
A Review of the Relationship between the Immune Response, Inflammation, Oxidative Stress, and the Pathogenesis of Sickle Cell Anaemia
 and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
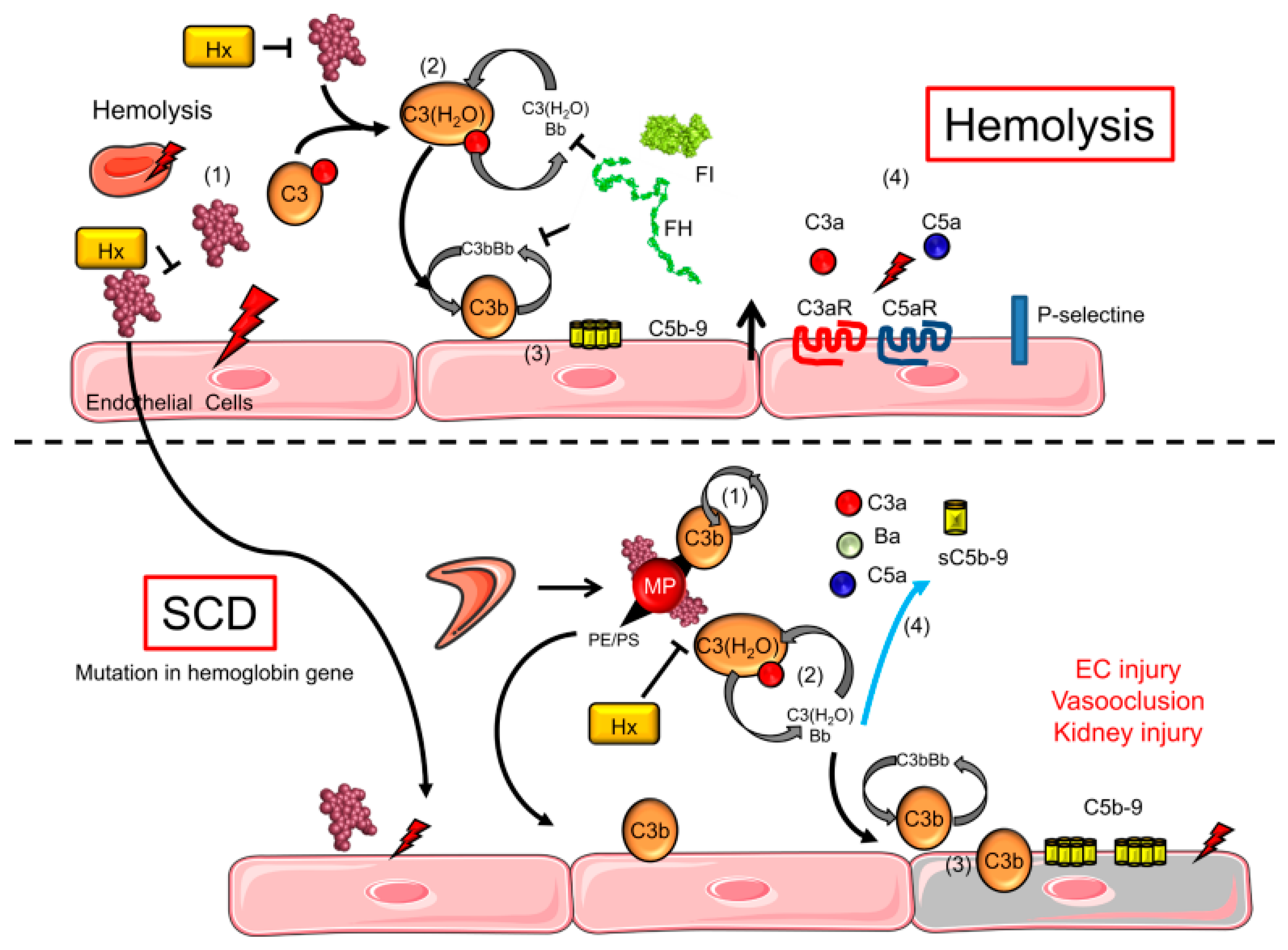
2. Immune Mechanisms Involved in the Pathogenesis of Sickle Cell Anaemia
3. Autoimmunity in Sickle Cell Disease
4. The Role of Oxidative Stress in the Pathogenesis of Sickle Cell Anaemia
5. The Role of Inflammation in the Pathogenesis of Sickle Cell Anaemia
6. The Relationship and Interdependence between Inflammation and Oxidative Stress in SCD
7. Inflammation and Blood Transfusion in SCD
8. The Cause of High Alloimmunisation in SCD Patients
9. Other Treatment Options
10. Conclusions
Funding
Conflicts of Interest
References
- Royal, C.D.M.; Babyak, M.; Shah, N.; Srivatsa, S.; Stewart, K.A.; Tanabe, P.; Wonkam, A.; Asnani, M. Sickle cell disease is a global prototype for integrative research and healthcare. Adv. Genet. 2021, 2, e10037. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle cell disease: Role of oxidative stress and antioxidant therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Adesina, O.A.; Opesade, A.O. Bibliometirc Analysis of Sickle Cell Anaemia Literature on Nigeria Listed in Pubmed between 2006 and 2016. Libr. Philos. Pract. 2018, 1–16. [Google Scholar]
- Fraiwan, A.; Hasan, M.N.; An, R.; Rezac, A.J.; Kocmich, N.J.; Oginni, T.; Olanipekun, G.M.; Hassan-Hanga, F.; Jibir, B.W.; Gambo, S.; et al. Advancing healthcare outcomes for sickle cell disease in Nigeria using mobile health tools. Blood 2019, 134, 2173. [Google Scholar] [CrossRef]
- Mwaiswelo, R.O.; Mawala, W.; Iversen, P.O.; de Montalembert, M.; Luzzatto, L.; Makani, J. Sickle cell disease and malaria: Decreased exposure and asplenia can modulate the risk from Plasmodium falciparum. Malar. J. 2020, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Nnodu, O.; Isa, H.; Nwegbu, M.; Ohiaeri, C.; Adegoke, S.; Chianumba, R.; Ugwu, N.; Brown, B.; Olaniyi, J.; Okocha, E.; et al. HemoTypeSC, a low-cost point-of-care testing device for sickle cell disease: Promises and challenges. Blood Cells Mol. Dis. 2019, 78, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Murphy, C.; Eakins, E.; Rondinelli, M.B.; Daves, M.; Vecchiato, C.; Wolf, D.; Mc Mahon, C.; Smith, O.P. Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. Eur. J. Haematol. 2019, 102, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Loggetto, S.; Veríssimo, M.; Darrigo-Junior, L.; Simões, R.; Bernardo, W.; Braga, J. Guidelines on sickle cell disease: Secondary stroke prevention in children and adolescents. Associação Brasileira de Hematologia, Hemoterapia e Terapia Celular guidelines project: Associação Médica Brasileira-2022. Haematol. Transfus. Cell Ther. 2022, 44, 246–255. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Connes, P. The red blood cell—Inflammation vicious circle in sickle cell disease. Front. Immunol. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Atiku, S.M.; Louise, N.; Kasozi, D.M. Kasozi, Severe oxidative stress in sickle cell disease patients with uncomplicated Plasmodium falciparum malaria in Kampala, Uganda. BMC Infect. Dis. 2019, 19, 600. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, D.L.; Anand, A.C. Sickle hepatopathy. J. Clin. Exp. Hepatol. 2021, 11, 82–96. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Pradhan-Sundd, T.; A Pritchard, K.; A Hillery, C. Redox signaling in sickle cell disease. Curr. Opin. Physiol. 2019, 9, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Quezado, Z.M.; Kamimura, S.; Smith, M.; Wang, X.; Heaven, M.R.; Jana, S.; Vogel, S.; Zerfas, P.; Combs, C.A.; Almeida, L.E.; et al. Mitapivat increases ATP and decreases oxidative stress and erythrocyte mitochondria retention in a SCD mouse model. Blood Cells Mol. Dis. 2022, 95, 102660. [Google Scholar] [CrossRef] [PubMed]
- Inusa, B.P.D.; Hsu, L.L.; Kohli, N.; Patel, A.; Ominu-Evbota, K.; Anie, K.A.; Atoyebi, W. Sickle cell disease—Genetics, pathophysiology, clinical presentation and treatment. Int. J. Neonatal Screen. 2019, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Bozza, M.T.; Jeney, V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front. Immunol. 2020, 11, 1323. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The worst things in life are free: The role of free heme in sickle cell disease. Front. Immunol. 2021, 11, 561917. [Google Scholar] [CrossRef]
