
NAFLD and AATD Are Two Diseases with Unbalanced Lipid Metabolism: Similarities and Differences
 ,
, 
Abstract
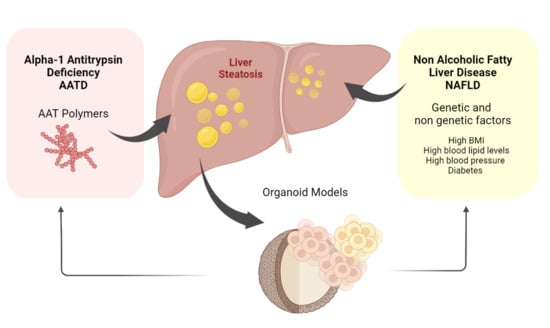
{kind=link}
{kind=link}
1. Non-Alcoholic Fatty Liver Disease (NAFLD)
2. Alpha-1 Antitrypsin Deficiency (AATD)
3. Meta-Inflammation in NAFLD and AATD
4. Features of Lipid Metabolism in NAFLD and AATD
5. Relationships between NAFLD, AATD, and Chronic Obstructive Pulmonary Disease (COPD)
6. Organoids to Model Liver Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatol. Baltim. Md. 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Qiu, S.; Zhao, Y.; Zhao, S.; Sun, D.; Hou, L.; Li, Y.; Zhou, K.; Yu, X.; Yang, C.; et al. Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7841. [Google Scholar] [CrossRef] [PubMed]
- Liebe, R.; Esposito, I.; Bock, H.H.; Vom Dahl, S.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and Management of Secondary Causes of Steatohepatitis. J. Hepatol. 2021, 74, 1455–1471. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatol. Baltim. Md. 2021, 73, 1194–1198. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Hadizadeh, F.; Faghihimani, E.; Adibi, P. Nonalcoholic Fatty Liver Disease: Diagnostic Biomarkers. World J. Gastrointest. Pathophysiol. 2017, 8, 11–26. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Tilg, H. NAFLD and Increased Risk of Cardiovascular Disease: Clinical Associations, Pathophysiological Mechanisms and Pharmacological Implications. Gut 2020, 69, 1691–1705. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated Endotoxin Levels in Non-Alcoholic Fatty Liver Disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [PubMed]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic Lipids Stored by Kupffer Cells Correlates with Their Pro-Inflammatory Phenotype at an Early Stage of Steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V. Lipid Droplets And Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic Lipid Droplets: A Balancing Act between Energy Storage and Metabolic Dysfunction in NAFLD. Mol. Metab. 2021, 50, 101115. [Google Scholar] [CrossRef]
- Yang, H.; Liu, J. Chapter 11-Structure and Function of Lipid Droplets. In Biochemistry of Lipids, Lipoproteins and Membranes, 7th ed.; Ridgway, N.D., McLeod, R.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 357–394. ISBN 978-0-12-824048-9. [Google Scholar]
- Schmidt, A.F.; Joshi, R.; Gordillo-Marañón, M.; Drenos, F.; Charoen, P.; Giambartolomei, C.; Bis, J.C.; Gaunt, T.R.; Hughes, A.D.; Lawlor, D.A.; et al. Biomedical Consequences of Elevated Cholesterol-Containing Lipoproteins and Apolipoproteins on Cardiovascular and Non-Cardiovascular Outcomes. Commun. Med. 2023, 3, 9. [Google Scholar] [CrossRef]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The Discovery of A1-Antitrypsin and Its Role in Health and Disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- Seixas, S.; Marques, P.I. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl. Clin. Genet. 2021, 14, 173–194. [Google Scholar] [CrossRef]
