
Detection of Single-Nucleotide and Copy Number Defects Underlying Hyperphenylalaninemia by Next-Generation Sequencing
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Design of a tNGS Panel
2.3. Library Preparation and Sequencing
2.4. Computational Analysis of NGS Data
2.5. Validation of Variants Identified by NGS
2.6. CNV Validation
3. Results
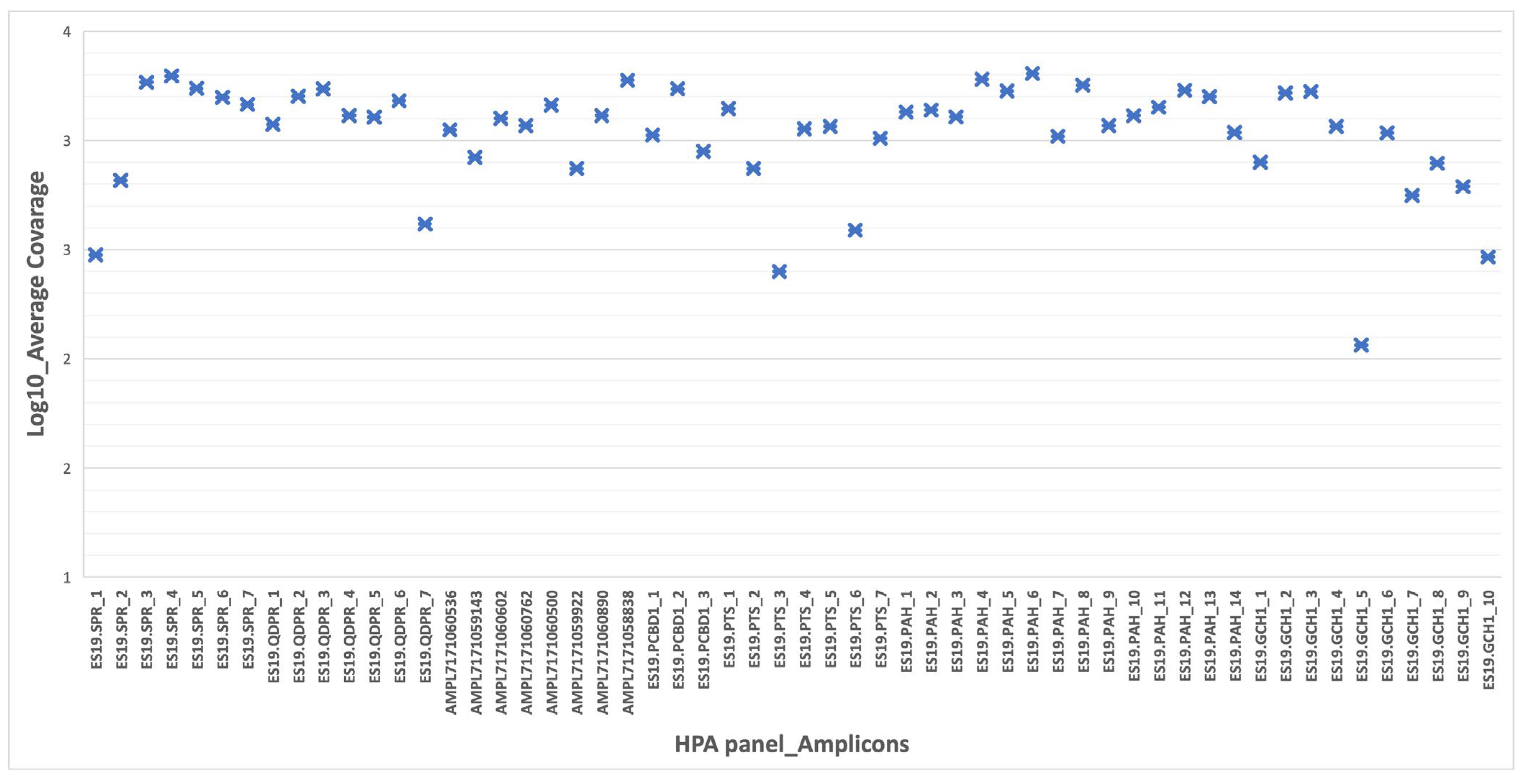
3.1. Performance and Coverage Analysis
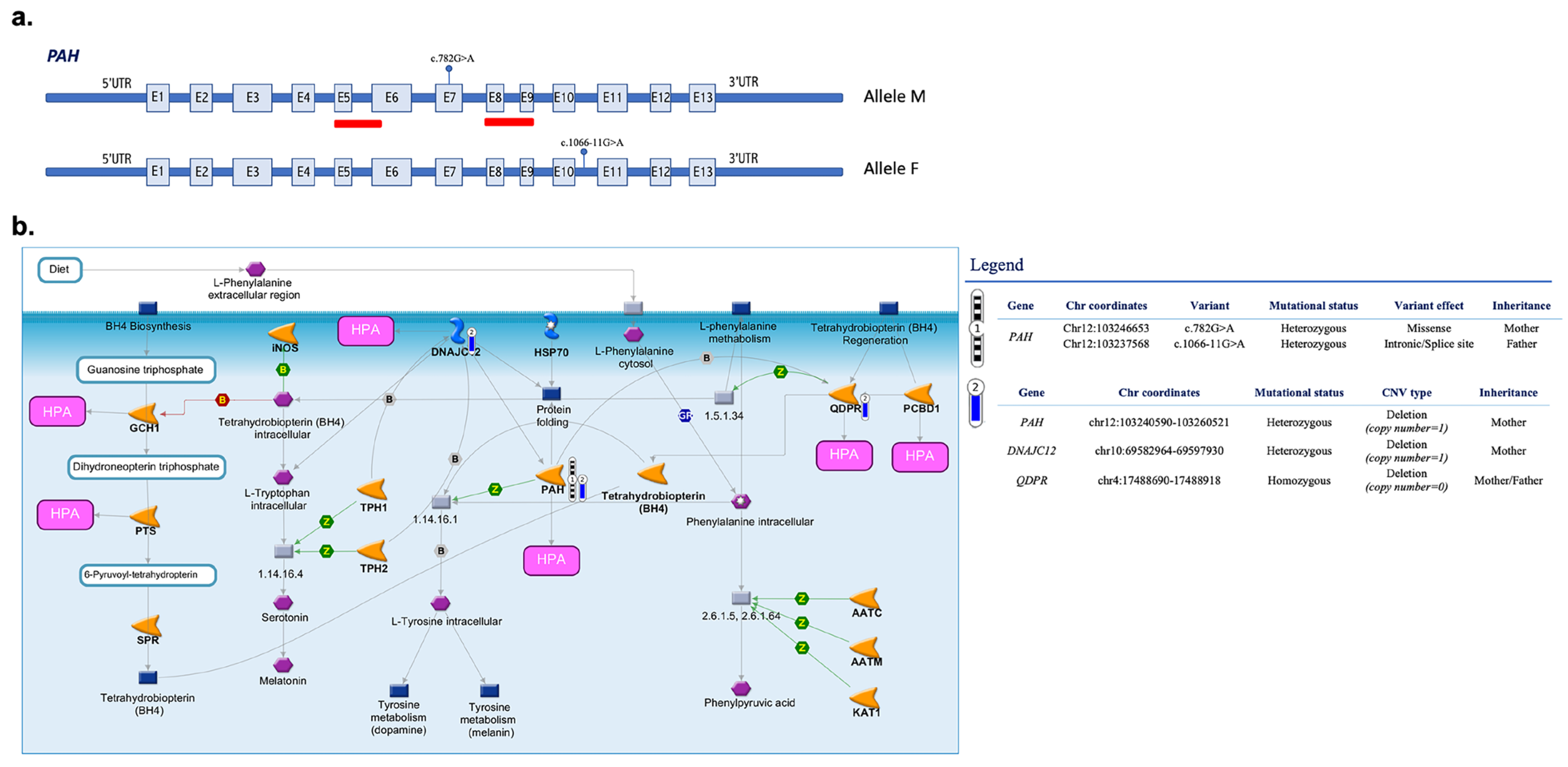
3.2. Identified Pathogenic Variants
3.3. CNVs in Targeted Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, R.A.; Mamotte, C.D.S.; Burnett, J.R. Phenylketonuria: An Inborn Error of Phenylalanine Metabolism. Clin. Biochem. Rev. 2008, 29, 31–41. [Google Scholar] [PubMed]
- Alsharhan, H.; Ficicioglu, C. Disorders of phenylalanine and tyrosine metabolism. Transl. Sci. Rare Dis. 2020, 5, 3–58. [Google Scholar] [CrossRef]
- Tendi, E.A.; Guarnaccia, M.; Morello, G.; Cavallaro, S. The Utility of Genomic Testing for Hyperphenylalaninemia. J. Clin. Med. 2022, 11, 1061. [Google Scholar] [CrossRef] [PubMed]
- Mazur, A.; Jarochowicz, S.; Sykut-Cegielska, J.; Gradowska, W.; Kwolek, A.; Ołtarzewski, M. Evaluation of Somatic Development in Adult Patients with Previously Undiagnosed and/or Untreated Phenylketonuria. Med. Princ. Pract. 2009, 19, 46–50. [Google Scholar] [CrossRef]
- Anjema, K.; Hofstede, F.C.; Bosch, A.M.; Rubio–Gozalbo, M.E.; de Vries, M.C.; Boelen, C.C.; van Rijn, M.; van Spronsen, F.J. The neonatal tetrahydrobiopterin loading test in phenylketonuria: What is the predictive value? Orphanet J. Rare Dis. 2016, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Anesthesia Analg. 2014, 16, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Thony, B.; Blau, N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum. Mutat. 2006, 27, 870–878. [Google Scholar] [CrossRef]
- Li, N.; Yu, P.; Rao, B.; Deng, Y.; Guo, Y.; Huang, Y.; Ding, L.; Zhu, J.; Yang, H.; Wang, J.; et al. Molecular genetics of tetrahydrobiopterin deficiency in Chinese patients. J. Pediatr. Endocrinol. Metab. 2018, 31, 911–916. [Google Scholar] [CrossRef]
- Vatanavicharn, N.; Kuptanon, C.; Liammongkolkul, S.; Liu, T.T.; Hsiao, K.J.; Ratanarak, P.; Blau, N.; Wasant, P. Novel mutation affecting the pterin-binding site of PTS gene and review of PTS mutations in Thai patients with 6-pyruvoyltetrahydropterin synthase deficiency. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S279–S282. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; Bonafé, L.; Thöny, B. Tetrahydrobiopterin Deficiencies without Hyperphenylalaninemia: Diagnosis and Genetics of DOPA-Responsive Dystonia and Sepiapterin Reductase Deficiency. Mol. Genet. Metab. 2001, 74, 172–185. [Google Scholar] [CrossRef]