- Adwas, A.A.; Elsayed, A.; Azab, A.E.; Quwaydir, F.A. Oxidative stress and antioxidant mechanisms in human body. J. Appl. Biotechnol. Bioeng. 2019, 6, 43–47. [Google Scholar]
- Tebuka, E.; Charles, M.; Bhuko, J.O. Prevalence and risk factors for red blood cell alloimmunisation among sickle cell patients in Mwanza City, Tanzania. Afr. J. Lab. Med. 2020, 9, 823. [Google Scholar] [CrossRef]
- Conrath, S.; Vantilcke, V.; Parisot, M.; Maire, F.; Selles, P.; Elenga, N. Increased prevalence of alloimmunization in sickle cell disease? Should we restore blood donation in French Guiana? Front. Med. 2021, 8, 681549. [Google Scholar] [CrossRef]
- Khatun, A.; Habibullah, M.; Biswas, D.; Quader, M.; Biswas, J. Frequency of alloantibody with their specification among multitransfused patients. Glob. J. Transfus. Med. 2020, 5, 178. [Google Scholar] [CrossRef]
- Fasano, R.M.; Meyer, E.K.; Branscomb, J.; White, M.S.; Gibson, R.W.; Eckman, J.R. Impact of red blood cell antigen matching on alloimmunization and transfusion complications in patients with sickle cell disease: A systematic review. Transfus. Med. Rev. 2019, 33, 12–23. [Google Scholar] [CrossRef]
- El Chaer, F.; Holtzman, N.G.; Baer, M.R.; Zimrin, A.B.; Law, J.Y. Sickle cell disease complicated by iron overload: An under-recognized risk factor for Vibrio vulnificus infection. Acta Haematol. 2018, 139, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Allali, S.; Maciel, T.T.; Hermine, O.; de Montalembert, M. Innate immune cells, major protagonists of sickle cell disease pathophysiology. Haematologica 2020, 105, 273. [Google Scholar] [CrossRef]
- de Azevedo, J.T.C.; Malmegrim, K.C.R. Immune mechanisms involved in sickle cell disease pathogenesis: Current knowledge and perspectives. Immunol. Lett. 2020, 224, 1–11. [Google Scholar] [CrossRef]
- Allali, S.; Dietrich, C.; Machavoine, F.; Rignault-Bricard, R.; Brousse, V.; de Montalembert, M.; Hermine, O.; Maciel, T.T.; Leite-De-Moraes, M. Innate-like T cells in children with sickle cell disease. PLoS ONE 2019, 14, e0219047. [Google Scholar] [CrossRef]
- Sesti-Costa, R.; Borges, M.D.; Lanaro, C.; de Albuquerque, D.M.; Saad, S.T.O.; Costa, F.F. Inflammatory Dendritic Cells Contribute to Regulate the Immune Response in Sickle Cell Disease. Front. Immunol. 2021, 11, 617962. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahsan, H. Biomarkers of inflammation and oxidative stress in ophthalmic disorders. J. Immunoass. Immunochem. 2020, 41, 257–271. [Google Scholar] [CrossRef]
- Engwa, G.A.; Okolie, A.; Chidili, J.P.C.; Okore, P.A.; Onu, P.C.; Ugwu, M.O.; Oko, D.E.; Ferdinand, P.U. Relationship of oxidative stress and antioxidant response with vaso-occlusive crisis in sickle cell anaemia. Afr. Health Sci. 2021, 21, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Ochocinski, D.; Dalal, M.; Black, L.V.; Carr, S.; Lew, J.; Sullivan, K.; Kissoon, N. Life-threatening infectious complications in sickle cell disease: A concise narrative review. Front. Pediatr. 2020, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Bourne, J.H.; Brill, A.; Watson, S.P. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res. Pract. Thromb. Haemost. 2020, 4, e12266. [Google Scholar] [CrossRef]
- Conran, N.; Belcher, J.D. Inflammation in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299. [Google Scholar] [CrossRef]
- Garcia, N.P.; Júnior, A.L.S.; Soares, G.A.S.; Costa, T.C.C.; Dos Santos, A.P.C.; Costa, A.G.; Tarragô, A.M.; Martins, R.N.; do Carmo Leão Pontes, F.; de Almeida, E.G.; et al. Sickle cell anemia patients display an intricate cellular and serum biomarker network highlighted by TCD4+ CD69+ lymphocytes, IL-17/MIP-1β, IL-12/VEGF, and IL-10/IP-10 axis. J. Immunol. Res. 2020, 2020, 4585704. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.F.; A Spring, F.; A Chasis, J.; Anstee, D.J. Erythroid cell adhesion molecules Lutheran and LW in health and disease. Best Pract. Res. Clin. Haematol. 1999, 12, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Wick, T.M.; Eckman, J.R. Activation of vascular endothelial cell adhesion molecule expression by sickle blood cells. Pediatr. Pathol. Mol. Med. 2001, 20, 47–72. [Google Scholar] [CrossRef]
