

Computational Guided Drug Targets Identification against Extended-Spectrum Beta-Lactamase-Producing Multi-Drug Resistant Uropathogenic Escherichia coli

Abstract
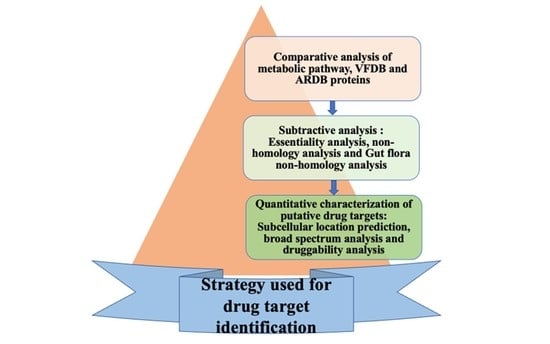
1. Introduction
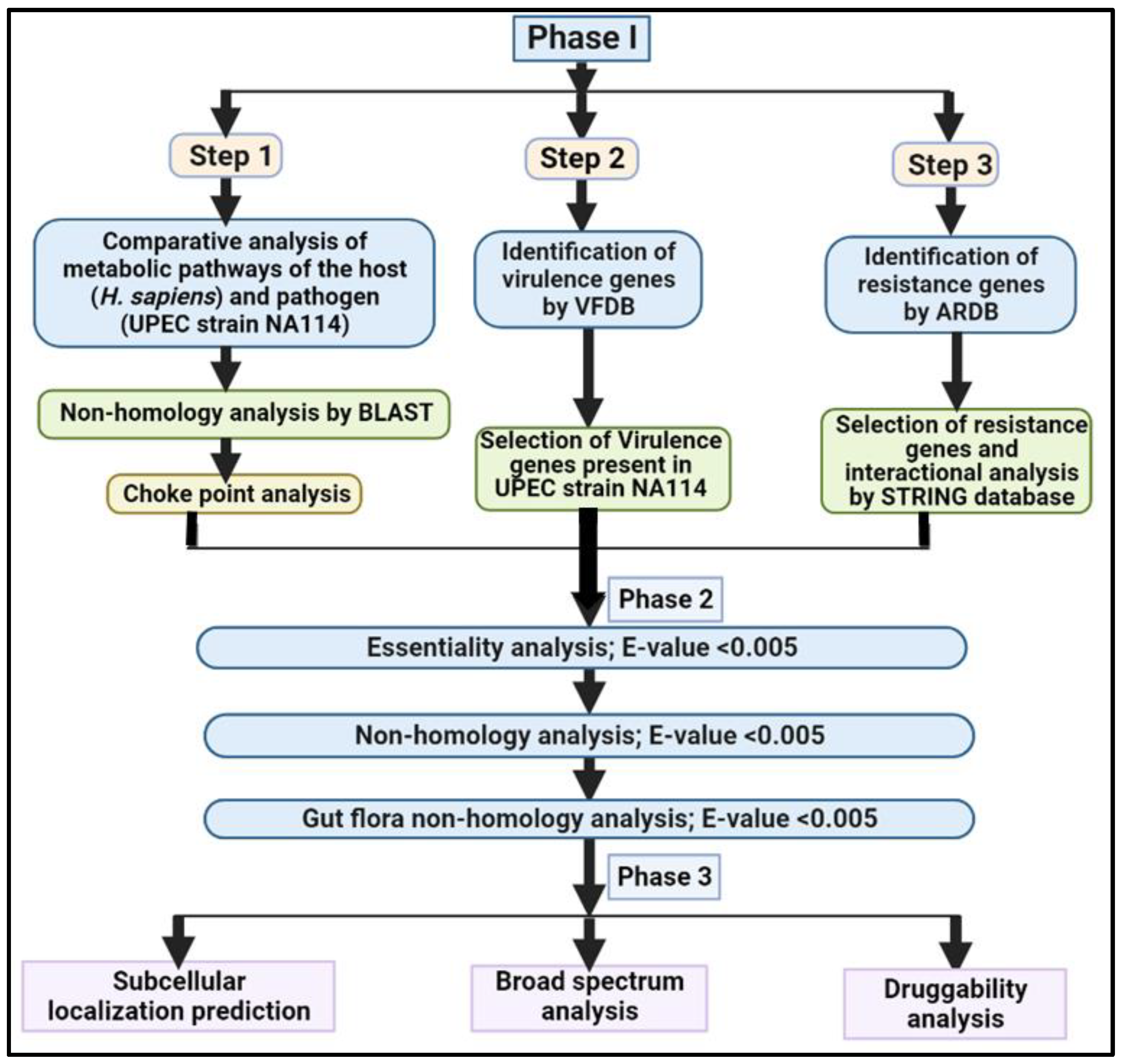
2. Material and Methods
2.1. Phase I: Comparative Analysis of Pathogen and Human Proteome
Step1: Non-Homology Analysis
2.2. Chokepoint Analysis
2.2.1. Step 2: Analysis of Virulence Genes
2.2.2. Step 3: Analysis of Resistance Genes
2.3. Phase II: Subtractive Analysis
2.3.1. Analysis of Essential Genes
2.3.2. Non-homology Analysis
2.3.3. Human Gut Flora Non-homology Analysis
2.4. Phase III: Quantitative Characterization of Putative Drug Targets
2.4.1. Subcellular Localization Prediction
2.4.2. Broad-Spectrum Analysis
2.4.3. Druggability Analysis
3. Results
3.1. Phase I: Comparative Analysis
3.1.1. Chokepoint Enzymes
3.1.2. Virulence Factors Analysis
3.1.3. Resistance Gene Analysis
3.2. Phase II: Subtractive Channel of Analysis
3.2.1. Analysis of Essential Genes
3.2.2. Non-homology Analysis
3.2.3. Gut Flora Non-homology Analysis
3.3. Phase III
3.3.1. Subcellular Localization Prediction of Putative Targets
3.3.2. Broad-Spectrum Analysis
3.3.3. Druggability Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Exner, M.; Bhattacharya, S.; Christiansen, B.; Gebel, J.; Goroncy-Bermes, P.; Hartemann, P.; Heeg, P.; Ilschner, C.; Kramer, A.; Larson, E.; et al. Antibiotic Resistance: What Is so Special about Multidrug-Resistant Gram-Negative Bacteria ? Antibiotikaresistenz: Was Ist so Besonders an Den Gram-Negativen. GMS Hyg. Infect. Control 2017, 12, Doc05. [Google Scholar]
- Kayastha, K.; Dhungel, B.; Karki, S.; Adhikari, B.; Banjara, M.R.; Rijal, K.R.; Ghimire, P. Extended-Spectrum β-Lactamase-Producing Escherichia Coli and Klebsiella Species in Pediatric Patients Visiting International Friendship Children’s Hospital, Kathmandu, Nepal. Infect. Dis. Res. Treat. 2020, 13, 117863372090979. [Google Scholar] [CrossRef]
- Taneja, N.; Rao, P.; Arora, J.; Dogra, A. Occurrence of ESBL & Amp-C b -Lactamases & Susceptibility to Newer Antimicrobial Agents in Complicated UTI. Indian J Med Res. 2008, 127, 85–88. [Google Scholar]
- Shaikh, S.; Fatima, J.; Shakil, S.; Rizvi, S.M.D.; Kamal, M.A. Antibiotic Resistance and Extended Spectrum Beta-Lactamases: Types, Epidemiology and Treatment. Saudi J. Biol. Sci. 2015, 22, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Kaza, P.; Mahindroo, J.; Veeraraghavan, B.; Mavuduru, R.S.; Mohan, B.; Taneja, N. Evaluation of Risk Factors for Colistin Resistance among Uropathogenic Isolates of Escherichia Coli and Klebsiella Pneumoniae: A Case–Control Study. J. Med. Microbiol. 