
Machine Learning-Based Virtual Screening and Molecular Simulation Approaches Identified Novel Potential Inhibitors for Cancer Therapy


Abstract
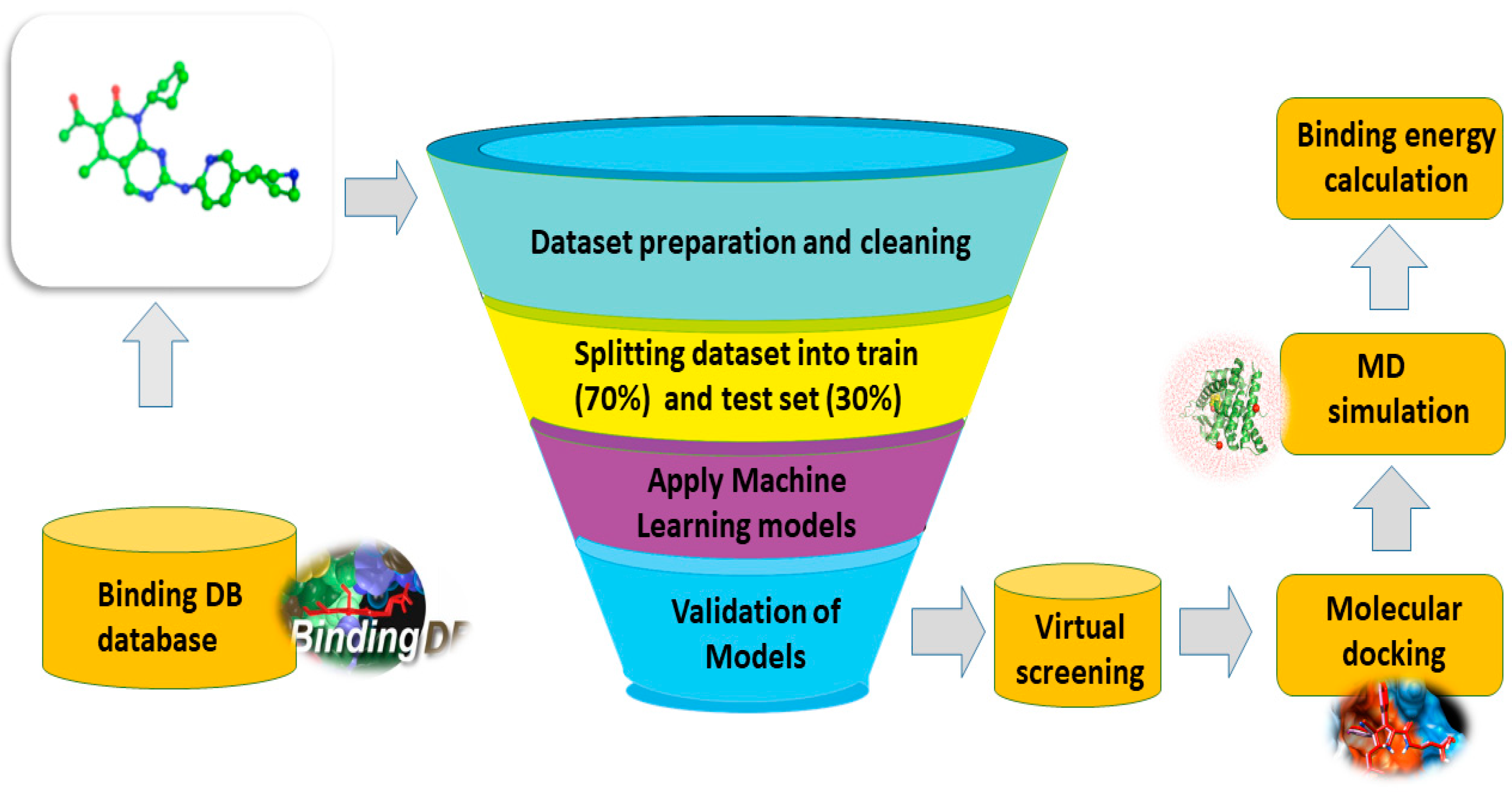
:1. Introduction
2. Material and Methods
2.1. Dataset Preparation
2.2. Descriptor Generation and Feature Selection
2.3. Machine Learning Algorithms
2.3.1. SVM
2.3.2. k-Nearest Neighbor (k-NN)
2.3.3. Random Forest
2.3.4. Gaussian Naïve Bayes
2.4. Performance Evaluation and Cross-Validation
2.5. Molecular Docking and Selection of the Final Hits
2.6. Molecular Dynamics Simulation (MDS)
2.7. Binding Free Energy Calculation
3. Results and Discussion
3.1. The ROC Curve of Each Algorithm
3.2. Binding Interactions of Final Selected Hits
3.3. Molecular Dynamics Simulation Analyses
3.3.1. Root Mean Square Deviation (RMSD) Analyses
3.3.2. Root Mean Square Fluctuation Analyses
3.3.3. Radius of Gyration
3.3.4. Hydrogen Bond (Polar Interactions) Analyses
3.3.5. Clustering of Protein Motion by PCA
3.3.6. Free Energy Landscape
3.3.7. Binding Free Energy Calculation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norbury, C.; Nurse, P. Animal cell cycles and their control. Annu. Rev. Biochem. 1992, 61, 441–470. [Google Scholar] [CrossRef]
- Volkart, P.A.; Bitencourt-Ferreira, G.; Souto, A.A.; de Azevedo, W.F. Cyclin-Dependent Kinase 2 in Cellular Senescence and Cancer. A Structural and Functional Review. Curr. Drug Targets 2019, 20, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Regan, K.M.; Lou, Z.; Chen, J.; Tindall, D.J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 2006, 314, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Bahram, F.; Su, Y.; Tronnersjö, S.; Högstrand, K.; von der Lehr, N.; Sharifi, H.R.; Lilischkis, R.; Hein, N.; Wu, S.; et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl. Acad. Sci. USA 2010, 107, 58–63. [Google Scholar] [CrossRef]
- Major, M.L.; Lepe, R.; Costa, R.H. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell Biol. 2004, 24, 2649–2661. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voit, R.; Grummt, I. Phosphorylation of UBF at serine 388 is required for interaction with RNA polymerase I and activation of rDNA transcription. Proc. Natl. Acad. Sci. USA 2001, 98, 13631–13636. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Chae, H.-D.; Choi, T.-S.; Kim, E.-H.; Bang, Y.-J.; Chung, J.; Choi, K.-S.; Mantovani, R.; Shin, D.Y. Cdk2-dependent phosphorylation of the NF-Y transcription factor and its involvement in the p53-p21 signaling pathway. J. Biol. Chem. 2003, 278, 36966–36972. [Google Scholar] [CrossRef] [Green Version]