- Pathare, A.; Al Kindi, S.; Daar, S.; Dennison, D. Cytokines in sickle cell disease. Hematology 2003, 8, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative profile of patients with sickle cell disease. Med. Sci. 2019, 7, 17. [Google Scholar] [CrossRef]
- Takeda, M.; Oami, T.; Hayashi, Y.; Shimada, T.; Hattori, N.; Tateishi, K.; Miura, R.E.; Yamao, Y.; Abe, R.; Kobayashi, Y.; et al. Prehospital diagnostic algorithm for acute coronary syndrome using machine learning: A prospective observational study. Sci. Rep. 2022, 12, 14593. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of oxidative stress and inflammation in sickle cell disease with the Nrf2 activator dimethyl fumarate. Antioxid. Redox Signal. 2017, 26, 748–762. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Darbari, D.S.; Sheehan, V.A.; Ballas, S.K. The vaso-occlusive pain crisis in sickle cell disease: Definition, pathophysiology, and management. Eur. J. Haematol. 2020, 105, 237–246. [Google Scholar] [CrossRef]
- Quintela-Carvalho, G.; Luz, N.F.; Celes, F.S.; Zanette, D.L.; Andrade, D.; Menezes, D.; Tavares, N.M.; Brodskyn, C.I.; Prates, D.B.; Gonçalves, M.S.; et al. Heme drives oxidative stress-associated cell death in human neutrophils infected with Leishmania infantum. Front. Immunol. 2017, 8, 1620. [Google Scholar] [CrossRef] [PubMed]
- Nasimuzzaman, M.; Malik, P. Role of the coagulation system in the pathogenesis of sickle cell disease. Blood Adv. 2019, 3, 3170–3180. [Google Scholar] [CrossRef]
- Annarapu, G.K.; Nolfi-Donegan, D.; Reynolds, M.; Wang, Y.; Shiva, S. Mitochondrial reactive oxygen species scavenging attenuates thrombus formation in a murine model of sickle cell disease. J. Thromb. Haemost. 2021, 19, 2256–2262. [Google Scholar] [CrossRef]
- Balandya, E.; Reynolds, T.; Aboud, S.; Obaro, S.; Makani, J. Increased memory phenotypes of CD4+ and CD8+ T cells in children with sickle cell anaemia in Tanzania. Tanzan. J. Health Res. 2017, 19. [Google Scholar] [CrossRef]
- Daltro, P.B.; Ribeiro, T.O.; Daltro, G.C.; Meyer, R.J.; Fortuna, V. CD4+ T cell profile and activation response in sickle cell disease patients with osteonecrosis. Mediat. Inflamm. 2020, 9, 1747894. [Google Scholar] [CrossRef]
- Zerra, P.E.; Patel, S.R.; Jajosky, R.P.; Arthur, C.M.; McCoy, J.W.; Allen, J.W.L.; Chonat, S.; Fasano, R.M.; Roback, J.D.; Josephson, C.D.; et al. Marginal zone B cells mediate a CD4 T-cell–dependent extrafollicular antibody response following RBC transfusion in mice. Blood 2021, 138, 706–721. [Google Scholar] [CrossRef]
- Bernaudin, F.; Djavidi, A.; Arnaud, C.; Kamdem, A.; Hau, I.; Pondarré, C.; Vernant, J.-P.; Kuentz, M.; Dhedin, N.; Dalle, J.-H.; et al. Immune reconstitution in 107 children with sickle cell anemia transplanted with bone marrow or cord blood from a matched-sibling donor after myeloablative conditioning regimen including 20mg/Kg ATG. Blood 2019, 134, 2253. [Google Scholar] [CrossRef]
- Shokrgozar, N.; Amirian, N.; Ranjbaran, R.; Bazrafshan, A.; Sharifzadeh, S. Evaluation of regulatory T cells frequency and FoxP3/GDF-15 gene expression in β-thalassemia major patients with and without alloantibody; correlation with serum ferritin and folate levels. Ann. Hematol. 2020, 99, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Fasola, F.; Adekanmi, A. Haematological profile and blood transfusion pattern of patients with sickle cell anaemia vary with spleen size. Ann. Ib. Postgrad. Med. 2019, 17, 30–38. [Google Scholar] [PubMed]
- Ojo, O.T.; Ibijola, A.; Shokunbi, W.; Busari, O.; Olatunji, P.; Ganiyu, A. Correlation between splenic size and CD4+ T lymphocytes in sickle cell anaemia patients in a Tertiary Hospital. Egypt. J. Haematol. 2018, 43, 85. [Google Scholar] [CrossRef]
- ElAlfy, M.S.; Adly, A.A.M.; Ebeid, F.S.E.; Eissa, D.S.; Ismail, E.A.R.; Mohammed, Y.H.; Ahmed, M.E.; Saad, A.S. Immunological role of CD4+CD28null T lymphocytes, natural killer cells, and interferon-gamma in pediatric patients with sickle cell disease: Relation to disease severity and response to therapy. Immunol. Res. 2018, 66, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Boulassel, M.-R.; Al-Naamani, A.; Al-Zubaidi, A.; Al-Qarni, Z.; Khan, H.; Oukil, A.; Al-Badi, A.; Al-Kaabi, J.; Al-Shekaili, J.; Al-Hashmi, S.; et al. Coexistence of sickle cell disease and systemic lupus erythematosus is associated with quantitative and qualitative impairments in circulating regulatory B cells. Hum. Immunol. 2022, 83, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Fichou, Y.; Berlivet, I.; Richard, G.; Tournamille, C.; Castilho, L.; Férec, C. Defining blood group gene reference alleles by long-read sequencing: Proof of concept in the ACKR1 gene encoding the Duffy antigens. Transfus. Med. Hemotherapy 2020, 47, 23–32. [Google Scholar] [CrossRef]