- Ogushi, F.; Fells, G.A.; Hubbard, R.C.; Straus, S.D.; Crystal, R.G. Z-Type Alpha 1-Antitrypsin Is Less Competent than M1-Type Alpha 1-Antitrypsin as an Inhibitor of Neutrophil Elastase. J. Clin. Investig. 1987, 80, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- de Serres, F.J.; Blanco, I. Prevalence of A1-Antitrypsin Deficiency Alleles PI*S and PI*Z Worldwide and Effective Screening for Each of the Five Phenotypic Classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A Comprehensive Review. Ther. Adv. Respir. Dis. 2012, 6, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Hawkins, H.K.; Finegold, M.J.; Woo, S.L. Multiple Tissues Express Alpha 1-Antitrypsin in Transgenic Mice and Man. J. Clin. Investig. 1988, 82, 26–36. [Google Scholar] [CrossRef]
- Sinden, N.J.; Baker, M.J.; Smith, D.J.; Kreft, J.-U.; Dafforn, T.R.; Stockley, R.A. α-1-Antitrypsin Variants and the Proteinase/Antiproteinase Imbalance in Chronic Obstructive Pulmonary Disease. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 308, L179–L190. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; Lipsker, D.; Lara, B.; Janciauskiene, S. Neutrophilic Panniculitis Associated with Alpha-1-Antitrypsin Deficiency: An Update. Br. J. Dermatol. 2016, 174, 753–762. [Google Scholar] [CrossRef]
- Sun, R.; Xu, Z.; Zhu, C.; Chen, T.; Muñoz, L.E.; Dai, L.; Zhao, Y. Alpha-1 Antitrypsin in Autoimmune Diseases: Roles and Therapeutic Prospects. Int. Immunopharmacol. 2022, 110, 109001. [Google Scholar] [CrossRef]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The Mechanism of Z Alpha 1-Antitrypsin Accumulation in the Liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Stoller, J.K.; Aboussouan, L.S. A Review of A1-Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259. [Google Scholar] [CrossRef]
- Santos, G.; Turner, A.M. Alpha-1 Antitrypsin Deficiency: An Update on Clinical Aspects of Diagnosis and Management. Fac. Rev. 2020, 9, 1. [Google Scholar] [CrossRef]
- Bouchecareilh, M. Alpha-1 Antitrypsin Deficiency-Mediated Liver Toxicity: Why Do Some Patients Do Poorly? What Do We Know So Far? Chronic Obstr. Pulm. Dis. J. COPD Found. 2020, 7, 172–181. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, L.R.; Lee, J.; Mohammad, N.S.; Aranyos, A.M.; Gould, C.; Khodayari, N.; Oshins, R.A.; Moneypenny, C.G.; Brantly, M.L. The Unfolded Protein Response to PI*Z Alpha-1 Antitrypsin in Human Hepatocellular and Murine Models. Hepatol. Commun. 2022, 6, 2354–2367. [Google Scholar] [CrossRef] [PubMed]
- Mela, M.; Smeeton, W.; Davies, S.E.; Miranda, E.; Scarpini, C.; Coleman, N.; Alexander, G.J.M. The Alpha-1 Antitrypsin Polymer Load Correlates With Hepatocyte Senescence, Fibrosis Stage and Liver-Related Mortality. Chronic Obstr. Pulm. Dis. Miami Fla 2020, 7, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Hupertz, V.; Aboussouan, L.S. Alpha-1 Antitrypsin Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Topic, A.; Alempijevic, T.; Milutinovic, A.S.; Kovacevic, N. Alpha-1-Antitrypsin Phenotypes in Adult Liver Disease Patients. Ups. J. Med. Sci. 2009, 114, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Hamesch, K.; Mandorfer, M.; Pereira, V.M.; Moeller, L.S.; Pons, M.; Dolman, G.E.; Reichert, M.C.; Schneider, C.V.; Woditsch, V.; Voss, J.; et al. Liver Fibrosis and Metabolic Alterations in Adults With Alpha-1-Antitrypsin Deficiency Caused by the Pi*ZZ Mutation. Gastroenterology 2019, 157, 705–719.e18. [Google Scholar] [CrossRef] [PubMed]
- Khodayari, N.; Wang, R.L.; Oshins, R.; Lu, Y.; Millett, M.; Aranyos, A.M.; Mostofizadeh, S.; Scindia, Y.; Flagg, T.O.; Brantly, M. The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease. Int. J. Mol. Sci. 2021, 22, 13255. [Google Scholar] [CrossRef]
