

Genetic Therapy Approaches for Ornithine Transcarbamylase Deficiency



Abstract
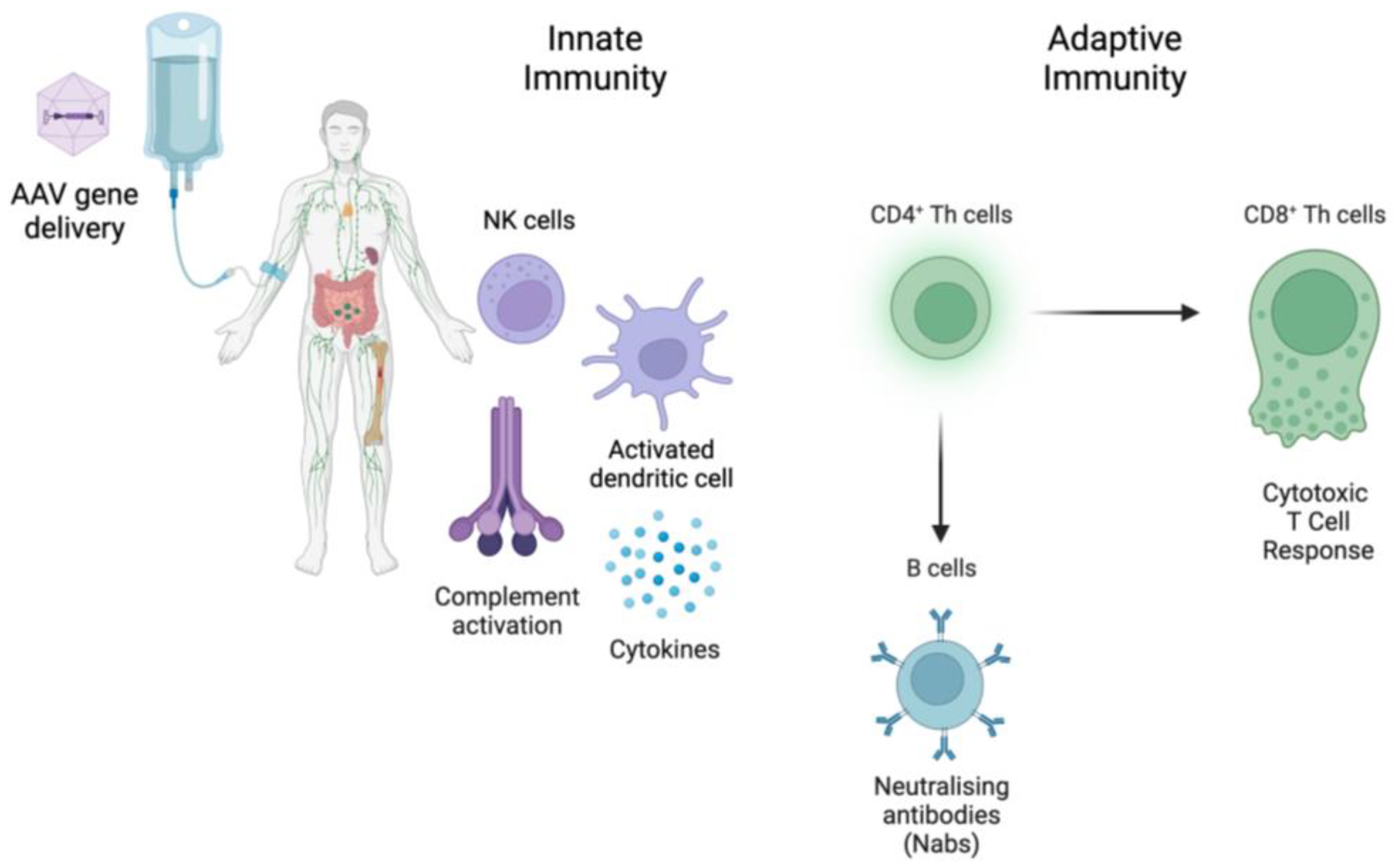
:1. Introduction
2. Why Is OTCD an Appealing Candidate for Gene Therapy?
3. Three Decades of Gene Therapy for OTCD
4. Current Status of Clinical Gene Therapy Trials for OTCD
5. Future Directions of Gene Therapy for OTCD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Seminara, J.; Tuchman, M.; Krivitzky, L.; Krischer, J.; Lee, H.-S.; LeMons, C.; Baumgartner, M.; Cederbaum, S.; Diaz, G.A.; Feigenbaum, A. Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium. Mol. Genet. Metab. 2010, 100, S97–S105. [Google Scholar] [CrossRef] [Green Version]
- Brusilow, S.W.; Maestri, N.E. Urea cycle disorders: Diagnosis, pathophysiology, and therapy. Adv. Pediatr. 1996, 43, 127–170. [Google Scholar] [PubMed]
- Nagata, N.; Matsuda, I.; Oyanagi, K. Estimated frequency of urea cycle enzymopathies in Japan. Am. J. Med. Genet. 1991, 39, 228–229. [Google Scholar] [CrossRef] [PubMed]
- Rapp, B.; Häberle, J.; Linnebank, M.; Wermuth, B.; Marquardt, T.; Harms, E.; Koch, H.G. Genetic analysis of carbamoylphosphate synthetase I and ornithine transcarbamylase deficiency using fibroblasts. Eur. J. Pediatr. 2001, 160, 283–287. [Google Scholar] [CrossRef]
- Hata, A.; Tsuzuki, T.; Shimada, K.; Takiguchi, M.; Mori, M.; Matsuda, I. Structure of the human ornithine transcarbamylase gene. J. Biochem. 1988, 103, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, V.; de Martinville, B.; Horwich, A.L.; Rosenberg, L.E.; Francke, U. Human ornithine transcarbamylase locus mapped to band Xp21.1 near the Duchenne muscular dystrophy locus. Science 1984, 226, 698–700. [Google Scholar] [CrossRef]
- Caldovic, L.; Abdikarim, I.; Narain, S.; Tuchman, M.; Morizono, H. Genotype-Phenotype Correlations in Ornithine Transcarbamylase Deficiency: A Mutation Update. J. Genet. Genom. 2015, 42, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yorifuji, T.; Muroi, J.; Uematsu, A.; Tanaka, K.; Kiwaki, K.; Endo, F.; Matsuda, I.; Nagasaka, H.; Furusho, K. X-inactivation pattern in the liver of a manifesting female with ornithine transcarbamylase (OTC) deficiency. Clin. Genet. 1998, 54, 349–353. [Google Scholar] [CrossRef]
- Matsumoto, S.; Häberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders—Update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef]
- Seker Yilmaz, B.; Baruteau, J.; Arslan, N.; Aydin, H.I.; Barth, M.; Bozaci, A.E.; Brassier, A.; Canda, E.; Cano, A.; Chronopoulou, E.; et al. Three-Country Snapshot of Ornithine Transcarbamylase Deficiency. Life 2022, 12, 1721. [Google Scholar] [CrossRef]