- Thompson, K.; Adams, F.; Davison, G.M. Elevated unidentified antibodies in sickle cell anaemia patients receiving blood transfusions in Cape Town, South Africa. South Afr. Med. J. 2019, 109, 872–875. [Google Scholar] [CrossRef]
- Lopez, G.H.; Hyland, C.A.; Flower, R.L. Glycophorins and the MNS blood group system: A narrative review. Ann. Blood 2021, 6, 39. [Google Scholar] [CrossRef]
- Seck, M.; Senghor, A.B.; Loum, M.; Touré, S.A.; Faye, B.F.; Diallo, A.B.; Keita, M.; Bousso, E.S.; Guèye, S.M.; Gadji, M.; et al. Transfusion practice, post-transfusion complications and risk factors in Sickle Cell Disease in Senegal, West Africa. Mediterr. J. Hematol. Infect. Dis. 2022, 14, e2022004. [Google Scholar] [CrossRef] [PubMed]
- Molina-Aguilar, R.; Gómez-Ruiz, S.; Vela-Ojeda, J.; Montiel-Cervantes, L.A.; Reyes-Maldonado, E. Pathophysiology of Alloimmunization. Transfus. Med. Hemotherapy 2020, 47, 152–159. [Google Scholar] [CrossRef]
- Pal, M.; Bao, W.; Wang, R.; Liu, Y.; An, X.; Mitchell, W.B.; Lobo, C.A.; Minniti, C.P.; Shi, P.A.; Manwani, D.; et al. Hemolysis inhibits humoral B-cell responses and modulates alloimmunization risk in patients with sickle cell disease. Blood 2021, 137, 269–280. [Google Scholar] [CrossRef]
- Li-Thiao-Te, V.; Uettwiller, F.; Quartier, P.; Lacaille, F.; Bader-Meunier, B.; Brousse, V.; de Montalembert, M. Coexistent sickle-cell anemia and autoimmune disease in eight children: Pitfalls and challenges. Pediatr. Rheumatol. 2018, 16, 5. [Google Scholar] [CrossRef]
- Wang, R.H.; Phillips, G.; E Medof, M.; Mold, C. Activation of the alternative complement pathway by exposure of phosphatidylethanolamine and phosphatidylserine on erythrocytes from sickle cell disease patients. J. Clin. Investig. 1993, 92, 1326–1335. [Google Scholar] [CrossRef]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.-L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 2018, 3, e96910. [Google Scholar] [CrossRef]
- Vercellotti, G.M.; Dalmasso, A.P.; Schaid, T.R.; Nguyen, J.; Chen, C.; Ericson, M.E.; Abdulla, F.; Killeen, T.; Lindorfer, M.A.; Taylor, R.P.; et al. Critical role of C5a in sickle cell disease. Am. J. Hematol. 2019, 94, 327–337. [Google Scholar] [CrossRef]
- Gkaliagkousi, E.; Gavriilaki, E.; Vasileiadis, I.; Nikolaidou, B.; Yiannaki, E.; Lazaridis, A.; Triantafyllou, A.; Anyfanti, P.; Markala, D.; Zarifis, I.; et al. Endothelial microvesicles circulating in peripheral and coronary circulation are associated with central blood pressure in coronary artery disease. Am. J. Hypertens. 2019, 32, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- De Vlam, K.; De Keyser, F.; Verbruggen, G.; Vandenbossche, M.; Vanneuville, B.; D’Haese, D.; Veys, E.M. Detection and identification of antinuclear autoantibodies in the serum of normal blood donors. Clin. Exp. Rheumatol. 1993, 11, 393–397. [Google Scholar] [PubMed]
- Adebajo, A.O.; Charles, P.; Maini, R.N.; Hazleman, B.L. Autoantibodies in malaria, tuberculosis and hepatitis B in a west African population. Clin. Exp. Immunol. 1993, 92, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Baethge, B.A.; Bordelon, T.R.; Mills, G.M.; Bowen, M.; Wolf, R.E.; Bairnsfather, L. Antinuclear antibodies in sickle cell disease. Acta Haematol. 1990, 84, 186–189. [Google Scholar] [CrossRef]
- Toly-Ndour, C.; Rouquette, A.-M.; Obadia, S.; M’bappe, P.; Lionnet, F.; Hagege, I.; Boussa-Khettab, F.; Tshilolo, L.; Girot, R. High titers of autoantibodies in patients with sickle-cell disease. J. Rheumatol. 2011, 38, 302–309. [Google Scholar] [CrossRef]
- Balsalobre, B.; Hernández-Godoy, J.; Planelles, D. Autoantibodies in splenectomized patients as a consequence of abdominal trauma. J. Investig. Allergol. Clin. Immunol. 1992, 2, 91–95. [Google Scholar]