2019, 68, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Asokan, G.V.; Ramadhan, T.; Ahmed, E.; Sanad, H. WHO Global Priority Pathogens List: A Bibliometric Analysis of Medline-Pubmed for Knowledge Mobilization to Infection Prevention and Control Practices in Bahrain. Oman. Med. J. 2019, 34, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.P.; Alam, A.S.M.R.U.; Shill, D.K.; Rahman, A. Identification and Qualitative Characterization of New Therapeutic Targets in Stenotrophomonas Maltophilia through in Silico Proteome Exploration. Microb. Pathog. 2020, 149, 104293. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Kalia, M.; Taneja, N. Identification of Novel Non-Homologous Drug Targets against Acinetobacter Baumannii Using Subtractive Genomics and Comparative Metabolic Pathway Analysis. Microb. Pathog. 2021, 152, 104608. [Google Scholar] [CrossRef]
- Kaur, H.; Kalia, M.; Singh, V.; Modgil, V.; Mohan, B.; Taneja, N. In Silico Identification and Characterization of Promising Drug Targets in Highly Virulent Uropathogenic Escherichia Coli Strain CFT073 by Protein-Protein Interaction Network Analysis. Inf. Med. Unlocked 2021, 25, 100704. [Google Scholar] [CrossRef]
- Shahid, F.; Ashfaq, U.A.; Saeed, S.; Munir, S.; Almatroudi, A.; Khurshid, M. In Silico Subtractive Proteomics Approach for Identification of Potential Drug Targets in Staphylococcus Saprophyticus. Int. J. Environ. Res. Public Health 2020, 17, 3644. [Google Scholar] [CrossRef] [PubMed]
- Solanki, V.; Tiwari, V. Subtractive Proteomics to Identify Novel Drug Targets and Reverse Vaccinology for the Development of Chimeric Vaccine against Acinetobacter Baumannii. Sci. Rep. 2018, 8, 9044. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Singh, S.K.; Ghosh, P.; Mukherjee, R.; Mitter, S.; Bandyopadhyay, D. In Silico Identification of Potential Therapeutic Targets in the Human Pathogen Helicobacter Pylori. Silico Biol. 2006, 6, 43–47. [Google Scholar]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef]
- Avasthi, T.S.; Kumar, N.; Baddam, R.; Hussain, A.; Nandanwar, N.; Jadhav, S.; Ahmed, N. Genome of Multidrug-Resistant Uropathogenic Escherichia Coli Strain NA114 from India. J. Bacteriol. 2011, 193, 4272–4273. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG Databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef]
- Anishetty, S.; Pulimi, M.; Pennathur, G. Potential Drug Targets in Mycobacterium Tuberculosis through Metabolic Pathway Analysis. Comput. Biol. Chem. 2005, 29, 368–378. [Google Scholar] [CrossRef]
- Singh, S.; Malik, B.K.; Sharma, D.K. Choke Point Analysis of Metabolic Pathways in E.Histolytica: A Computational Approach for Drug Target Identification. Bioinformation 2007, 2, 68–72. [Google Scholar] [CrossRef]
- Sc, S.K.M.; Phil, M. Protocol of Rice Genome Annotation through Comparative Functional Genomics Approach. Genome 2009, 4, 1–8. [Google Scholar]