- Bonafé, L.; Thöny, B.; Penzien, J.M.; Czarnecki, B.; Blau, N. Mutations in the Sepiapterin Reductase Gene Cause a Novel Tetrahydrobiopterin-Dependent Monoamine-Neurotransmitter Deficiency without Hyperphenylalaninemia. Am. J. Hum. Genet. 2001, 69, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Roca, I.; González-Castro, L.; Fernandez-Lopez, H.; Couce, M.L.; Fernández-Marmiesse, A. Free-access copy-number variant detection tools for targeted next-generation sequencing data. Mutat. Res. Mol. Mech. Mutagen. 2019, 779, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Pinkel, D.; Segraves, R.; Sudar, D.; Clark, S.; Poole, I.; Kowbel, D.; Collins, C.; Kuo, W.-L.; Chen, C.; Zhai, Y.; et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998, 20, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Lucito, R.; Healy, J.; Alexander, J.; Reiner, A.; Esposito, D.; Chi, M.; Rodgers, L.; Brady, A.; Sebat, J.; Troge, J.; et al. Representational Oligonucleotide Microarray Analysis: A High-Resolution Method to Detect Genome Copy Number Variation. Genome Res. 2003, 13, 2291–2305. [Google Scholar] [CrossRef] [Green Version]
- Gulilat, M.; Lamb, T.; Teft, W.A.; Wang, J.; Dron, J.S.; Robinson, J.F.; Tirona, R.G.; Hegele, R.A.; Kim, R.B.; Schwarz, U.I. Targeted next generation sequencing as a tool for precision medicine. BMC Med. Genom. 2019, 12, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Anesthesia Analg. 2015, 17, 444–451. [Google Scholar] [CrossRef] [Green Version]
- La Cognata, V.; Cavallaro, S. A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases. Genes 2021, 12, 1750. [Google Scholar] [CrossRef]
- La Cognata, V.; Guarnaccia, M.; Morello, G.; Ruggieri, M.; Polizzi, A.; Cavallaro, S. Design and Validation of a Custom NGS Panel Targeting a Set of Lysosomal Storage Diseases Candidate for NBS Applications. Int. J. Mol. Sci. 2021, 22, 10064. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Calì, F.; Ruggeri, G.; Vinci, M.; Meli, C.; Carducci, C.; Leuzzi, V.; Pozzessere, S.; Schinocca, P.; Ragalmuto, A.; Chiavetta, V.; et al. Exon deletions of the phenylalanine hydroxylase gene in Italian hyperphenylalaninemics. Exp. Mol. Med. 2010, 42, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Anikster, Y.; Haack, T.B.; Vilboux, T.; Pode-Shakked, B.; Thöny, B.; Shen, N.; Guarani, V.; Meissner, T.; Mayatepek, E.; Trefz, F.K.; et al. Biallelic Mutations in DNAJC12 Cause Hyperphenylalaninemia, Dystonia, and Intellectual Disability. Am. J. Hum. Genet. 