- Ziebold, U.; Bartsch, O.; Marais, R.; Ferrari, S.; Klempnauer, K.-H. Phosphorylation and activation of B-Myb by cyclin A-Cdk2. Curr. Biol. 1997, 7, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef]
- Akli, S.; Van Pelt, C.S.; Bui, T.; Meijer, L.; Keyomarsi, K. Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin E. Cancer Res. 2011, 71, 3377–3386. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Fang, D.; Chen, H.; Lu, Y.; Dong, Z.; Ding, H.-F.; Jing, Q.; Su, S.-B.; Huang, S. Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget 2015, 6, 20801–20812. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gan, Y.; Li, H.; Yin, J.; He, X.; Lin, L.; Xu, S.; Fang, Z.; Kim, B.-W.; Gao, L.; et al. Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat. Commun. 2022, 13, 2835. [Google Scholar] [CrossRef]
- Łukasik, P.; Baranowska-Bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 2806. [Google Scholar] [CrossRef] [PubMed]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef]
- Rafique, R.; Islam, S.R.; Kazi, J.U. Machine learning in the prediction of cancer therapy. Comput. Struct. Biotechnol. J. 2021, 19, 4003–4017. [Google Scholar] [CrossRef] [PubMed]
- Bhinder, B.; Gilvary, C.; Madhukar, N.S.; Elemento, O. Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov. 2021, 11, 900–915. [Google Scholar] [CrossRef]
- Yang, J.; Cai, Y.; Zhao, K.; Xie, H.; Chen, X. Concepts and applications of chemical fingerprint for hit and lead screening. Drug Discov. Today 2022, 27, 103356. [Google Scholar] [CrossRef]
- Singla, R.K.; De, R.; Efferth, T.; Mezzetti, B.; Uddin, S.; Sanusi; Ntie-Kang, F.; Wang, D.; Schultz, F.; Kharat, K.R.; et al. The International Natural Product Sciences Taskforce (INPST) and the power of Twitter networking exemplified through# INPST hashtag analysis. Phytomedicine 2023, 108, 154520. [Google Scholar]
- Sandhu, H.; Kumar, R.N.; Garg, P. Machine learning-based modeling to predict inhibitors of acetylcholinesterase. Mol. Divers. 2022, 26, 331–340. [Google Scholar] [CrossRef]
- Dos Santos, R.P.; Beko, M.; Leithardt, V.R. Package Proposal for Data Pre-Processing for Machine Learning Applied to Precision Irrigation. In Proceedings of the 2023 6th Conference on Cloud and Internet of Things (CIoT), Lisbon, Portugal, 20–22 March 2023. [Google Scholar]
- Gholizadeh, M.; Jamei, M.; Ahmadianfar, I.; Pourrajab, R. Prediction of nanofluids viscosity using random forest (RF) approach. Chemom. Intell. Lab. Syst. 2020, 201, 104010. [Google Scholar] [CrossRef]