- Baron, C.; Coombes, B. Targeting Bacterial Secretion Systems: Benefits of Disarmament in the Microcosm. Infect. Disord. Drug Targets 2007, 7, 19–27. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis--10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, V.; Malini, A. Antimicrobial Resistance Pattern in Escherichia Coli Causing Urinary Tract Infection among Inpatients. Indian J. Med. Res. 2014, 139, 945–948. [Google Scholar] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein–Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.T.; Zhang, R. DEG 10, an Update of the Database of Essential Genes That Includes Both Protein-Coding Genes and Noncoding Genomic Elements. Nucleic Acids Res 2014, 42, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Maganti, L.; Ghoshal, N.; Dutta, C. In Silico Quest for Putative Drug Targets in Helicobacter Pylori HPAG1: Molecular Modeling of Candidate Enzymes from Lipopolysaccharide Biosynthesis Pathway. J. Mol. Model 2012, 18, 1855–1865. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Fujimura, K.E.; Slusher, N.A.; Cabana, M.D.; Lynch, S.V. Role of the Gut Microbiota in Defining Human Health. Expert Rev. Anti. Infect. Ther. 2010, 8, 435–454. [Google Scholar] [CrossRef]
- Rabizadeh, S.; Sears, C. New Horizons for the Infectious Diseases Specialist: How Gut Microflora Promote Health and Disease. Curr. Infect. Dis. Rep. 2008, 10, 92–98. [Google Scholar] [CrossRef]
- Shanmugham, B.; Pan, A. Identification and Characterization of Potential Therapeutic Candidates in Emerging Human Pathogen Mycobacterium Abscessus: A Novel Hierarchical In Silico Approach. PLoS ONE 2013, 8, e59126. [Google Scholar] [CrossRef]
- Barh, D.; Tiwari, S.; Jain, N.; Ali, A.; Santos, A.R.; Misra, A.N.; Azevedo, V.; Kumar, A. In Silico Subtractive Genomics for Target Identification in Human Bacterial Pathogens. Drug Dev. Res. 2011, 72, 162–177. [Google Scholar] [CrossRef]
- Yu, C.; Lin, C.; Hwang, J.-K. Predicting Subcellular Localization of Proteins for Gram-Negative Bacteria by Support Vector Machines Based on n -Peptide Compositions. Proteins Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Cenk Sahinalp, S.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A Knowledgebase for Drugs, Drug Actions and Drug Targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Butt, A.M.; Nasrullah, I.; Tahir, S.; Tong, Y. Comparative Genomics Analysis of Mycobacterium Ulcerans for the Identification of Putative Essential Genes and Therapeutic Candidates. PloS ONE 2012, 7, e43080. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, S.; Akter, A. Identification of Potential Drug Targets by Subtractive Genome Analysis of Escherichia Coli O157: H7: An in Silico Approach. Adv. Appl. Bioinform. Chem. 2015, 8, 49–63. [Google Scholar]
- Yeh, I. Computational Analysis of Plasmodium Falciparum Metabolism: Organizing Genomic Information to Facilitate Drug Discovery. Genome Res. 2004, 14, 917–924. [Google Scholar] [CrossRef]
- Martínez, O.F.; Cardoso, M.H.; Ribeiro, S.M.; Franco, O.L. Recent Advances in Anti-Virulence Therapeutic Strategies with a Focus on Dismantling Bacterial Membrane Microdomains, Toxin Neutralization, Quorum-Sensing Interference and Biofilm Inhibition. Front. Cell Infect. Microbiol. 2019, 9, 74. [Google Scholar] [CrossRef]