- Nistala, K.; Murray, K.J. Co-existent sickle cell disease and juvenile rheumatoid arthritis. Two cases with delayed diagnosis and severe destructive arthropathy. J. Rheumatol. 2001, 28, 2125–2128. [Google Scholar]
- Saxena, V.R.; Mina, R.; Moallem, H.J.; Rao, S.P.; Miller, S.T. Systemic lupus erythematosus in children with sickle cell disease. J. Pediatr. Hematol./Oncol. 2003, 25, 668–671. [Google Scholar] [CrossRef]
- Lykavieris, P.; Benichou, J.-J.; Benkerrou, M.; Feriot, J.-P.; Bernard, O.; Debray, D. Autoimmune liver disease in three children with sickle cell disease. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Bernini, J.C.; Rogers, Z.R.; Sandler, E.S.; Reisch, J.S.; Quinn, C.T.; Buchanan, G.R. Beneficial effect of intravenous dexamethasone in children with mild to moderately severe acute chest syndrome complicating sickle cell disease. Blood J. Am. Soc. Hematol. 1998, 92, 3082–3089. [Google Scholar]
- Michel, M.; Habibi, A.; Godeau, B.; Bachir, D.; Lahary, A.; Galacteros, F.; Fifi-Mah, A.; Arfi, S. Characteristics and outcome of connective tissue diseases in patients with sickle-cell disease: Report of 30 cases. Semin. Arthritis Rheum. 2008, 38, 228–240. [Google Scholar] [CrossRef]
- Solovey, A.; Somani, A.; Chen, C.; Kiley, J.; Marker, P.; Abdulla, F.; O’Sullivan, M.G.; Kelm Robert, J., Jr.; Belcher, J.D.; Vercellotti, G.M.; et al. Interference with TNFα using long-term etanercept in S+ SAntilles sickle transgenic mice ameliorates abnormal endothelial activation, vasoocclusion, and pulmonary hypertension including its pulmonary arterial wall remodeling. Blood 2013, 122, 728. [Google Scholar] [CrossRef]
- Wang, Q.; Zennadi, R. The role of RBC oxidative stress in sickle cell disease: From the molecular basis to pathologic implications. Antioxidants 2021, 10, 1608. [Google Scholar] [CrossRef]
- Cao, H.; Vickers, M.A. Oxidative stress, malaria, sickle cell disease, and innate immunity. Trends Immunol. 2021, 42, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhou, X. Octamer-binding transcription factor 4 correlates with complex karyotype, FLT3-ITD mutation and poorer risk stratification, and predicts unfavourable prognosis in patients with acute myeloid leukaemia. Hematology 2018, 23, 721–728. [Google Scholar] [CrossRef]
- Bernard, K.F.C.; Cabral, B.N.P.; Bernard, C.; Flora, N.L.; Anatole, P.C.; Donatien, G. Electrolytic and oxidative stress profile of sickle cell anaemia patients in Cameroon: The effect of some extrinsic factors. Asian Hematol. Res J. 2018, 1, 13–23. [Google Scholar]
- Beri, D.; Singh, M.; Rodriguez, M.; Yazdanbakhsh, K.; Lobo, C.A. Sickle cell anemia and Babesia infection. Pathogens 2021, 10, 1435. [Google Scholar] [CrossRef]
- Bou-Fakhredin, R.; De Franceschi, L.; Motta, I.; Eid, A.A.; Taher, A.T.; Cappellini, M.D. Redox Balance in β-Thalassemia and Sickle Cell Disease: A Love and Hate Relationship. Antioxidants 2022, 11, 967. [Google Scholar] [CrossRef]
- Soomro, S. Oxidative stress and inflammation. Open J. Immunol. 2019, 9, 1. [Google Scholar] [CrossRef]
- Pedrosa, A.M.; Leal, L.K.A.; Lemes, R.P.G. Effects of hydroxyurea on cytotoxicity, inflammation and oxidative stress markers in neutrophils of patients with sickle cell anemia: Dose-effect relationship. Hematol. Transfus. Cell Ther. 2021, 43, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Glennon-Alty, L.; Hackett, A.P.; Chapman, E.A.; Wright, H.L. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free. Radic. Biol. Med. 2018, 125, 25–35. [Google Scholar] [CrossRef]
- Ito, F.; Sono, Y.; Ito, T. Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: Oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Piacenza, L.; Trujillo, M.; Radi, R. Reactive species and pathogen antioxidant networks during phagocytosis. J. Exp. Med. 2019, 216, 501–516. [Google Scholar] [CrossRef]
- Cervantes-Gracia, K.; Raja, K.; Llanas-Cornejo, D.; Cobley, J.N.; Megson, I.L.; Chahwan, R.; Husi, H. Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy. Vessel. Plus 2020, 4, 27. [Google Scholar] [CrossRef]
- Ojongnkpot, T.A.; Sofeu-Feugaing, D.D.; Jugha, V.T.; Taiwe, G.S.; Kimbi, H.K. Implication of Oxidative Stress and Antioxidant Defence Systems in Symptomatic and Asymptomatic Plasmodium falciparum Malaria Infection among Children Aged1 to 15 Years in the Mount Cameroon Area. J. Biosci. Med. 2023, 11, 124–145. [Google Scholar]
