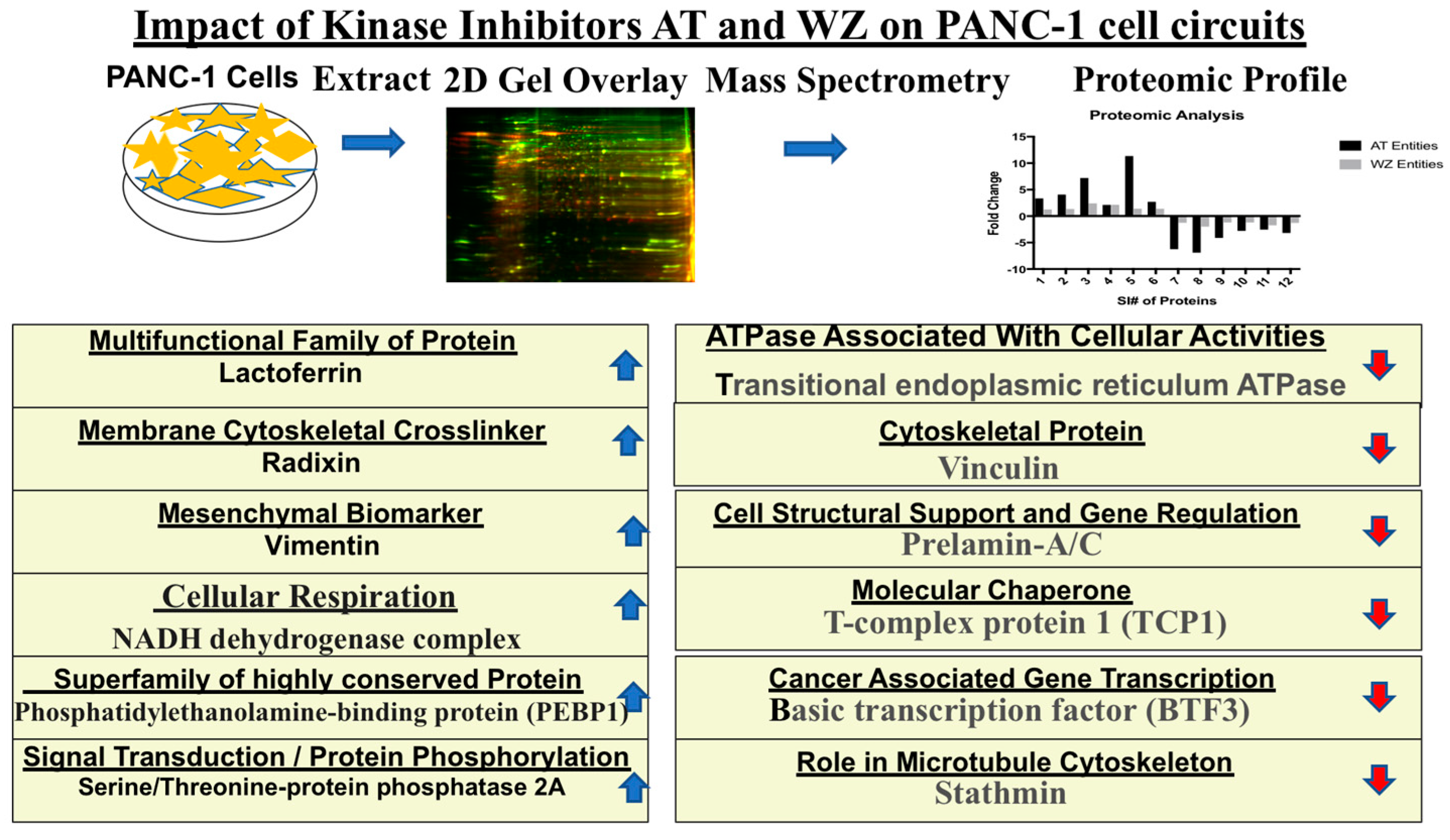
 ) and downregulated (
) and downregulated ( ) entities in PANC-1 pancreatic cancer cells upon treatment with AT and WZ.
) and downregulated () entities in PANC-1 pancreatic cancer cells upon treatment with AT and WZ.
) entities in PANC-1 pancreatic cancer cells upon treatment with AT and WZ.
) and downregulated () entities in PANC-1 pancreatic cancer cells upon treatment with AT and WZ.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SI # | Spot # | Fold Change in | Key Entities Impacted by Aurora Kinase Inhibitor (AT) and EGFR Kinase Inhibitor (WZ) | |
|---|---|---|---|---|
| AT | WZ | |||
| 1 | 7 | +3.35 | +1.25 | Lactotransferrin or lactoferrin (LTF) |
| 2 | 9 | +4.07 | +1.35 | Radixin (RDX) |
| 3 | 12 | +7.2 | +2.4 | Serine/threonine-protein phosphatase 2A (PPP2R1A) |
| 4 | 26 | +2.12 | +2.14 | Vimentin (VIM) |
| 5 | 46 | +11.34 | +1.4 | Phosphatidylethanolamine-binding protein 1(PEBP1) |
| 6 | 48 | +2.69 | +1.39 | NADH dehydrogenase (Ubiquinone)1 beta subcomplex subunit 10 (NDUFB10) |
| 7 | 2 | −6.26 | −1.26 | Transitional endoplasmic reticulum ATPase (VCP) |
| 8 | 4 | −6.9 | −1.98 | Vinculin (VCL) |
| 9 | 18 | −4.11 | −1.2 | Prelamin-A/C (LMNA) |
| 10 | 22 | −2.79 | −1.19 | T-complex protein 1 (TCP1 subunit beta) (CCT2) |
| 11 | 51 | −2.55 | −1.74 | Basic transcription factor (BTF3) |
| 12 | 53 | −3.17 | −1.29 | Stathmin (STMN1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.N.; Patil, K.; Ma, J.; Dufek, G.A.; Pai, S.B. Multifaceted Effects of Kinase Inhibitors on Pancreatic Cancer Cells Reveals Pivotal Entities with Therapeutic Implications. Biomedicines 2023, 11, 1716. https://doi.org/10.3390/biomedicines11061716
Kim YN, Patil K, Ma J, Dufek GA, Pai SB. Multifaceted Effects of Kinase Inhibitors on Pancreatic Cancer Cells Reveals Pivotal Entities with Therapeutic Implications. Biomedicines. 2023; 11(6):1716. https://doi.org/10.3390/biomedicines11061716
Chicago/Turabian StyleKim, Yoo Na, Ketki Patil, Jeonghwa Ma, Griffin A. Dufek, and S. Balakrishna Pai. 2023. "Multifaceted Effects of Kinase Inhibitors on Pancreatic Cancer Cells Reveals Pivotal Entities with Therapeutic Implications" Biomedicines 11, no. 6: 1716. https://doi.org/10.3390/biomedicines11061716
APA StyleKim, Y. N., Patil, K., Ma, J., Dufek, G. A., & Pai, S. B. (2023). Multifaceted Effects of Kinase Inhibitors on Pancreatic Cancer Cells Reveals Pivotal Entities with Therapeutic Implications. Biomedicines, 11(6), 1716. https://doi.org/10.3390/biomedicines11061716