2017, 100, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, Y.; Nygaard, T.G.; Gutlich, M.; Rajput, A.H.; Pifl, C.; Distefano, L.; Chang, L.J.; Price, K.; Shimadzu, M.; Hornykiewicz, O.; et al. Striatal biopterin and tyrosine hydroxylase protein reduction in dopa-responsive dystonia. Neurology 1999, 53, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Roze, E.; Abdenur, J.E.; Chang, R.; Gasperini, S.; Saletti, V.; Wali, G.M.; Eiroa, H.; Neville, B.; Felice, A.; et al. Sepiapterin reductase deficiency: A treatable mimic of cerebral palsy. Ann. Neurol. 2012, 71, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Directors ABo. Clinical utility of genetic and genomic services: A position statement of the American College of Medical Genetics and Genomics. Genet. Med. 2015, 17, 505–507. [Google Scholar] [CrossRef] [Green Version]
- Trujillano, D.; Perez, B.; González, J.; Tornador, C.; Navarrete, R.; Escaramis, G.; Ossowski, S.; Armengol, L.; Cornejo, V.; Desviat, L.R.; et al. Accurate molecular diagnosis of phenylketonuria and tetrahydrobiopterin-deficient hyperphenylalaninemias using high-throughput targeted sequencing. Eur. J. Hum. Genet. 2013, 22, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Guldberg, P.; Rey, F.; Zschocke, J.; Romano, V.; François, B.; Michiels, L.; Ullrich, K.; Hoffmann, G.F.; Burgard, P.; Schmidt, H.; et al. A European Multicenter Study of Phenylalanine Hydroxylase Deficiency: Classification of 105 Mutations and a General System for Genotype-Based Prediction of Metabolic Phenotype. Am. J. Hum. Genet. 1998, 63, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, N. Genetics of Phenylketonuria: Then and Now. Hum. Mutat. 2016, 37, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Gersting, S.W.; Kemter, K.F.; Staudigl, M.; Messing, D.D.; Danecka, M.K.; Lagler, F.B.; Sommerhoff, C.P.; Roscher, A.A.; Muntau, A.C. Loss of Function in Phenylketonuria Is Caused by Impaired Molecular Motions and Conformational Instability. Am. J. Hum. Genet. 