- Winther, S.V.; Ahmed, D.; Al-Shuweli, S.; Landt, E.M.; Nordestgaard, B.G.; Seersholm, N.; Dahl, M. Severe A1-Antitrypsin Deficiency Associated with Lower Blood Pressure and Reduced Risk of Ischemic Heart Disease: A Cohort Study of 91,540 Individuals and a Meta-Analysis. Respir. Res. 2022, 23, 55. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Obin, M.S. Obesity and the Role of Adipose Tissue in Inflammation and Metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 Contributes to Macrophage Infiltration into Adipose Tissue, Insulin Resistance, and Hepatic Steatosis in Obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W. CCR2 Modulates Inflammatory and Metabolic Effects of High-Fat Feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.-M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two Distinct Interstitial Macrophage Populations Coexist across Tissues in Specific Subtissular Niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage Polarization and Metainflammation. Transl. Res. J. Lab. Clin. Med. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and Pathogenic Functions of Macrophage Subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Alternative Macrophage Activation and Metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.-P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic Macrophages but Not Dendritic Cells Contribute to Liver Fibrosis by Promoting the Survival of Activated Hepatic Stellate Cells in Mice. Hepatol. Baltim. Md. 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Rautou, P.-E. Role of Liver Sinusoidal Endothelial Cells in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef]
- Pigeolet, E.; Corbisier, P.; Houbion, A.; Lambert, D.; Michiels, C.; Raes, M.; Zachary, M.-D.; Remacle, J. Glutathione Peroxidase, Superoxide Dismutase, and Catalase Inactivation by Peroxides and Oxygen Derived Free Radicals. Mech. Ageing Dev. 1990, 51, 283–297. [Google Scholar] [CrossRef]
- Clare, K.; Dillon, J.F.; Brennan, P.N. Reactive Oxygen Species and Oxidative Stress in the Pathogenesis of MAFLD. J. Clin. Transl. Hepatol. 2022, 10, 939–946. [Google Scholar] [CrossRef]
- Song, M.J.; Malhi, H. The Unfolded Protein Response and Hepatic Lipid Metabolism in Non Alcoholic Fatty Liver Disease. Pharmacol. Ther. 2019, 203, 107401. [Google Scholar] [CrossRef]
- Febbraio, M.; Guy, E.; Coburn, C.; Knapp, F.F.; Beets, A.L.; Abumrad, N.A.; Silverstein, R.L. The Impact of Overexpression and Deficiency of Fatty Acid Translocase (FAT)/CD36. Mol. Cell. Biochem. 2002, 239, 193–197. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Rey, E.; del Pozo-Maroto, E.; Marañón, P.; Beeler, B.; García-García, Y.; Landete, P.; Isaza, S.C.; Farré, R.; García-Monzón, C.; Almendros, I.; et al. Intrahepatic Expression of Fatty Acid Translocase CD36 Is Increased in Obstructive Sleep Apnea. Front. Med. 2020, 7, 450. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, A.M.; O’Neill, S.J.; McElvaney, N.G. Evidence for Unfolded Protein Response Activation in Monocytes from Individuals with Alpha-1 Antitrypsin Deficiency. J. Immunol. Baltim. Md 1950 2010, 184, 4538–4546. [Google Scholar] [CrossRef]