- Topa, S.H.; Palombo, E.A.; Kingshott, P.; Blackall, L.L. Activity of Cinnamaldehyde on Quorum Sensing and Biofilm Susceptibility to Antibiotics in Pseudomonas Aeruginosa. Microorganisms 2020, 8, 455. [Google Scholar] [CrossRef]
- Butt, A.M.; Tahir, S.; Nasrullah, I.; Idrees, M.; Lu, J.; Tong, Y. Mycoplasma Genitalium: A Comparative Genomics Study of Metabolic Pathways for the Identification of Drug and Vaccine Targets. Infect. Genet. Evol. 2012, 12, 53–62. [Google Scholar] [CrossRef]
- Prosser, G.A.; Carvalho, L.P.S. De Kinetic Mechanism and Inhibition of Mycobacterium Tuberculosis D—Alanine: D—Alanine Ligase by the Antibiotic D—Cycloserine. FEBS J. 2013, 280, 1150–1166. [Google Scholar] [CrossRef]
- Bruning, J.B.; Murillo, A.C.; Chacon, O.; Barletta, R.G.; Sacchettini, J.C. Structure of the Mycobacterium Tuberculosis D-Alanine:D-Alanine Ligase, a Target of the Antituberculosis Drug D-Cycloserine. Antimicrob. Agents Chemother. 2011, 55, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Howden, B.P.; Davies, J.K.; Johnson, P.D.R.; Stinear, T.P.; Grayson, M.L. Reduced Vancomycin Susceptibility in Staphylococcus Aureus, Including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: Resistance Mechanisms, Laboratory Detection, and Clinical Implications. Clin. Microbiol. Rev. 2010, 23, 99–139. [Google Scholar] [CrossRef]
- Kovač, A.; Konc, J.; Vehar, B.; Bostock, J.M.; Chopra, I.; Janežič, D.; Gobec, S. Discovery of New Inhibitors of D-Alanine:D-Alanine Ligase by Structure-Based Virtual Screening. J. Med. Chem. 2008, 51, 7442–7448. [Google Scholar] [CrossRef] [PubMed]
- Pandeya, A.; Ojo, I.; Alegun, O.; Wei, Y. Periplasmic Targets for the Development of Effective Antimicrobials against Gram-Negative Bacteria. ACS Infect. Dis. 2020, 6, 2337–2354. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Azevedo, C.; Desfougères, Y.; Portela-Torres, P.; Wilson, M.S.C. Microbial Inositol Polyphosphate Metabolic Pathway as Drug Development Target. Adv. Biol. Regul. 2018, 67, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.; Nagendrappa, J.H.; Shivashankara, S.K.H. Comparative Analysis of Rosetta Stone Events in Klebsiella Pneumoniae and Streptococcus Pneumoniae for Drug Target Identification. Beni. Suef. Univ. J. Basic Appl. Sci. 2021, 10, 37. [Google Scholar] [CrossRef]
- Pirruccello, M.; Nandez, R.; Idevall-Hagren, O.; Alcazar-Roman, A.; Abriola, L.; Berwick, S.A.; Lucast, L.; Morel, D.; De Camilli, P. Identification of Inhibitors of Inositol 5-Phosphatases through Multiple Screening Strategies. ACS Chem. Biol. 2014, 9, 1359–1368. [Google Scholar] [CrossRef]
- Liao, J.; Wang, Q.; Wu, F.; Huang, Z. In Silico Methods for Identification of Potential Active Sites of Therapeutic Targets. Molecules 2022, 27, 7103. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Enzyme ID | Total No. of Pathways of the Enzyme |
|---|---|