2008, 83, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Dworniczak, B.; Aulehla-Scholz, C.; Kalaydjieva, L.; Bartholomé, K.; Grudda, K.; Horst, J. Aberrant splicing of phenylalanine hydroxylase mRNA: The major cause for phenylketonuria in parts of southern Europe. Genomics 1991, 11, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Vela-Amieva, M.; Alcántara-Ortigoza, M.A.; Ibarra-González, I.; González-del Angel, A.; Fernández-Hernández, L.; Guillén-López, S.; López-Mejía, L.; Carrillo-Nieto, R.I.; Belmont-Martínez, L.; Fernández-Lainez, C. An Updated PAH Mutational Spectrum of Phenylketonuria in Mexican Patients Attending a Single Center: Biochemical, Clinical-Genotyping Correlations. Genes 2021, 12, 1676. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.; Azevedo, L.; Neiva, R.; Sousa, C.; Fonseca, H.; Marcão, A.; Rocha, H.; Carmona, C.; Ramos, S.; Bandeira, A.; et al. Phenylketonuria in Portugal: Genotype-phenotype correlations using molecular, biochemical, and haplotypic analyses. Mol. Genet. Genomic Med. 2021, 9, e1559. [Google Scholar] [CrossRef]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef]
- Danecka, M.K.; Woidy, M.; Zschocke, J.; Feillet, F.; Muntau, A.C.; Gersting, S.W. Mapping the functional landscape of frequent phenylalanine hydroxylase (PAH) genotypes promotes personalised medicine in phenylketonuria. J. Med. Genet. 2015, 52, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Heintz, C.; Cotton, R.G.; Blau, N. Tetrahydrobiopterin, its Mode of Action on Phenylalanine Hydroxylase, and Importance of Genotypes for Pharmacological Therapy of Phenylketonuria. Hum. Mutat. 2013, 34, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Zurflüh, M.R.; Zschocke, J.; Lindner, M.; Feillet, F.; Chery, C.; Burlina, A.; Stevens, R.C.; Thöny, B.; Blau, N. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008, 29, 167–175. [Google Scholar] [CrossRef]
- Shi, Z.; Sellers, J.; Moult, J. Protein stability and in vivo concentration of missense mutations in phenylalanine hydroxylase. Proteins Struct. Funct. Bioinform. 2011, 80, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Couce, M.; Bóveda, M.; Fernández-Marmiesse, A.; Mirás, A.; Pérez, B.; Desviat, L.; Fraga, J.M. Molecular epidemiology and BH4-responsiveness in patients with phenylalanine hydroxylase deficiency from Galicia region of Spain. Gene 2013, 521, 100–104. [Google Scholar] [CrossRef]
- Jeannesson-Thivisol, E.; Feillet, F.; Chéry, C.; Perrin, P.; Battaglia-Hsu, S.-F.; Herbeth, B.; Cano, A.; Barth, M.; Fouilhoux, A.; Mention, K.; et al. Genotype-phenotype associations in French patients with phenylketonuria and importance of genotype for full assessment of tetrahydrobiopterin responsiveness. Orphanet J. Rare Dis. 2015, 10, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enacán, R.E.; Miñana, M.N.; Fernandez, L.; Valle, M.G.; Salerno, M.; Fraga, C.I.; Santos-Simarro, F.; Prieto, L.; Lapunzina, P.; Specola, N.; et al. Phenylalanine Hydroxylase (PAH) Genotyping in PKU Argentine Patients. J. Inborn Errors Metab. Screen. 2019, 7, e20190012. [Google Scholar] [CrossRef] [Green Version]