- Bazzan, E.; Tinè, M.; Biondini, D.; Benetti, R.; Baraldo, S.; Turato, G.; Fagiuoli, S.; Sonzogni, A.; Rigobello, C.; Rea, F.; et al. A1-Antitrypsin Polymerizes in Alveolar Macrophages of Smokers With and Without A1-Antitrypsin Deficiency. Chest 2018, 154, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Belchamber, K.B.R.; Walker, E.M.; Stockley, R.A.; Sapey, E. Monocytes and Macrophages in Alpha-1 Antitrypsin Deficiency. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Khodayari, N.; Oshins, R.; Aranyos, A.M.; Duarte, S.; Mostofizadeh, S.; Lu, Y.; Brantly, M. Characterization of Hepatic Inflammatory Changes in a C57BL/6J Mouse Model of Alpha1-Antitrypsin Deficiency. Am. J. Physiol.-Gastrointest. Liver Physiol. 2022, 323, G594–G608. [Google Scholar] [CrossRef]
- Daemen, S.; Gainullina, A.; Kalugotla, G.; He, L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2021, 34, 108626. [Google Scholar] [CrossRef]
- Pott, G.B.; Chan, E.D.; Dinarello, C.A.; Shapiro, L. Alpha-1-Antitrypsin Is an Endogenous Inhibitor of Proinflammatory Cytokine Production in Whole Blood. J. Leukoc. Biol. 2009, 85, 886–895. [Google Scholar] [CrossRef]
- Lee, J.; Mohammad, N.; Lu, Y.; Kang, K.; Han, K.; Brantly, M. Alu RNA Induces NLRP3 Expression through TLR7 Activation in α-1-Antitrypsin-Deficient Macrophages. JCI Insight 2022, 7, e158791. [Google Scholar] [CrossRef]
- Callea, F.; Francalanci, P.; Giovannoni, I. Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases. Int. J. Mol. Sci. 2021, 22, 5778. [Google Scholar] [CrossRef]
- Anand, P.K. Lipids, Inflammasomes, Metabolism, and Disease. Immunol. Rev. 2020, 297, 108–122. [Google Scholar] [CrossRef]
- Krahmer, N.; Farese, R.V.; Walther, T.C. Balancing the Fat: Lipid Droplets and Human Disease. EMBO Mol. Med. 2013, 5, 905–915. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD). European Association for the Study of Obesity (EASO) EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Tulenko, T. The Physiology of Lipoproteins. J. Nucl. Cardiol. 2002, 9, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Diffenderfer, M.R.; Schaefer, E.J. The Composition and Metabolism of Large and Small LDL. Curr. Opin. Lipidol. 2014, 25, 221–226. [Google Scholar] [CrossRef]
- Olofsson, S.O.; Stillemark-Billton, P.; Asp, L. Intracellular Assembly of VLDL: Two Major Steps in Separate Cell Compartments. Trends Cardiovasc. Med. 2000, 10, 338–345. [Google Scholar] [CrossRef]
- Stillemark-Billton, P.; Beck, C.; Borén, J.; Olofsson, S.-O. Relation of the Size and Intracellular Sorting of ApoB to the Formation of VLDL 1 and VLDL 2. J. Lipid Res. 2005, 46, 104–114. [Google Scholar] [CrossRef]
- Björkegren, J.; Beigneux, A.; Bergo, M.O.; Maher, J.J.; Young, S.G. Blocking the Secretion of Hepatic Very Low Density Lipoproteins Renders the Liver More Susceptible to Toxin-Induced Injury. J. Biol. Chem. 2002, 277, 5476–5483. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.-O.; Taskinen, M.-R.; Borén, J. Overproduction of Very Low-Density Lipoproteins Is the Hallmark of the Dyslipidemia in the Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1225–1236. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744.e7. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Ståhlman, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of TM6SF2 E167K on Hepatic Lipid and Very Low-Density Lipoprotein Metabolism in Humans. JCI Insight 2020, 5, e144079. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Henricsson, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of PNPLA3 I148M on Hepatic Lipid and Very-Low-Density Lipoprotein Metabolism in Humans. J. Intern. Med. 2022, 291, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K Genetic Variant Induces Lipid Biosynthesis and Reduces Apolipoprotein B Secretion in Human Hepatic 3D Spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef] [PubMed]