- Moreira, G.C.; Carneiro, C.N.; dos Anjos, G.L.; da Silva, F.; Santos, J.L.; Dias, F.d.S. Support vector machine and PCA for the exploratory analysis of Salvia officinalis samples treated with growth regulators based in the agronomic parameters and multielement composition. Food Chem. 2022, 373, 131345. [Google Scholar] [CrossRef] [PubMed]
- Ali, L.; Khan, S.U.; Golilarz, N.A.; Yakubu, I.; Qasim, I.; Noor, A.; Nour, R. A feature-driven decision support system for heart failure prediction based on statistical model and Gaussian naive bayes. Comput. Math. Methods Med. 2019, 2019, 6314328. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Yao, Y. Study on an adaptive thermal comfort model with K-nearest-neighbors (KNN) algorithm. Build. Environ. 2021, 202, 108026. [Google Scholar] [CrossRef]
- Zhang, F.; O’Donnell, L.J. Support vector regression. In Machine Learning; Elsevier: Amsterdam, The Netherlands, 2020; pp. 123–140. [Google Scholar]
- Xiong, L.; Yao, Y. Machine learning-based drug design for identification of thymidylate kinase inhibitors as a potential anti-Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2023, 202, 108026. [Google Scholar]
- Bouallegue, G.; Djemal, R. EEG person identification using Facenet, LSTM-RNN and SVM. In Proceedings of the 2020 17th International Multi-Conference on Systems, Signals & Devices (SSD), Sfax, Tunisia, 20–23 July 2020. [Google Scholar]
- Mohebbian, M.R.; HMarateb, R.; Wahid, K.A. Semi-supervised active transfer learning for fetal ECG arrhythmia detection. Comput. Methods Programs Biomed. Update 2023, 3, 100096. [Google Scholar] [CrossRef]
- Granitto, P.M.; Furlanello, C.; Biasioli, F.; Gasperi, F. Recursive feature elimination with random forest for PTR-MS analysis of agroindustrial products. Chemom. Intell. Lab. Syst. 2006, 83, 83–90. [Google Scholar] [CrossRef]
- Svetnik, V.; Wang, T.-C.; Xu, Y.; Hansen, B.J.; Fox, S.V. A deep learning approach for automated sleep-wake scoring in pre-clinical animal models. J. Neurosci. Methods 2020, 337, 108668. [Google Scholar] [CrossRef]
- Kabir, M.; Arif, M.; Ahmad, S.; Ali, Z.; Swati, Z.N.K.; Yu, D.-J. Intelligent computational method for discrimination of anticancer peptides by incorporating sequential and evolutionary profiles information. Chemom. Intell. Lab. Syst. 2018, 182, 158–165. [Google Scholar] [CrossRef]
- Palko, N.; Potemkin, V.; Grishina, M. Decision tree for mechanism of antitumor drugs action prediction. Bull. South Ural. State Univ. Ser. Chem. 2019, 11, 18–24. [Google Scholar] [CrossRef]
- Ali, M.; Aittokallio, T. Machine learning and feature selection for drug response prediction in precision oncology applications. Biophys. Rev. 2019, 11, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghori, K.M.; Abbasi, R.A.; Awais, M.; Imran, M.; Ullah, A.; Szathmary, L. Performance analysis of different types of machine learning classifiers for non-technical loss detection. IEEE Access 2019, 8, 16033–16048. [Google Scholar] [CrossRef]