- Bohn, T. Carotenoids and markers of oxidative stress in human observational studies and intervention trials: Implications for chronic diseases. Antioxidants 2019, 8, 179. [Google Scholar] [CrossRef]
- Detterich, J.A.; Liu, H.; Suriany, S.; Kato, R.M.; Chalacheva, P.; Tedla, B.; Shah, P.M.; Khoo, M.C.; Wood, J.C.; Coates, T.D.; et al. Erythrocyte and plasma oxidative stress appears to be compensated in patients with sickle cell disease during a period of relative health, despite the presence of known oxidative agents. Free. Radic. Biol. Med. 2019, 141, 408–415. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Guillot, N.; Fort, R.; Stauffer, E.; Lemonne, N.; Garnier, Y.; Skinner, S.C.; Etienne-Julan, M.; Robert, M.; et al. Association between nitric oxide, oxidative stress, eryptosis, red blood cell microparticles, and vascular function in sickle cell anemia. Front. Immunol. 2020, 11, 551441. [Google Scholar] [CrossRef]
- El Azab, E.F.; Saleh, A.M.; Yousif, S.O.; Mazhari, B.B.Z.; Abu Alrub, H.; Elfaki, E.M.; Hamza, A.; Abdulmalek, S. New insights into geraniol’s antihemolytic, anti-inflammatory, antioxidant, and anticoagulant potentials using a combined biological and in silico screening strategy. Inflammopharmacology 2022, 30, 1811–1833. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zennadi, R. Oxidative stress and thrombosis during aging: The roles of oxidative stress in RBCs in venous thrombosis. Int. J. Mol. Sci. 2020, 21, 4259. [Google Scholar] [CrossRef] [PubMed]
- Abboud, M.R. Standard management of sickle cell disease complications. Hematol./Oncol. Stem Cell Ther. 2020, 13, 85–90. [Google Scholar] [CrossRef] [PubMed]
- McMahon, T.J. Red blood cell deformability, vasoactive mediators, and adhesion. Front. Physiol. 2019, 10, 1417. [Google Scholar] [CrossRef]
- Kucukal, E.; Man, Y.; Hill, A.; Liu, S.; Bode, A.; An, R.; Kadambi, J.; Little, J.A.; Gurkan, U.A. Whole blood viscosity and red blood cell adhesion: Potential biomarkers for targeted and curative therapies in sickle cell disease. Am. J. Hematol. 2020, 95, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W. Heme oxygenase-1: An anti-inflammatory effector in cardiovascular, lung, and related metabolic disorders. Antioxidants 2022, 11, 555. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W. Therapeutic potential of heme oxygenase-1 and carbon monoxide in acute organ injury, critical illness, and inflammatory disorders. Antioxidants 2020, 9, 1153. [Google Scholar] [CrossRef]
- Kim, H.; Moore, C.M.; Mestre-Fos, S.; A Hanna, D.; Williams, L.D.; Reddi, A.R.; Torres, M.P. Depletion Assisted Hemin Affinity (DAsHA) Proteomics Reveals an Expanded Landscape of Heme Binding Proteins in the Human Proteome. Metallomics 2023, 15, mfad004. [Google Scholar] [CrossRef]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme oxygenase-1 signaling and redox homeostasis in physiopathological conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef]
- Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of heme oxygenase as a modulator of heme-mediated pathways. Antioxidants 2019, 8, 475. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Beauvillain, C.; Delneste, Y.; Scotet, M.; Peres, A.; Gascan, H.; Guermonprez, P.; Barnaba, V.; Jeannin, P. Neutrophils efficiently cross-prime naive T cells in vivo. Blood J. Am. Soc. Hematol. 2007, 110, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Cassatella, M.A. The defensive alliance between neutrophils and NK cells as a novel arm of innate immunity. J. Leukoc. Biol. 2011, 89, 221–233. [Google Scholar] [CrossRef]
- Lee, S.-K.; Goh, S.-Y.; Wong, Y.-Q.; Ding, J.-L. Response of Neutrophils to Extracellular Haemoglobin and LTA in Human Blood System. EBioMedicine 2015, 2, 225–233. [Google Scholar] [CrossRef]
- Toledo, S.L.d.O.; Ladeira, V.S.; Nogueira, L.S.; Ferreira, L.G.R.; Oliveira, M.M.; Renó, C.d.O.; dos Santos, H.L.; Coelho-Dos-Reis, J.G.A.; Campi-Azevedo, A.C.; Teixeira-Carvalho, A.; et al. Plasma immune mediators as laboratorial biomarkers for Sickle Cell Disease patients according to the hydroxyurea therapy and disease severity. Blood Cells Mol. Dis. 2023, 98, 102703. [Google Scholar] [CrossRef]