- Obermayer, G.; Afonyushkin, T.; Binder, C.J. Oxidized Low-Density Lipoprotein in Inflammation-Driven Thrombosis. J. Thromb. Haemost. 2018, 16, 418–428. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger Receptors Class A-I/II and CD36 Are the Principal Receptors Responsible for the Uptake of Modified Low Density Lipoprotein Leading to Lipid Loading in Macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Florance, I.; Ramasubbu, S. Current Understanding on the Role of Lipids in Macrophages and Associated Diseases. Int. J. Mol. Sci. 2022, 24, 589. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 Links Hyperlipidemia, Oxidant Stress and a Prothrombotic Phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef]
- He, J.; Lee, J.H.; Febbraio, M.; Xie, W. The Emerging Roles of Fatty Acid Translocase/CD36 and the Aryl Hydrocarbon Receptor in Fatty Liver Disease. Exp. Biol. Med. Maywood NJ 2011, 236, 1116–1121. [Google Scholar] [CrossRef]
- Bonen, A.; Chabowski, A.; Luiken, J.J.F.P.; Glatz, J.F.C. Is Membrane Transport of FFA Mediated by Lipid, Protein, or Both? Mechanisms and Regulation of Protein-Mediated Cellular Fatty Acid Uptake: Molecular, Biochemical, and Physiological Evidence. Physiol. Bethesda Md 2007, 22, 15–29. [Google Scholar] [CrossRef]
- Devereux, C.J.; Bayliss, J.; Keenan, S.N.; Montgomery, M.K.; Watt, M.J. Investigating Dual Inhibition of ACC and CD36 for the Treatment of Non-Alcoholic Fatty Liver Disease in Mice. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E187–E198. [Google Scholar] [CrossRef] [PubMed]
- Samovski, D.; Abumrad, N.A. Regulation of Lipophagy in NAFLD by Cellular Metabolism and CD36. J. Lipid Res. 2019, 60, 755–757. [Google Scholar] [CrossRef]
- Feng, Y.; Sun, W.; Sun, F.; Yin, G.; Liang, P.; Chen, S.; Liu, X.; Jiang, T.; Zhang, F. Biological Mechanisms and Related Natural Inhibitors of CD36 in Nonalcoholic Fatty Liver. Drug Des. Devel. Ther. 2022, 16, 3829–3845. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 Plays a Negative Role in the Regulation of Lipophagy in Hepatocytes through an AMPK-Dependent Pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotti, I.; Corsico, A.G.; Stolk, J.; Ottaviani, S.; Fumagalli, M.; Janciauskiene, S.; Iadarola, P. Advances in Identifying Urine/Serum Biomarkers in Alpha-1 Antitrypsin Deficiency for More Personalized Future Treatment Strategies. COPD 2017, 14, 56–65. [Google Scholar] [CrossRef]
- Siebers, K.; Fink, B.; Zakrzewicz, A.; Agné, A.; Richter, K.; Konzok, S.; Hecker, A.; Zukunft, S.; Küllmar, M.; Klein, J.; et al. Alpha-1 Antitrypsin Inhibits ATP-Mediated Release of Interleukin-1β via CD36 and Nicotinic Acetylcholine Receptors. Front. Immunol. 2018, 9, 877. [Google Scholar] [CrossRef]
- Nati, M.; Haddad, D.; Birkenfeld, A.L.; Koch, C.A.; Chavakis, T.; Chatzigeorgiou, A. The Role of Immune Cells in Metabolism-Related Liver Inflammation and Development of Non-Alcoholic Steatohepatitis (NASH). Rev. Endocr. Metab. Disord. 2016, 17, 29–39. [Google Scholar] [CrossRef]