- Gundorova, P.; Stepanova, A.A.; Kuznetsova, I.A.; Kutsev, S.I.; Polyakov, A.V. Genotypes of 2579 patients with phenylketonuria reveal a high rate of BH4 non-responders in Russia. PLoS ONE 2019, 14, e0211048. [Google Scholar] [CrossRef] [Green Version]
- Bik-Multanowski, M.; Kaluzny, L.; Mozrzymas, R.; Oltarzewski, M.; Starostecka, E.; Lange, A.; Didycz, B.; Giżewska, M.; Ulewicz-Filipowicz, J.; Chrobot, A.; et al. Molecular genetics of PKU in Poland and potential impact of mutations on BH4 responsiveness. Acta Biochim. Pol. 2013, 60, 613–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biglari, A.; Saffari, F.; Rashvand, Z.; Alizadeh, S.; Najafipour, R.; Sahmani, M. Mutations of the phenylalanine hydroxylase gene in Iranian patients with phenylketonuria. Springerplus 2015, 4, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintz, C.; Dobrowolski, S.F.; Andersen, H.S.; Demirkol, M.; Blau, N.; Andresen, B.S. Splicing of phenylalanine hydroxylase (PAH) exon 11 is vulnerable: Molecular pathology of mutations in PAH exon 11. Mol. Genet Metab. 2012, 106, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Shen, N.; Heintz, C.; Thiel, C.; Okun, J.G.; Hoffmann, G.F.; Blau, N. Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol. Genet. Metab. 2016, 117, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Heintz, C.; Troxler, H.; Martinez, A.; Thöny, B.; Blau, N. Quantification of phenylalanine hydroxylase activity by isotope-dilution liquid chromatography-electrospray ionization tandem mass spectrometry. Mol. Genet. Metab. 2012, 105, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Okano, Y.; Wang, T.; Eisensmith, R.C.; Longhi, R.; Riva, E.; Giovannini, M.; Cerone, R.; Romano, C.; Woo, S.L. Phenylketonuria missense mutations in the Mediterranean. Genomics 1991, 9, 96–103. [Google Scholar] [CrossRef]
- Wang, W.; Xin, B.; Wang, H. Dopa-Responsive Dystonia: A Male Patient Inherited a Novel GCH1 Deletion from an Asymptomatic Mother. J. Mov. Disord. 2020, 13, 150–153. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene | Cytogenetic Location | Number of Exons | Number of Amplicons | Pathology | Phenotype |
|---|---|---|---|---|---|
| OMIM No. | |||||
| PAH | 12q23.2 | 13 | 14 | Phenylketonuria (homozygous) | 261600 |
| Hyperphenylalaninemia, non-PKU mild (heterozygous) | |||||