- Gallagher, R.C.; Lam, C.; Wong, D.; Cederbaum, S.; Sokol, R.J. Significant hepatic involvement in patients with ornithine transcarbamylase deficiency. J. Pediatr. 2014, 164, 720–725.e6. [Google Scholar] [CrossRef] [Green Version]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.V.; McKiernan, P.J. The role of liver transplantation in urea cycle disorders. Mol. Genet. Metab. 2004, 81, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Kido, J.; Matsumoto, S.; Häberle, J.; Inomata, Y.; Kasahara, M.; Sakamoto, S.; Horikawa, R.; Tanemura, A.; Okajima, H.; Suzuki, T.; et al. Role of liver transplantation in urea cycle disorders: Report from a nationwide study in Japan. J. Inherit. Metab. Dis. 2021, 44, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Meyburg, J.; Opladen, T.; Spiekerkötter, U.; Schlune, A.; Schenk, J.P.; Schmidt, J.; Weitz, J.; Okun, J.; Bürger, F.; Omran, T.B.; et al. Human heterologous liver cells transiently improve hyperammonemia and ureagenesis in individuals with severe urea cycle disorders. J. Inherit. Metab. Dis. 2018, 41, 81–90. [Google Scholar] [CrossRef]
- Yilmaz, B.S.; Gurung, S.; Perocheau, D.; Counsell, J.; Baruteau, J. Gene therapy for inherited metabolic diseases. J. Mother Child 2020, 24, 53–64. [Google Scholar] [CrossRef]
- Batshaw, M.L.; Tuchman, M.; Summar, M.; Seminara, J. A longitudinal study of urea cycle disorders. Mol. Genet. Metab. 2014, 113, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Dionisi-Vici, C.; Rizzo, C.; Burlina, A.B.; Caruso, U.; Sabetta, G.; Uziel, G.; Abeni, D. Inborn errors of metabolism in the Italian pediatric population: A national retrospective survey. J. Pediatr. 2002, 140, 321–327. [Google Scholar] [CrossRef]
- Keskinen, P.; Siitonen, A.; Salo, M. Hereditary urea cycle diseases in Finland. Acta Paediatr. 2008, 97, 1412–1419. [Google Scholar] [CrossRef]
- Cavicchi, C.; Malvagia, S.; la Marca, G.; Gasperini, S.; Donati, M.A.; Zammarchi, E.; Guerrini, R.; Morrone, A.; Pasquini, E. Hypocitrullinemia in expanded newborn screening by LC-MS/MS is not a reliable marker for ornithine transcarbamylase deficiency. J. Pharm. Biomed. Anal. 2009, 49, 1292–1295. [Google Scholar] [CrossRef]
- Rüfenacht, V.; Häberle, J. Mini-Review: Challenges in Newborn Screening for Urea Cycle Disorders. Int. J. Neonatal Screen. 2015, 1, 27–35. [Google Scholar] [CrossRef]
- Brassier, A.; Gobin, S.; Arnoux, J.B.; Valayannopoulos, V.; Habarou, F.; Kossorotoff, M.; Servais, A.; Barbier, V.; Dubois, S.; Touati, G.; et al. Long-term outcomes in Ornithine Transcarbamylase deficiency: A series of 90 patients. Orphanet J. Rare Dis. 2015, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Han, F.; Qiu, W.; Zhang, H.; Ye, J.; Liang, L.; Wang, Y.; Ji, W.; Zhan, X.; Gu, X.; et al. Clinical and molecular characteristics of 69 Chinese patients with ornithine transcarbamylase deficiency. Orphanet J. Rare Dis. 2020, 15, 340. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, S.C.; Kok, C.Y.; Spinoulas, A.; Carpenter, K.H.; Alexander, I.E. AAV-encoded OTC activity persisting to adulthood following delivery to newborn spf(ash) mice is insufficient to prevent shRNA-induced hyperammonaemia. Gene Ther. 2013, 20, 1184–1187. [Google Scholar] [CrossRef] [Green Version]
- Foschi, F.G.; Morelli, M.C.; Savini, S.; Dall’Aglio, A.C.; Lanzi, A.; Cescon, M.; Ercolani, G.; Cucchetti, A.; Pinna, A.D.; Stefanini, G.F. Urea cycle disorders: A case report of a successful treatment with liver transplant and a literature review. World J. Gastroenterol. 2015, 21, 4063–4068. [Google Scholar] [CrossRef]