- Ye, W.-L.; Shen, C.; Xiong, G.-L.; Ding, J.-J.; Lu, A.-P.; Hou, T.-J.; Cao, D.-S. Improving docking-based virtual screening ability by integrating multiple energy auxiliary terms from molecular docking scoring. J. Chem. Inf. Model. 2020, 60, 4216–4230. [Google Scholar] [CrossRef]
- Yang, C.; Yang, Z.; Tong, K.; Wang, J.; Yang, W.; Yu, R.; Jiang, F.; Ji, Y. Homology modeling and molecular docking simulation of martentoxin as a specific inhibitor of the BK channel. Ann. Transl. Med. 2022, 10, 71. [Google Scholar] [CrossRef]
- Vassetti, D.; Pagliai, M.; Procacci, P. Assessment of GAFF2 and OPLS-AA general force fields in combination with the water models TIP3P, SPCE, and OPC3 for the solvation free energy of druglike organic molecules. J. Chem. Theory Comput. 2019, 15, 1983–1995. [Google Scholar] [CrossRef] [Green Version]
- Indrakumar, S.; Zalar, M.; Pohl, C.; Nørgaard, A.; Streicher, W.; Harris, P.; Golovanov, A.P.; Peters, G.H.J. Conformational stability study of a therapeutic peptide plectasin using molecular dynamics simulations in combination with NMR. J. Phys. Chem. B 2019, 123, 4867–4877. [Google Scholar] [CrossRef]
- Shahab, M.; Danial, M.; Khan, T.; Liang, C.; Duan, X.; Wang, D.; Gao, H.; Zheng, G. In Silico Identification of Lead Compounds for Pseudomonas Aeruginosa PqsA Enzyme: Computational Study to Block Biofilm Formation. Biomedicines 2023, 11, 961. [Google Scholar] [CrossRef]
- Fang, Y.; Lin, S.; Dou, Q.; Gui, J.; Li, W.; Tan, H.; Wang, Y.; Zeng, J.; Khan, A.; Wei, D.-Q. Network pharmacology-and molecular simulation-based exploration of therapeutic targets and mechanisms of heparin for the treatment of sepsis/COVID-19. J. Biomol. Struct. Dyn. 2023, 1–13. [Google Scholar] [CrossRef]
- Clyde, A.R. Artificial Intelligence and High-Performance Computing for Accelerating Structure-Based Drug Discovery. Ph.D. Thesis, The University of Chicago, Chicago, IL, USA, 2022. [Google Scholar]
- Aljuaid, A.; Salam, A.; Almehmadi, M.; Baammi, S.; Alshabrmi, F.M.; Allahyani, M.; Al-Zaydi, K.M.; Izmirly, A.M.; Almaghrabi, S.; Baothman, B.K.; et al. Structural homology-based drug repurposing approach for targeting NSP12 SARS-CoV-2. Molecules 2022, 27, 7732. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithms | F1_Score | ACC | roc_auc Score | Specificity | Sensitivity |
|---|---|---|---|---|---|
| SVM | 0.753 | 0.940 | 0.940 | 0.998 | 0.489 |
| k-NN | 0.653 | 0.609 | 0.743 | 0.998 | 0.489 |