- Moon, S.W.; Kim, S.Y.; Jung, J.Y.; Kang, Y.A.; Park, M.S.; Kim, Y.S.; Chang, J.; Ro, J.S.; Lee, Y.-H.; Lee, S.H. Relationship between Obstructive Lung Disease and Non-Alcoholic Fatty Liver Disease in the Korean Population: Korea National Health and Nutrition Examination Survey, 2007–2010. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 2603–2611. [Google Scholar] [CrossRef]
- Viglino, D.; Plazanet, A.; Bailly, S.; Benmerad, M.; Jullian-Desayes, I.; Tamisier, R.; Leroy, V.; Zarski, J.-P.; Maignan, M.; Joyeux-Faure, M.; et al. Impact of Non-Alcoholic Fatty Liver Disease on Long-Term Cardiovascular Events and Death in Chronic Obstructive Pulmonary Disease. Sci. Rep. 2018, 8, 16559. [Google Scholar] [CrossRef]
- Viglino, D.; Jullian-Desayes, I.; Minoves, M.; Aron-Wisnewsky, J.; Leroy, V.; Zarski, J.-P.; Tamisier, R.; Joyeux-Faure, M.; Pépin, J.-L. Nonalcoholic Fatty Liver Disease in Chronic Obstructive Pulmonary Disease. Eur. Respir. J. 2017, 49, 1601923. [Google Scholar] [CrossRef]
- Rodríguez-Roisin, R.; Krowka, M.J. Hepatopulmonary Syndrome--a Liver-Induced Lung Vascular Disorder. N. Engl. J. Med. 2008, 358, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Cheeney, G.; Pac, L.J.; Gopal, P.; Landis, C.S.; Konnick, E.Q.; Swanson, P.E.; Greene, D.N.; Lockwood, C.M.; Westerhoff, M. Increased Frequency of Heterozygous Alpha-1-Antitrypsin Deficiency in Liver Explants From Nonalcoholic Steatohepatitis Patients. Liver Transpl. 2020, 26, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lockett, A.D.; Kimani, S.; Ddungu, G.; Wrenger, S.; Tuder, R.M.; Janciauskiene, S.M.; Petrache, I. A₁-Antitrypsin Modulates Lung Endothelial Cell Inflammatory Responses to TNF-α. Am. J. Respir. Cell Mol. Biol. 2013, 49, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Madyaningrana, K.; Vijayan, V.; Nikolin, C.; Aljabri, A.; Tumpara, S.; Korenbaum, E.; Shah, H.; Stankov, M.; Fuchs, H.; Janciauskiene, S.; et al. Alpha1-Antitrypsin Counteracts Heme-Induced Endothelial Cell Inflammatory Activation, Autophagy Dysfunction and Death. Redox Biol. 2021, 46, 102060. [Google Scholar] [CrossRef]
- Nakagiri, T.; Wrenger, S.; Sivaraman, K.; Ius, F.; Goecke, T.; Zardo, P.; Grau, V.; Welte, T.; Haverich, A.; Knöfel, A.-K.; et al. A1-Antitrypsin Attenuates Acute Rejection of Orthotopic Murine Lung Allografts. Respir. Res. 2021, 22, 295. [Google Scholar] [CrossRef]
- Jedicke, N.; Struever, N.; Aggrawal, N.; Welte, T.; Manns, M.P.; Malek, N.P.; Zender, L.; Janciauskiene, S.; Wuestefeld, T. α-1-Antitrypsin Inhibits Acute Liver Failure in Mice. Hepatol. Baltim. Md. 2014, 59, 2299–2308. [Google Scholar] [CrossRef]
- Grander, C.; Schaefer, B.; Schwärzler, J.; Grabherr, F.; de Graaf, D.M.; Enrich, B.; Oberhuber, G.; Mayr, L.; Sangineto, M.; Jaschke, N.; et al. Alpha-1 Antitrypsin Governs Alcohol-Related Liver Disease in Mice and Humans. Gut 2021, 70, 585–594. [Google Scholar] [CrossRef]
- Cho, J.-H.; Ryu, H.-M.; Oh, E.-J.; Yook, J.-M.; Ahn, J.-S.; Jung, H.-Y.; Choi, J.-Y.; Park, S.-H.; Kim, Y.-L.; Kwak, I.S.; et al. Alpha1-Antitrypsin Attenuates Renal Fibrosis by Inhibiting TGF-Β1-Induced Epithelial Mesenchymal Transition. PLoS ONE 2016, 11, e0162186. [Google Scholar] [CrossRef]
- Kalis, M.; Kumar, R.; Janciauskiene, S.; Salehi, A.; Cilio, C.M. α 1-Antitrypsin Enhances Insulin Secretion and Prevents Cytokine-Mediated Apoptosis in Pancreatic β-Cells. Islets 2010, 2, 185–189. [Google Scholar] [CrossRef]