- Campeau, P.M.; Pivalizza, P.J.; Miller, G.; McBride, K.; Karpen, S.; Goss, J.; Lee, B.H. Early orthotopic liver transplantation in urea cycle defects: Follow up of a developmental outcome study. Mol. Genet. Metab. 2010, 100 (Suppl. S1), S84–S87. [Google Scholar] [CrossRef] [Green Version]
- Perito, E.R.; Rhee, S.; Roberts, J.P.; Rosenthal, P. Pediatric liver transplantation for urea cycle disorders and organic acidemias: United Network for Organ Sharing data for 2002–2012. Liver Transpl. 2014, 20, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, S.S.; Alonso, E.M.; Whitington, P.F. Liver transplantation in neonates. Liver Transpl. 2003, 9, 783–788. [Google Scholar] [CrossRef]
- Stevenson, T.; Millan, M.T.; Wayman, K.; Berquist, W.E.; Sarwal, M.; Johnston, E.E.; Esquivel, C.O.; Enns, G.M. Long-term outcome following pediatric liver transplantation for metabolic disorders. Pediatr. Transplant. 2010, 14, 268–275. [Google Scholar] [CrossRef]
- Yeowell, G.; Burns, D.S.; Fatoye, F. The burden of pharmacological treatment on health-related quality of life in people with a urea cycle disorder: A qualitative study. J. Patient-Rep. Outcomes 2021, 5, 110. [Google Scholar] [CrossRef]
- Li, M.; Dick, A.; Montenovo, M.; Horslen, S.; Hansen, R. Cost-effectiveness of liver transplantation in methylmalonic and propionic acidemias. Liver Transpl. 2015, 21, 1208–1218. [Google Scholar] [CrossRef]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Granelli-Piperno, A.; Choi, Y.; Steinman, R.M. Recombinant adenovirus is an efficient and non-perturbing genetic vector for human dendritic cells. Eur. J. Immunol. 1999, 29, 964–972. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Raper, S.E.; Yudkoff, M.; Chirmule, N.; Gao, G.P.; Nunes, F.; Haskal, Z.J.; Furth, E.E.; Propert, K.J.; Robinson, M.B.; Magosin, S.; et al. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum. Gene Ther. 2002, 13, 163–175. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Wilson, J.M.; Shchelochkov, O.A.; Gallagher, R.C.; Batshaw, M.L. Hepatocellular carcinoma in a research subject with ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2012, 105, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Li, S.; Li, M.; Xie, J.; Zhang, Y.; Lee, B.; Batshaw, M.L.; Wilson, J.M.; Gao, G. Vector sequences are not detected in tumor tissue from research subjects with ornithine transcarbamylase deficiency who previously received adenovirus gene transfer. Hum. Gene Ther. 2013, 24, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Amalfitano, A.; Parks, R.J. Separating fact from fiction: Assessing the potential of modified adenovirus vectors for use in human gene therapy. Curr. Gene Ther. 2002, 2, 111–133. [Google Scholar] [CrossRef]