- Hendrickson, J.E. Red blood cell alloimmunization and sickle cell disease: A narrative review on antibody induction. Ann. Blood 2020, 5, 33. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Yildirim, A.; Granger, D.N.; Gavins, F.N. Novel Role of T Cells and IL-6 (Interleukin-6) in angiotensin II–induced microvascular dysfunction. Hypertension 2019, 73, 829–838. [Google Scholar] [CrossRef]
- Wasnik, R.R.; Akarte, N.R.; Dhoke, A.; Anjankar, A.P. Impact of Oxidative stress on Sickle cell anaemia patients: A Review. Nveo-Nat. Volatiles Essent. Oils J.|NVEO 2021, 8, 1128–1134. [Google Scholar]
- Connes, P.; Möckesch, B.; Sock, E.T.N.; Hardy-Dessources, M.; Reminy, K.; Skinner, S.; Billaud, M.; Nader, E.; Tressieres, B.; Etienne-Julan, M.; et al. Oxidative stress, inflammation, blood rheology, and microcirculation in adults with sickle cell disease: Effects of hydroxyurea treatment and impact of sickle cell syndrome. Eur. J. Haematol. 2021, 106, 800–807. [Google Scholar] [CrossRef]
- Netea, M.G.; Schlitzer, A.; Placek, K.; Joosten, L.A.; Schultze, J.L. Innate and adaptive immune memory: An evolutionary continuum in the host’s response to pathogens. Cell Host Microbe 2019, 25, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Tavares, W.R.; Seca, A.M. Inula L. secondary metabolites against oxidative stress-related human diseases. Antioxidants 2019, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Ouyang, Y.; Lu, N.; Li, N. The NF-κB signaling pathway, the microbiota, and gastrointestinal tumorigenesis: Recent advances. Front. Immunol. 2020, 11, 1387. [Google Scholar] [CrossRef]
- Renó, C.O.; Barbosa, A.R.; de Carvalho, S.S.; Pinheiro, M.B.; Rios, D.R.; Cortes, V.F.; Barbosa, L.A.; Santos, H.L. Oxidative stress assessment in sickle cell anemia patients treated with hydroxyurea. Ann. Hematol. 2020, 99, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From subclinical condition to pathological biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef]
- Trevelin, S.C.; Shah, A.M.; Lombardi, G. Beyond bacterial killing: NADPH oxidase 2 is an immunomodulator. Immunol. Lett. 2020, 221, 39–48. [Google Scholar] [CrossRef]
- Gan, A.M.; Tracz-Gaszewska, Z.; Ellert-Miklaszewska, A.; Navrulin, V.O.; Ntambi, J.M.; Dobrzyn, P. Stearoyl-CoA Desaturase Regulates Angiogenesis and Energy Metabolism in Ischemic Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 10459. [Google Scholar] [CrossRef]
- Chou, S.T.; Alsawas, M.; Fasano, R.M.; Field, J.J.; Hendrickson, J.E.; Howard, J.; Kameka, M.; Kwiatkowski, J.L.; Pirenne, F.; Shi, P.A.; et al. American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support. Blood Adv. 2020, 4, 327–355. [Google Scholar] [CrossRef]
- Balbuena-Merle, R.; Santhanakrishnan, M.; Devine, L.; Gibb, D.R.; Tormey, C.A.; Siddon, A.J.; Curtis, S.A.; Gallagher, P.G.; Weinstein, J.S.; Hendrickson, J.E. Characterization of circulating and cultured Tfh-like cells in sickle cell disease in relation to red blood cell alloimmunization status. Transfus. Apher. Sci. 2020, 59, 102778. [Google Scholar] [CrossRef]
- Meda, E.; Magesa, P.M.; Marlow, T.; Reid, C.; Roberts, D.J.; Makani, J. Red blood cell alloimmunization in sickle cell disease patients in Tanzania. East Afr. J. Public Health 2014, 11, 775. [Google Scholar]
- Firmansyah, M.; Abduh, M. Production of protein hydrolysate containing antioxidant activity from Hermetia illucens. Heliyon 2019, 5, e02005. [Google Scholar] [CrossRef] [PubMed]
- Cherif-Alami, S.; Hau, I.; Arnaud, C.; Kamdem, A.; Coulon, B.; Idoux, E.; Bechet, S.; Creidy, R.; Bernaudin, F.; Epaud, R.; et al. Serum immunoglobulin levels in children with sickle cell disease: A large prospective study. J. Clin. Med. 2019, 8, 1688. [Google Scholar] [CrossRef]
- Boateng, L.A.; Ngoma, A.M.; Bates, I.; Schonewille, H. Red blood cell alloimmunization in transfused patients with sickle cell disease in sub-Saharan Africa; a systematic review and meta-analysis. Transfus. Med. Rev. 2019, 33, 162–169. [Google Scholar] [CrossRef] [PubMed]
- El Fetouh, R.M.A.; Elmoniem, G.M.A.; Allam, R.M.; Sobeih, M.E.; Kamel, M.M.; Radwan, S.M. Frequency and specificity of Red blood cell alloantibodies in multitransfused Egyptian patients with hematological and nonhematological malignancies. Transfus. Apher. Sci. 2020, 59, 102909. [Google Scholar] [CrossRef] [PubMed]