- Abiru, S.; Migita, K.; Maeda, Y.; Daikoku, M.; Ito, M.; Ohata, K.; Nagaoka, S.; Matsumoto, T.; Takii, Y.; Kusumoto, K.; et al. Serum Cytokine and Soluble Cytokine Receptor Levels in Patients with Non-Alcoholic Steatohepatitis. Liver Int. Off. J. Int. Assoc. Study Liver 2006, 26, 39–45. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Huch, M. Disease Modelling in Human Organoids. Dis. Model. Mech. 2019, 12, dmm039347. [Google Scholar] [CrossRef] [PubMed]
- Marsee, A.; Roos, F.J.M.; Verstegen, M.M.A.; Gehart, H.; de Koning, E.; Lemaigre, F.; Forbes, S.J.; Peng, W.C.; Huch, M.; Takebe, T.; et al. Building Consensus on Definition and Nomenclature of Hepatic, Pancreatic, and Biliary Organoids. Cell Stem Cell 2021, 28, 816–832. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, C.N.; Singhania, R. A Review of Protocols for Brain Organoids and Applications for Disease Modeling. STAR Protoc. 2022, 4, 101860. [Google Scholar] [CrossRef] [PubMed]
- Yanakiev, M.; Soper, O.; Berg, D.A.; Kang, E. Modelling Alzheimer’s Disease Using Human Brain Organoids: Current Progress and Challenges. Expert Rev. Mol. Med. 2022, 25, e3. [Google Scholar] [CrossRef]
- Harada, K.; Sakamoto, N. Cancer Organoid Applications to Investigate Chemotherapy Resistance. Front. Mol. Biosci. 2022, 9, 1067207. [Google Scholar] [CrossRef]
- Riedel, N.C.; de Faria, F.W.; Alfert, A.; Bruder, J.M.; Kerl, K. Three-Dimensional Cell Culture Systems in Pediatric and Adult Brain Tumor Precision Medicine. Cancers 2022, 14, 5972. [Google Scholar] [CrossRef]
- Chen, J.; Na, F. Organoid Technology and Applications in Lung Diseases: Models, Mechanism Research and Therapy Opportunities. Front. Bioeng. Biotechnol. 2022, 10, 1066869. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321. [Google Scholar] [CrossRef]
- Peng, L.; Gao, L.; Wu, X.; Fan, Y.; Liu, M.; Chen, J.; Song, J.; Kong, J.; Dong, Y.; Li, B.; et al. Lung Organoids as Model to Study SARS-CoV-2 Infection. Cells 2022, 11, 2758. [Google Scholar] [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to Model Liver Disease. JHEP Rep. Innov. Hepatol. 2021, 3, 100198. [Google Scholar] [CrossRef]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; Bowen, W.C.; Mulè, K.; Stolz, D.B. Histological Organization in Hepatocyte Organoid Cultures. Am. J. Pathol. 2001, 159, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.W.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In Vitro Expansion of Single Lgr5+ Liver Stem Cells Induced by Wnt-Driven Regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and Functional Human Liver from an IPSC-Derived Organ Bud Transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver Organoids: From Basic Research to Therapeutic Applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef]
- Gómez-Mariano, G.; Matamala, N.; Martínez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzón, S.; Cuesta, I.; Garfia, C.; Martínez, M.T.; et al. Liver Organoids Reproduce Alpha-1 Antitrypsin Deficiency-Related Liver Disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Tao, T.; Liu, H.; Wu, S.; Chen, W.; Qin, J. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743. [Google Scholar] [CrossRef]