- Mian, A.; McCormack, W.M., Jr.; Mane, V.; Kleppe, S.; Ng, P.; Finegold, M.; O’Brien, W.E.; Rodgers, J.R.; Beaudet, A.L.; Lee, B. Long-term correction of ornithine transcarbamylase deficiency by WPRE-mediated overexpression using a helper-dependent adenovirus. Mol. Ther. 2004, 10, 492–499. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Clarke, C.; Mane, V.; Palmer, D.J.; Lanpher, B.; Sun, Q.; O’Brien, W.; Lee, B. Phenotypic correction of ornithine transcarbamylase deficiency using low dose helper-dependent adenoviral vectors. J. Gene Med. 2008, 10, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Stapleton, G.E.; Palmer, D.J.; Zuo, Y.; Mane, V.P.; Finegold, M.J.; Beaudet, A.L.; Leland, M.M.; Mullins, C.E.; Ng, P. Pseudo-hydrodynamic delivery of helper-dependent adenoviral vectors into non-human primates for liver-directed gene therapy. Mol. Ther. 2007, 15, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Deplazes, S.; Schlegel, A.; Song, Z.; Allegri, G.; Rimann, N.; Scherer, T.; Willimann, M.; Opitz, L.; Cunningham, S.C.; Alexander, I.E.; et al. Intrabiliary infusion of naked DNA vectors targets periportal hepatocytes in mice. Mol. Ther. Methods Clin. Dev. 2022, 27, 352–367. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Gissen, P. A retrograde approach for liver gene transfer. Mol. Ther. Methods Clin. Dev. 2022, 27, 488–490. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Jayandharan, G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014, 14, 86–100. [Google Scholar] [CrossRef]
- Servellita, V.; Sotomayor Gonzalez, A.; Lamson, D.M.; Foresythe, A.; Huh, H.J.; Bazinet, A.L.; Bergman, N.H.; Bull, R.L.; Garcia, K.Y.; Goodrich, J.S.; et al. Adeno-associated virus type 2 in US children with acute severe hepatitis. Nature 2023, 617, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Morfopoulou, S.; Buddle, S.; Torres Montaguth, O.E.; Atkinson, L.; Guerra-Assunção, J.A.; Moradi Marjaneh, M.; Zennezini Chiozzi, R.; Storey, N.; Campos, L.; Hutchinson, J.C.; et al. Genomic investigations of unexplained acute hepatitis in children. Nature 2023, 617, 564–573. [Google Scholar] [CrossRef]
- Ho, A.; Orton, R.; Tayler, R.; Asamaphan, P.; Herder, V.; Davis, C.; Tong, L.; Smollett, K.; Manali, M.; Allan, J.; et al. Adeno-associated virus 2 infection in children with non-A-E hepatitis. Nature 2023, 617, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Grieger, J.C.; Samulski, R.J. Adeno-associated virus vectorology, manufacturing, and clinical applications. Methods Enzymol. 2012, 507, 229–254. [Google Scholar]
- Pupo, A.; Fernández, A.; Low, S.H.; François, A.; Suárez-Amarán, L.; Samulski, R.J. AAV vectors: The Rubik’s cube of human gene therapy. Mol. Ther. 2022, 30, 3515–3541. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Lisowski, L.; Kay, M.A. rAAV-mediated tumorigenesis: Still unresolved after an AAV assault. Mol. Ther. 2012, 20, 2014–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther.—Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.L.; Martino, A.T.; Aslanidi, G.V.; Jayandharan, G.R.; Srivastava, A.; Herzog, R.W. Innate Immune Responses to AAV Vectors. Front. Microbiol. 2011, 2, 194. [Google Scholar] [CrossRef] [Green Version]