| DNAJC12 | 10q21.3 | 6 | 8 | Hyperphenylalaninemia, mild, non-BH4-deficient | 617384 |
| QDPR | 4p15.32 | 7 | 7 | Hyperphenylalaninemia, BH4-deficient, C | 261630 |
| PTS | 11q23.1 | 6 | 7 | Hyperphenylalaninemia, BH4-deficient, A | 261640 |
| PCBD1 | 10q22.1 | 6 | 3 | Hyperphenylalaninemia, BH4-deficient, D | 264070 |
| GCH1 | 14q22.2 | 7 | 10 | Dystonia, DOPA-responsive, with or without hyperphenylalaninemia | 128230 |
| Hyperphenylalaninemia, BH4-deficient, B | 233910 | ||||
| SPR | 2p13.2 | 3 | 7 | Dystonia, dopa-responsive, due to sepiapterin reductase deficiency | 612716 |
| Nucleotide Aberration | Protein Change | Location | Type | dbSNP | Probands Carrying Variant |
|---|---|---|---|---|---|
| c.121C>T | L41F | E2 | Miss | rs62642928 | 1 |
| c.143T>C | L48S | E2 | Miss | rs5030841 | 1 |
| c.165delT | F55fs | E2 | Del | rs199475566 | 1 |
| c.442-5C>G | - | I4 | Unkn | rs62514909 | 1 |
| c.526C>T | R176 * | E6 | Non | rs199475575 | 2 |
| c.781C>T | R261 * | E7 | Non | rs5030850 | 1 |
| c.782G>A | R261Q | E7 | Miss | rs5030849 | 4 |
| c.842C>T | P281L | E7 | Miss | rs5030851 | 1 |
| c.848T>A | p.Ile283Asn | E8 | Miss | rs62508693 | 1 |
| c.898G>T | A300S | E8 | Miss | rs5030853 | 5 |
| c.1028A>G | Y343C | E10 | Miss | rs62507265 | 1 |
| c.1045T>C | S349P | E10 | Miss | rs62508646 | 1 |
| c.1066-11G>A | - | I10 | Splic | rs5030855 | 7 |
| c.1139C>T | T380M | E11 | Miss | rs62642937 | 1 |
| c.1169A>G | E390G | E11 | Miss | rs5030856 | 2 |
| c.1208C>T | A403V | E12 | Miss | rs5030857 | 4 |
| c.1241A>G | Y414C | E12 | Miss | rs5030860 | 1 |
| Proband | Sex | Age at On-Set/First Event | Phe Blood Conc. (μM) at Diagnosis | Detected Mutation | Detected Mutation | ||||
|---|---|---|---|---|---|---|---|---|---|
| Systematic Name | FI | MI | Systematic Name | FI | MI | ||||
| #1 | M | birth | 726 | c.782G>A | X | c.782G>A | X | ||
| #2 | F | birth | 678 | c.842C>T | n.a. | n.a. | c.1028A>G | n.a. | n.a. |
| #3 | M | birth | 119 | c.442-5C>G | n.a. | n.a. | c.1208C>T | n.a. | n.a. |
| #4 | F | birth | 1117 | c.1066-11G>A | n.a. | n.a. | c.165delT | n.a. | n.a. |
| #5 | M | birth | 400 | c.1066-11G>A | n.a. | n.a. | c.143T>C | n.a. | n.a. |
| #6 | F | birth | 154 | c.1241A>G | X | c.1139C>T | X | ||
| #7 | F | birth | 193 | c.898G>T | X | c.1066-11G>A | X | ||
| #8 | F | birth | 514 | c.526C>T | X | c.1169A>G | X | ||
| #9 | M | birth | 115 | c.1169A>G | n.a. | c.1208C>T | X | ||
| #10 | M | birth | 194 | c.898G>T | X | c.1066-11G>A | X | ||