- Carter, J.K.; Friedman, S.L. Hepatic Stellate Cell-Immune Interactions in NASH. Front. Endocrinol. 2022, 13, 867940. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yu, H.; Li, Q.-Y.; Wei, Y.-T.; Fu, J.; Dong, H.; Cao, D.; Guo, L.-N.; Chen, L.; Yang, Y.; et al. Hepatocyte-Derived VEGFA Accelerates the Progression of Non-Alcoholic Fatty Liver Disease to Hepatocellular Carcinoma via Activating Hepatic Stellate Cells. Acta Pharmacol. Sin. 2022, 43, 2917–2928. [Google Scholar] [CrossRef]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Lindén, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [PubMed]
- Gwag, T.; Ma, E.; Zhou, C.; Wang, S. Anti-CD47 Antibody Treatment Attenuates Liver Inflammation and Fibrosis in Experimental Non-Alcoholic Steatohepatitis Models. Liver Int. Off. J. Int. Assoc. Study Liver 2022, 42, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Sendi, H.; Mead, I.; Wan, M.; Mehrab-Mohseni, M.; Koch, K.; Atala, A.; Bonkovsky, H.L.; Bishop, C.E. MiR-122 Inhibition in a Human Liver Organoid Model Leads to Liver Inflammation, Necrosis, Steatofibrosis and Dysregulated Insulin Signaling. PLoS ONE 2018, 13, e0200847. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef] [PubMed]
- Collin de l’Hortet, A.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401.e9. [Google Scholar] [CrossRef]
- Uygun, B.E.; Soto-Gutierrez, A.; Yagi, H.; Izamis, M.-L.; Guzzardi, M.A.; Shulman, C.; Milwid, J.; Kobayashi, N.; Tilles, A.; Berthiaume, F.; et al. Organ Reengineering through Development of a Transplantable Recellularized Liver Graft Using Decellularized Liver Matrix. Nat. Med. 2010, 16, 814–820. [Google Scholar] [CrossRef]
- Park, Y.; Thadasina, D.; Bolujo, I.; Isidan, A.; Cross-Najafi, A.A.; Lopez, K.; Li, P.; Dahlem, A.M.; Kennedy, L.; Sato, K.; et al. Three-Dimensional Organoids as a Model to Study Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2022, 42, 423–433. [Google Scholar] [CrossRef]
- Anant, S.; Davidson, N.O. Identification and Regulation of Protein Components of the Apolipoprotein B MRNA Editing Enzyme. A Complex Event. Trends Cardiovasc. Med. 2002, 12, 311–317. [Google Scholar] [CrossRef]
- Schneider, C.V.; Hamesch, K.; Gross, A.; Mandorfer, M.; Moeller, L.S.; Pereira, V.; Pons, M.; Kuca, P.; Reichert, M.C.; Benini, F.; et al. Liver Phenotypes of European Adults Heterozygous or Homozygous for Pi∗Z Variant of AAT (Pi∗MZ vs Pi∗ZZ Genotype) and Noncarriers. Gastroenterology 2020, 159, 534–548.e11. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Luz, S.; Matamala, N.; Gomez-Mariano, G.; Janciauskiene, S.; Martínez-Delgado, B. NAFLD and AATD Are Two Diseases with Unbalanced Lipid Metabolism: Similarities and Differences. Biomedicines 2023, 11, 1961. https://doi.org/10.3390/biomedicines11071961
Perez-Luz S, Matamala N, Gomez-Mariano G, Janciauskiene S, Martínez-Delgado B. NAFLD and AATD Are Two Diseases with Unbalanced Lipid Metabolism: Similarities and Differences. Biomedicines. 2023; 11(7):1961. https://doi.org/10.3390/biomedicines11071961
Chicago/Turabian StylePerez-Luz, Sara, Nerea Matamala, Gema Gomez-Mariano, Sabina Janciauskiene, and Beatriz Martínez-Delgado. 2023. "NAFLD and AATD Are Two Diseases with Unbalanced Lipid Metabolism: Similarities and Differences" Biomedicines 11, no. 7: 1961. https://doi.org/10.3390/biomedicines11071961
APA StylePerez-Luz, S., Matamala, N., Gomez-Mariano, G., Janciauskiene, S., & Martínez-Delgado, B. (2023). NAFLD and AATD Are Two Diseases with Unbalanced Lipid Metabolism: Similarities and Differences. Biomedicines, 11(7), 1961. https://doi.org/10.3390/biomedicines11071961