- Adewoyin, A.; Daramola, O.; Ogbenna, A.; Adeyemo, T. Immune erythrocyte antibodies in adult patients with sickle cell disease and blood donors in Lagos, Nigeria: A comparative study. Immunohematology 2021, 37, 131–137. [Google Scholar] [CrossRef]
- Subramaniyan, R. Serological characteristics of Lewis antibodies and their clinical significance–A case series. Hematol. Transfus. Cell Ther. 2021, 45, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Lamarre, Y.; Nader, E.; Connes, P.; Romana, M.; Garnier, Y. Extracellular Vesicles in Sickle Cell Disease: A Promising Tool. Bioengineering 2022, 9, 439. [Google Scholar] [CrossRef]
- McGann, P.T.; Tshilolo, L.; Santos, B.; Tomlinson, G.A.; Stuber, S.; Latham, T.; Aygun, B.; Obaro, S.K.; Olupot-Olupot, P.; Williams, T.N.; et al. Hydroxyurea therapy for children with sickle cell anemia in sub-saharan africa: Rationale and design of the REACH trial. Pediatr. Blood Cancer 2016, 63, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Barbu, E.A.; Dominical, V.M.; Mendelsohn, L.; Thein, S.L. Neutrophils remain detrimentally active in hydroxyurea-treated patients with sickle cell disease. PLoS ONE 2019, 14, e0226583. [Google Scholar] [CrossRef]
- Hutchaleelaha, A.; Patel, M.; Washington, C.; Siu, V.; Allen, E.; Oksenberg, D.; Gretler, D.D.; Mant, T.; Lehrer-Graiwer, J. Pharmacokinetics and pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. Br. J. Clin. Pharmacol. 2019, 85, 1290–1302. [Google Scholar] [CrossRef]
- Zaidi, A.U.; Estepp, J.; Shah, N.; Alkindi, S.; Ezzat, H.; Lam, H.; Minniti, C.P. A reanalysis of pain crises data from the pivotal l-glutamine in sickle cell disease trial. Contemp. Clin. Trials 2021, 110, 106546. [Google Scholar] [CrossRef] [PubMed]
- Dick, M.H.; Abdelgadir, A.; Kulkarni, V.V.; Akram, H.; Chatterjee, A.; Pokhrel, S.; Khan, S. Comparing the safety and efficacy of L-glutamine, voxelotor, and crizanlizumab for reducing the frequency of vaso-occlusive crisis in sickle cell disease: A systematic review. Cureus 2022, 14, e24920. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Zhang, B.-S.; Chen, Y.-P.; Lv, J.-L.; Yang, Y. Comparison of the efficacy of nilotinib and imatinib in the treatment of chronic myeloid leukemia. J. Coll. Physicians Surg. Pak. 2019, 29, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Kulturoglu, G.; Ergil, J.; O Kulturoglu, M.; Yalcindag, A.; Onder, E.O. The effects of hydroxyurea on proinflammatory cytokine and tissue histopathology in an experimental sepsis model. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 526–533. [Google Scholar]
- Jayasinghe, C.D.; Ratnasooriya, W.D.; Premakumara, S.; Udagama, P.V. Platelet augmentation activity of mature leaf juice of Sri Lankan wild type cultivar of Carica papaya L: Insights into potential cellular mechanisms. J. Ethnopharmacol. 2022, 296, 115511. [Google Scholar] [CrossRef]
- Cominal, J.G.; Cacemiro, M.D.C.; Berzoti-Coelho, M.G.; Pereira, I.E.G.; Frantz, F.G.; Souto, E.X.; Covas, D.T.; de Figueiredo-Pontes, L.L.; Oliveira, M.C.; Malmegrim, K.C.R.; et al. Bone marrow soluble mediator signatures of patients with philadelphia chromosome-negative myeloproliferative neoplasms. Front. Oncol. 2021, 11, 665037. [Google Scholar] [CrossRef]
- Cacciola, R.; Cacciola, E.G.; Vecchio, V.; Cacciola, E. Impact of Anti-Endothelial Cell Antibodies (AECAs) in Patients with Polycythemia Vera and Thrombosis. Diagnostics 2022, 12, 1077. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboderin, F.I.; Oduola, T.; Davison, G.M.; Oguntibeju, O.O. A Review of the Relationship between the Immune Response, Inflammation, Oxidative Stress, and the Pathogenesis of Sickle Cell Anaemia. Biomedicines 2023, 11, 2413. https://doi.org/10.3390/biomedicines11092413
Aboderin FI, Oduola T, Davison GM, Oguntibeju OO. A Review of the Relationship between the Immune Response, Inflammation, Oxidative Stress, and the Pathogenesis of Sickle Cell Anaemia. Biomedicines. 2023; 11(9):2413. https://doi.org/10.3390/biomedicines11092413
Chicago/Turabian StyleAboderin, Florence Ifechukwude, Taofeeq Oduola, Glenda Mary Davison, and Oluwafemi Omoniyi Oguntibeju. 2023. "A Review of the Relationship between the Immune Response, Inflammation, Oxidative Stress, and the Pathogenesis of Sickle Cell Anaemia" Biomedicines 11, no. 9: 2413. https://doi.org/10.3390/biomedicines11092413