- Chand, D.H.; Zaidman, C.; Arya, K.; Millner, R.; Farrar, M.A.; Mackie, F.E.; Goedeker, N.L.; Dharnidharka, V.R.; Dandamudi, R.; Reyna, S.P. Thrombotic Microangiopathy Following Onasemnogene Abeparvovec for Spinal Muscular Atrophy: A Case Series. J. Pediatr. 2021, 231, 265–268. [Google Scholar] [CrossRef]
- Smith, C.J.; Ross, N.; Kamal, A.; Kim, K.Y.; Kropf, E.; Deschatelets, P.; Francois, C.; Quinn, W.J., 3rd; Singh, I.; Majowicz, A.; et al. Pre-existing humoral immunity and complement pathway contribute to immunogenicity of adeno-associated virus (AAV) vector in human blood. Front. Immunol. 2022, 13, 999021. [Google Scholar] [CrossRef]
- Perocheau, D.P.; Cunningham, S.C.; Lee, J.; Antinao Diaz, J.; Waddington, S.N.; Gilmour, K.; Eaglestone, S.; Lisowski, L.; Thrasher, A.J.; Alexander, I.E. Age-related seroprevalence of antibodies against AAV-LK03 in a UK population cohort. Hum. Gene Ther. 2019, 30, 79–87. [Google Scholar] [CrossRef] [Green Version]
- George, L.A.; Ragni, M.V.; Rasko, J.E.; Raffini, L.J.; Samelson-Jones, B.J.; Ozelo, M.; Hazbon, M.; Runowski, A.R.; Wellman, J.A.; Wachtel, K. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Mol. Ther. 2020, 28, 2073–2082. [Google Scholar] [CrossRef]
- Earley, J.; Piletska, E.; Ronzitti, G.; Piletsky, S. Evading and overcoming AAV neutralization in gene therapy. Trends Biotechnol. 2022, 41, 836–845. [Google Scholar] [CrossRef]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C. Adenovirus-associated virus vector–mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [Green Version]
- Konkle, B.A.; Walsh, C.E.; Escobar, M.A.; Josephson, N.C.; Young, G.; von Drygalski, A.; McPhee, S.W.J.; Samulski, R.J.; Bilic, I.; de la Rosa, M.; et al. BAX 335 hemophilia B gene therapy clinical trial results: Potential impact of CpG sequences on gene expression. Blood 2021, 137, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Bortolussi, G.; Zentillin, L.; Vaníkova, J.; Bockor, L.; Bellarosa, C.; Mancarella, A.; Vianello, E.; Tiribelli, C.; Giacca, M.; Vitek, L.; et al. Life-long correction of hyperbilirubinemia with a neonatal liver-specific AAV-mediated gene transfer in a lethal mouse model of Crigler-Najjar Syndrome. Hum. Gene Ther. 2014, 25, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Barzel, A.; Paulk, N.K.; Shi, Y.; Huang, Y.; Chu, K.; Zhang, F.; Valdmanis, P.N.; Spector, L.P.; Porteus, M.H.; Gaensler, K.M.; et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature 2015, 517, 360–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Lisowski, L.; Finegold, M.J.; Nakai, H.; Kay, M.A.; Grompe, M. AAV vectors containing rDNA homology display increased chromosomal integration and transgene persistence. Mol. Ther. 2012, 20, 1902–1911. [Google Scholar] [CrossRef] [Green Version]
- Bell, P.; Wang, L.; Lebherz, C.; Flieder, D.B.; Bove, M.S.; Wu, D.; Gao, G.P.; Wilson, J.M.; Wivel, N.A. No evidence for tumorigenesis of AAV vectors in a large-scale study in mice. Mol. Ther. 2005, 12, 299–306. [Google Scholar] [CrossRef]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathwani, A.C.; McIntosh, J.; Sheridan, R. Liver Gene Therapy. Hum. Gene Ther. 2022, 33, 879–888. [Google Scholar] [CrossRef]