| ECNA114_0085 | ena00473, ena00550, ena01100, ena01502 |
| ECNA114_3261 | ena00550 |
| ECNA114_1004, ECNA114_3778 | ena00540, ena01100 |
| ECNA114_2045 | ena01053 |
| ECNA114_0627 | ena00010, ena00030, ena00052, ena00230, ena00500, ena00520, ena00521, ena01100, ena01110, ena01120, ena01130 |
| ECNA114_2137, ECNA114_2136 | ena00521, ena00523, ena01100, ena01130 |
| ECNA114_2904, ECNA114_2317 | ena00071, ena00280, ena00310, ena00362,00380, ena00620, ena00630, ena00640,ena00650, ena00900, ena01100, ena01110, ena01120, ena01130, ena01200, ena01220, ena01212 |
| ECNA114_1060 | ena00562, ena00627, ena01120 |
| ECNA114_3742 | ena00910, ena01120, ena02020 |
| ECNA114_1411 | ena00010, ena00071, ena00350, ena00625, ena00626, ena00650, ena01100, ena01110, ena01120, ena01130, ena01220 |
| ECNA114_1698, ECNA114_1052, ECNA114_3080 | ena00633, ena01120 |
| ECNA114_3742, ECNA114_3652 | ena00010, ena0071, ena00350,ena00625, ena00626, ena01100, ena01110, ena01120, ena01130, ena01150 |
| ECNA114_4463 | ena02060, ena00500 |
| ECNA114_2504 | ena00520, ena02060 |
| ECNA114_1862 | ena00051, ena00520, ena01100, ena02060 |
| ECNA114_4175, ECNA114_4173, ECNA114_4172, ECNA114_2735, ECNA114_3748, ECNA114_2977 | ena00051, ena02060 |
| ECNA114_3218, ECNA114_3216, ECNA114_3217, ECNA114_3224, | ena00052, ena02060 |
| Virulence Factors UPEC | Genes Name | Found in UPEC Strain NA114 and Not in Human |
|---|---|---|
| Iron uptake | iutA, iucA, iucB, iucC, iucD | iucA, iucB, iucD, iucC |
| Chu (E. coli hemin uptake) | chuA, chuS, chuT, chuU, chuV, chuW, chuX, and chuY | chuU, chuW |
| Enterobactin | entA, entB, entC, entD, entE, entF, fepA, fepB, fepC, fepD, fepE, and fepG | fepA, fepB, fepC, fepD, fepG, entE, entA, entB, entF, entC |
| IroN | iroN | iroN |
| Hemolysin | hlyA, hlyB, hlyC, and hlyD | hlyB, hlyD |
| Resistance Gene Found in String after ARDB Tool | Found UPEC Strain NA114 | Found in Human | Enzyme No. |
|---|---|---|---|
| acrB | Yes | No | c0580 |
| acrA | Yes | No | c0581 |
| macB | Yes | No | c1016 |
| arnA | Yes | No | c2797 |
| tolC | Yes | No | c3781 |
| bacA | Yes | No | c3807 |
| Sr. No. | Query Protein | No. of a Homolog in DEG | DEG Accession Number |
|---|---|---|---|
| 1 | ECNA114_0085 | 1 | DEG10180021 |
| 2 | ECNA114_1004 | 2 | DEG10190079, DEG10180150 |
| 3 | ECNA114_3778 | 3 | DEG10480267,DEG10180536, DEG10190246 |
| 4 | ECNA114_2045 | 1 | DEG10180357 |
| 5 | ECNA114_2137 | 1 | DEG10180210 |
| 6 | ECNA114_3652 | 1 | DEG10180338 |
| 7 | ECNA114_1052 | 1 | DEG10180159 |
| 8 | ECNA114_1862 | 1 | DEG10180464 |
| 9 | ECNA114_3218 | 1 | DEG10180032 |
| 10 | entD | 1 | DEG10190060 |
| 11 | entE | 1 | DEG10180357 |
| 12 | fepB | 1 | DEG10180106 |
| 13 | fepC | 2 | DEG10190239; DEG10480100 |
| 14 | arnAc2797 | 3 | DEG10180489, DEG10480227, DEG10190203 |
| 15 | ECNA114_0580 | 1 | DEG10480311 |
| Protein Enzyme Code | Cello | PSORTb | ProtCompb | Subcellular Location |
|---|---|---|---|---|