| #11 | M | birth | 363 | c.526C>T | n.a. | n.a. | c.781C>T | n.a. | n.a. |
| #12 | F | birth | 1930 | c.121C>T | n.a. | n.a. | c.782G>A | n.a. | n.a. |
| #13 | F | birth | 1271 | c.1045T>C | X | c.1066-11G>A | X | ||
| #14 | F | birth | 173 | c.782G>A | X | c.898G>T | X | ||
| #15 | M | birth | 224 | c.898G>T | X | c.1066-11G>A | X | ||
| #16 | F | birth | 701 | c.782G>A | X | c.1066-11G>A | X | ||
| #17 | M | birth | 149 | c.898G>T | X | c.1208C>T | X | ||
| #18 | F | birth | 242 | c.848T>A | X | c.1208C>T | n.a. | ||
| Locus | Length | Gene | CNV | Exon(s) | Functional Interpretation (ClinVar) | Proband(s) |
|---|---|---|---|---|---|---|
| chr 2:73114493-73114894 | 401 | SPR | Heterozygous | 1 | Pathogenic (SPR deficiency; Dopa-responsive dystonia) | #9 |
| Deletion | ||||||
| chr12:103240590-103260521 | 19,931 | PAH | Heterozygous | 5, 8–9 | Pathogenic | #8, #16 |
| Deletion | (HPA) | |||||
| chr10:69556778-69597930 | 41,152 | DNAJC12 | Heterozygous | 4–5 | Pathogenic | #8 |
| Deletion | (HPA, mild, non-BH4-deficient) | |||||
| chr10:69582964-69597930 | 14,966 | DNAJC12 | Heterozygous | 1–2 | Pathogenic | #8, #16 |
| Deletion | (HPA, mild, non-BH4-deficient) | |||||
| chr14:55312420-55313886 | 1466 | GCH1 | Heterozygous | 4–5 | Pathogenic | #8 |
| Deletion | (GTP cyclohydrolase I deficiency) | |||||
| chr4:17488690-17488918 | 228 | QDPR | Heterozygous/Homozygous Deletion | 7 | Uncertain significance (BH4-deficient HPA) | #8, #16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tendi, E.A.; Morello, G.; Guarnaccia, M.; La Cognata, V.; Petralia, S.; Messina, M.A.; Meli, C.; Fiumara, A.; Ruggieri, M.; Cavallaro, S. Detection of Single-Nucleotide and Copy Number Defects Underlying Hyperphenylalaninemia by Next-Generation Sequencing. Biomedicines 2023, 11, 1899. https://doi.org/10.3390/biomedicines11071899
Tendi EA, Morello G, Guarnaccia M, La Cognata V, Petralia S, Messina MA, Meli C, Fiumara A, Ruggieri M, Cavallaro S. Detection of Single-Nucleotide and Copy Number Defects Underlying Hyperphenylalaninemia by Next-Generation Sequencing. Biomedicines. 2023; 11(7):1899. https://doi.org/10.3390/biomedicines11071899
Chicago/Turabian StyleTendi, Elisabetta Anna, Giovanna Morello, Maria Guarnaccia, Valentina La Cognata, Salvatore Petralia, Maria Anna Messina, Concetta Meli, Agata Fiumara, Martino Ruggieri, and Sebastiano Cavallaro. 2023. "Detection of Single-Nucleotide and Copy Number Defects Underlying Hyperphenylalaninemia by Next-Generation Sequencing" Biomedicines 11, no. 7: 1899. https://doi.org/10.3390/biomedicines11071899