- Moscioni, D.; Morizono, H.; McCarter, R.J.; Stern, A.; Cabrera-Luque, J.; Hoang, A.; Sanmiguel, J.; Wu, D.; Bell, P.; Gao, G.P.; et al. Long-term correction of ammonia metabolism and prolonged survival in ornithine transcarbamylase-deficient mice following liver-directed treatment with adeno-associated viral vectors. Mol. Ther. 2006, 14, 25–33. [Google Scholar] [CrossRef]
- Cunningham, S.C.; Spinoulas, A.; Carpenter, K.H.; Wilcken, B.; Kuchel, P.W.; Alexander, I.E. AAV2/8-mediated correction of OTC deficiency is robust in adult but not neonatal Spf(ash) mice. Mol. Ther. 2009, 17, 1340–1346. [Google Scholar] [CrossRef]
- Cunningham, S.C.; Kok, C.Y.; Dane, A.P.; Carpenter, K.; Kizana, E.; Kuchel, P.W.; Alexander, I.E. Induction and prevention of severe hyperammonemia in the spfash mouse model of ornithine transcarbamylase deficiency using shRNA and rAAV-mediated gene delivery. Mol. Ther. 2011, 19, 854–859. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.; Morizono, H.; Bell, P.; Jones, D.; Lin, J.; McMenamin, D.; Yu, H.; Batshaw, M.L.; Wilson, J.M. Sustained correction of OTC deficiency in spf( ash) mice using optimized self-complementary AAV2/8 vectors. Gene Ther. 2012, 19, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, P.; Wang, L.; Chen, S.J.; Yu, H.; Zhu, Y.; Nayal, M.; He, Z.; White, J.; Lebel-Hagan, D.; Wilson, J.M. Effects of Self-Complementarity, Codon Optimization, Transgene, and Dose on Liver Transduction with AAV8. Hum. Gene Ther. Methods 2016, 27, 228–237. [Google Scholar] [CrossRef]
- Wang, L.; Bell, P.; Morizono, H.; He, Z.; Pumbo, E.; Yu, H.; White, J.; Batshaw, M.L.; Wilson, J.M. AAV gene therapy corrects OTC deficiency and prevents liver fibrosis in aged OTC-knock out heterozygous mice. Mol. Genet. Metab. 2017, 120, 299–305. [Google Scholar] [CrossRef] [Green Version]
- De Sabbata, G.; Boisgerault, F.; Guarnaccia, C.; Iaconcig, A.; Bortolussi, G.; Collaud, F.; Ronzitti, G.; Sola, M.S.; Vidal, P.; Rouillon, J.; et al. Long-term correction of ornithine transcarbamylase deficiency in Spf-Ash mice with a translationally optimized AAV vector. Mol. Ther. Methods Clin. Dev. 2021, 20, 169–180. [Google Scholar] [CrossRef]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.-S.; Theisen, M.; Hong, S.-J.; Zhou, J.; Rajendran, R. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.O.; Geberhiwot, T.; Couce, M.L.; Tan, W.-H.; Khan, A.; Hualde, L.C.; Diaz, G.A.; Konczal, L.; Thomas, J.; Guffon, N. Safety and Efficacy of DTX301 in Adults with Late-Onset Ornithine Transcarbamylase (OTC) Deficiency: A Phase 1/2 Trial. In Molecular Therapy; Cell Press: Cambridge, MA, USA, 2022; p. 219. [Google Scholar]
- Ultragenyx Pharmaceutical Inc. Ultragenyx Announces Positive Longer-Term Durability Data from Two Phase 1/2 Gene Therapy Studies at American Society of Gene & Cell Therapy (ASGCT) 2022 Annual Meeting; Ultragenyx Pharmaceutical Inc.: Novato, CA, USA, 2022. [Google Scholar]
- Available online: https://www.clinicaltrials.gov (accessed on 1 July 2023).