| ECNA114_4463 | Innermembrane | Cytoplasmic Membrane | Innermembrane | Innermembrane |
| ECNA114_4172 | Innermembrane | Cytoplasmic membrane | Innermembrane | Innermembrane |
| ECNA114_2735 | Innermembrane | Cytoplasmic membrane | Innermembrane | Innermembrane |
| ECNA114_3216 | Innermembrane | Cytoplasmic membrane | Innermembrane | Innermembrane |
| ECNA114_3224 | Innermembrane | Cytoplasmic membrane | Innermembrane | Innermembrane |
| ECNA114_0085 | Cytoplasmic | Cytoplasmic | Cytoplasmic | Cytoplasmic |
| ECNA114_1060 | Periplasmic | Cytoplasmic | Periplasmic | Periplasmic |
| Putative Targets ID’s | Function | Pathways Involved | Druggability Analysis | Subcellular Location |
|---|---|---|---|---|
| ECNA114_4463 | treB, PTS system, | Phosphotransferase system, Starch and sucrose metabolism | No | Innermembrane |
| ECNA114_4172 | PTS system | Mannose and fructose metabolism, Metabolic pathways, Phosphotransferase system | No | Innermembrane |
| ECNA114_2735 | srlE, PTS system | Mannose and fructose metabolism, Metabolic pathways, Phosphotransferase system | No | Innermembrane |
| ECNA114_3216 | agaW, component of PTS system | Galactose metabolism, Metabolic pathways, Phosphotransferase system | No | Innermembrane |
| ECNA114_3224 | agaD, a component of the PTS system | Galactose metabolism, Metabolic pathways, Phosphotransferase system | No | Innermembrane |
| ECNA114_0085 | D-alanine-D-alanine ligase | D-alanine metabolism, Metabolic pathways, Vancomycin resistance, Peptidoglycan biosynthesis | Yes | Cytoplasmic |
| ECNA114_1060 | appA, Phosphoanhydride phosphohydrolase | Inositol phosphate and riboflavin metabolism, Metabolic pathways | No | Periplasmic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, H.; Modgil, V.; Chaudhary, N.; Mohan, B.; Taneja, N. Computational Guided Drug Targets Identification against Extended-Spectrum Beta-Lactamase-Producing Multi-Drug Resistant Uropathogenic Escherichia coli. Biomedicines 2023, 11, 2028. https://doi.org/10.3390/biomedicines11072028
Kaur H, Modgil V, Chaudhary N, Mohan B, Taneja N. Computational Guided Drug Targets Identification against Extended-Spectrum Beta-Lactamase-Producing Multi-Drug Resistant Uropathogenic Escherichia coli. Biomedicines. 2023; 11(7):2028. https://doi.org/10.3390/biomedicines11072028
Chicago/Turabian StyleKaur, Harpreet, Vinay Modgil, Naveen Chaudhary, Balvinder Mohan, and Neelam Taneja. 2023. "Computational Guided Drug Targets Identification against Extended-Spectrum Beta-Lactamase-Producing Multi-Drug Resistant Uropathogenic Escherichia coli" Biomedicines 11, no. 7: 2028. https://doi.org/10.3390/biomedicines11072028
APA StyleKaur, H., Modgil, V., Chaudhary, N., Mohan, B., & Taneja, N. (2023). Computational Guided Drug Targets Identification against Extended-Spectrum Beta-Lactamase-Producing Multi-Drug Resistant Uropathogenic Escherichia coli. Biomedicines, 11(7), 2028. https://doi.org/10.3390/biomedicines11072028