| RF | 0.653 | 0.893 | 0.893 | 0.998 | 0.489 |
| GNB | 0.93 | 0.986 | 0.888 | 0.992 | 0.482 |
| Compound | Score | Interaction Details | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand | Receptor | Interaction | Distance | E (kcal/mol) | |||||
| ZINC00498962 | −8.00 | N1 | 3 | OD1 | ASP | 145 | H–donor | 3.25 | −2.5 |
| S1 | 17 | OD2 | ASP | 86 | H–donor | 3.72 | −2.5 | ||
| C12 | 19 | OD2 | ASP | 145 | H–donor | 3.41 | −0.2 | ||
| O2 | 12 | CD2 | LEU | 134 | H–acceptor | 3.47 | −0.1 | ||
| S1 | 17 | CD2 | LEU | 134 | H–acceptor | 3.05 | −0.2 | ||
| N3 | 10 | 6-ring | PHE | 82 | H–pi | 3.47 | −1.2 | ||
| C9 | 14 | 6-ring | PHE | 82 | H–pi | 4.49 | −0.1 | ||
| 5-RING | NZ | LYS | 89 | pi–cation | 4.34 | −0.5 | |||
| ZINC01612669 | −7.90 | C10 | 12 | O | LEU | 83 | H–donor | 3.26 | −0.3 |
| N2 | 15 | OD2 | ASP | 86 | H–donor | 3.43 | −1.8 | ||
| N3 | 17 | CA | GLN | 85 | H–acceptor | 3.63 | −0.3 | ||
| N3 | 17 | NZ | LYS | 89 | H–acceptor | 3.09 | −7.6 | ||
| N6 | 23 | N | ASP | 145 | H–acceptor | 3.82 | −0.4 | ||
| C8 | 10 | 6-RING | PHE | 82 | H–pi | 4.51 | −0.4 | ||
| ZINC23543539 | −7.34 | N4 | 9 | OD2 | ASP | 86 | H–donor | 3.19 | −2.4 |
| N5 | 12 | O | LEU | 83 | H–donor | 3.56 | −0.6 | ||
| S1 | 19 | O | GLU | 81 | H–donor | 3.77 | −1.3 | ||
| O1 | 11 | NZ | LYS | 89 | H–acceptor | 3.26 | −3.7 | ||
| N6 | 15 | N | LEU | 83 | H–acceptor | 3.38 | −1.1 | ||
| N7 | 16 | CA | PHE | 82 | H–acceptor | 3.55 | −0.5 | ||
| N7 | 16 | N | LEU | 83 | H–acceptor | 3.35 | −0.8 | ||
| Control drug (Dalpiciclib) | −6.34 | N23 | 23 | OD1 | ASP | 145 | H–donor | 2.64 | −9.7 |
| O1 | 1 | NZ | LYS | 129 | H–donor | 3.37 | −1.7 | ||
| O32 | 32 | NZ | LYS | 129 | H–acceptor | 2.77 | −0.5 | ||
| 6-RING | CG | GLN | 145 | pi–H | 3.17 | −2.2 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahab, M.; Zheng, G.; Khan, A.; Wei, D.; Novikov, A.S. Machine Learning-Based Virtual Screening and Molecular Simulation Approaches Identified Novel Potential Inhibitors for Cancer Therapy. Biomedicines 2023, 11, 2251. https://doi.org/10.3390/biomedicines11082251
Shahab M, Zheng G, Khan A, Wei D, Novikov AS. Machine Learning-Based Virtual Screening and Molecular Simulation Approaches Identified Novel Potential Inhibitors for Cancer Therapy. Biomedicines. 2023; 11(8):2251. https://doi.org/10.3390/biomedicines11082251
Chicago/Turabian StyleShahab, Muhammad, Guojun Zheng, Abbas Khan, Dongqing Wei, and Alexander S. Novikov. 2023. "Machine Learning-Based Virtual Screening and Molecular Simulation Approaches Identified Novel Potential Inhibitors for Cancer Therapy" Biomedicines 11, no. 8: 2251. https://doi.org/10.3390/biomedicines11082251