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Bell, P.; McMenamin, D.; He, Z.; White, J.; Yu, H.; Xu, C.; Morizono, H.; Musunuru, K.; et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016, 34, 334–338. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Y.; Breton, C.; Bell, P.; Li, M.; Zhang, J.; Che, Y.; Saveliev, A.; He, Z.; White, J.; et al. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020, 6, eaax5701. [Google Scholar] [CrossRef] [Green Version]
- Ginn, S.L.; Amaya, A.K.; Liao, S.H.Y.; Zhu, E.; Cunningham, S.C.; Lee, M.; Hallwirth, C.V.; Logan, G.J.; Tay, S.S.; Cesare, A.J.; et al. Efficient in vivo editing of OTC-deficient patient-derived primary human hepatocytes. JHEP Rep. 2020, 2, 100065. [Google Scholar] [CrossRef] [Green Version]
- Zabulica, M.; Srinivasan, R.C.; Akcakaya, P.; Allegri, G.; Bestas, B.; Firth, M.; Hammarstedt, C.; Jakobsson, T.; Jakobsson, T.; Ellis, E.; et al. Correction of a urea cycle defect after ex vivo gene editing of human hepatocytes. Mol. Ther. 2021, 29, 1903–1917. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Sponsor | Trial Number (clinicaltrials.gov) | Phase | Status | Vector | Number of Participants | Age Group of Participants (Years) |
|---|---|---|---|---|---|---|
| Ultragenyx (Novato, CA, USA) | NCT02991144 | I, II | Completed | AAV8 | 16 | ≥18 |
| Ultragenyx | NCT05345171 | III | Recruiting | AAV8 | 50 | ≥12 |
| University College London (London, UK) | NCT05092685 | I, II | Not yet recruiting | AAVLK03 | 12 | 0–16 |
| Arcturus Therapeutics, Inc. (San Diego, CA, USA) | NCT04416126 | Ia | Completed | mRNA | 30 | 18–65 |
| Arcturus Therapeutics, Inc. | NCT04442347 | Ib | Active, not recruiting | mRNA | 12 | ≥18 |
| Arcturus Therapeutics, Inc. | NCT05526066 | III | Recruiting | mRNA | 24 | 12–65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seker Yilmaz, B.; Gissen, P. Genetic Therapy Approaches for Ornithine Transcarbamylase Deficiency. Biomedicines 2023, 11, 2227. https://doi.org/10.3390/biomedicines11082227
Seker Yilmaz B, Gissen P. Genetic Therapy Approaches for Ornithine Transcarbamylase Deficiency. Biomedicines. 2023; 11(8):2227. https://doi.org/10.3390/biomedicines11082227
Chicago/Turabian StyleSeker Yilmaz, Berna, and Paul Gissen. 2023. "Genetic Therapy Approaches for Ornithine Transcarbamylase Deficiency" Biomedicines 11, no. 8: 2227. https://doi.org/10.3390/biomedicines11082227
APA StyleSeker Yilmaz, B., & Gissen, P. (2023). Genetic Therapy Approaches for Ornithine Transcarbamylase Deficiency. Biomedicines, 11(8), 2227. https://doi.org/10.3390/biomedicines11082227