
Stiff Person Spectrum Disorders—An Update and Outlook on Clinical, Pathophysiological and Treatment Perspectives

Abstract
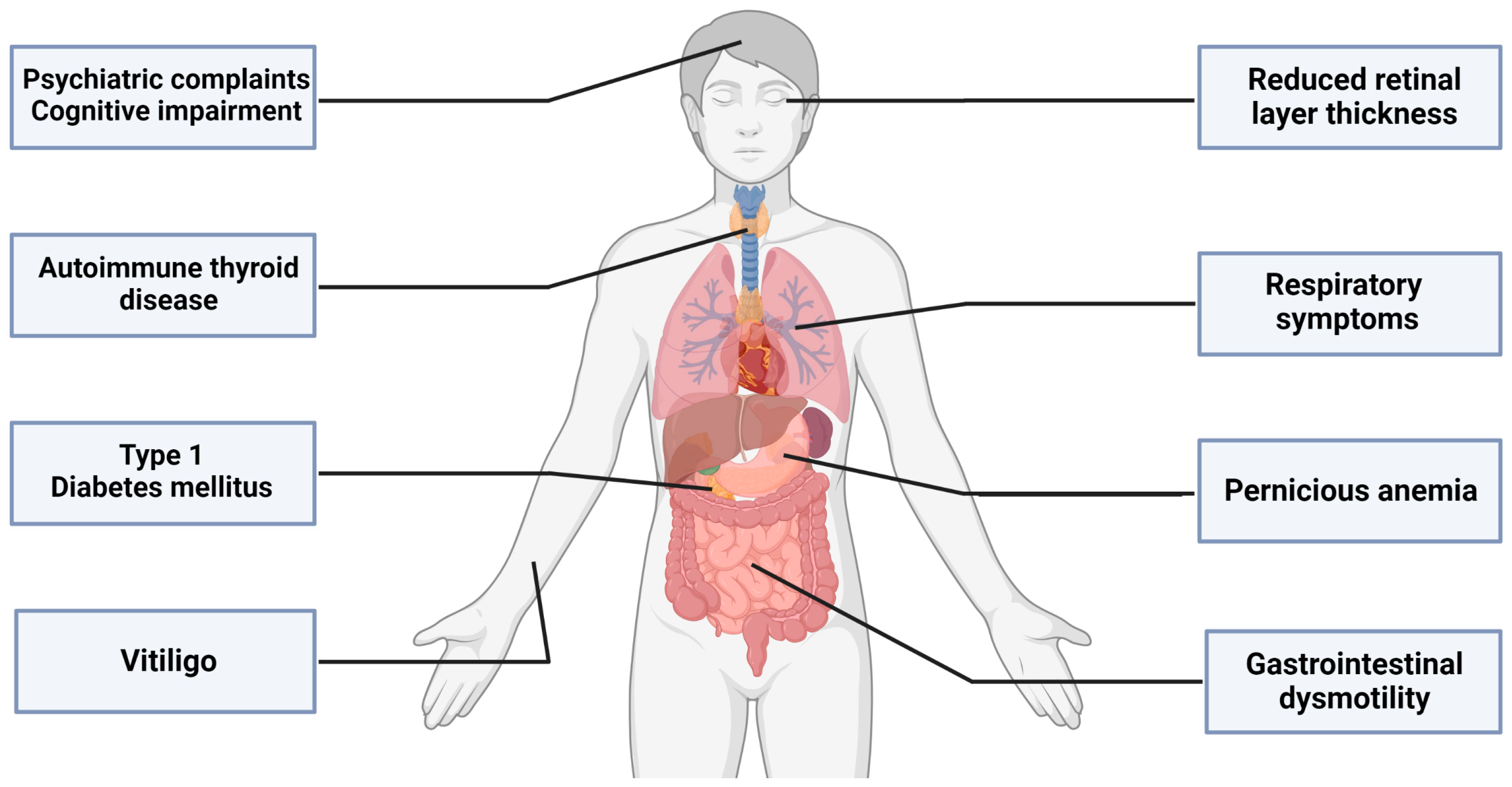
:1. Introduction
2. The Emerging Clinical Spectrum beyond the Motor Symptoms of SPSD
2.1. More than Meets the Eye: The Retina
2.2. Difficult to Swallow, Hard to Digest: The Gastrointestinal System
2.3. Gather your Breath: The Respiratory System
2.4. Mind Matters—Psychiatric and Cognitive Aspects
3. The Diagnosis—About Swallows, Summers and Antibodies
4. About Dogs, Horses, Mice and Men in SPSD
5. The Hen and the Egg: B Cells and T Cells in the Anti-GAD65 Immunopathophysiology
6. Anti-GAD65-Associated Neurological Syndromes and Genetics
7. Relevance of the Different Immunopathophysiologies for Therapeutic Strategies
8. Immunomodulatory Therapies—State of the Art and Future Outlook
9. Special Treatment Considerations: Pregnancy with SPSD and SPSD in Childhood
10. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Celine Dion’s Diagnosis Raises Awareness of Stiff Person Syndrome. 2023. Available online: https://www.brainandlife.org/articles/celine-dions-diagnosis-stiff-person-syndrome (accessed on 31 July 2023).
- Moersch, F.P.; Woltman, H.W. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome); report of a case and some observations in 13 other cases. Proc. Staff. Meet. Mayo Clin. 1956, 31, 421–427. [Google Scholar]
- Solimena, M.; Folli, F.; Denis-Donini, S.; Comi, G.C.; Pozza, G.; De Camilli, P.; Vicari, A.M. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N. Engl. J. Med. 1988, 318, 1012–1020. [Google Scholar] [CrossRef]
- Newsome, S.D.; Johnson, T. Stiff person syndrome spectrum disorders; more than meets the eye. J. Neuroimmunol. 2022, 369, 577915. [Google Scholar] [CrossRef]
- Dalakas, M.C. Therapies in Stiff-Person Syndrome: Advances and Future Prospects Based on Disease Pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gorin, F.; Baldwin, B.; Tait, R.; Pathak, R.; Seyal, M.; Mugnaini, E. Stiff-man syndrome: A GABAergic autoimmune disorder with autoantigenic heterogeneity. Ann. Neurol. 1990, 28, 711–714. [Google Scholar] [CrossRef] [PubMed]
- De Camilli, P.; Thomas, A.; Cofiell, R.; Folli, F.; Lichte, B.; Piccolo, G.; Meinck, H.M.; Austoni, M.; Fassetta, G.; Bottazzo, G.; et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of Stiff-Man syndrome with breast cancer. J. Exp. Med. 1993, 178, 2219–2223. [Google Scholar] [CrossRef] [PubMed]
- Dropcho, E.J. Antiamphiphysin antibodies with small-cell lung carcinoma and paraneoplastic encephalomyelitis. Ann. Neurol. 1996, 39, 659–667. [Google Scholar] [CrossRef]
- Hutchinson, M.; Waters, P.; McHugh, J.; Gorman, G.; O’Riordan, S.; Connolly, S.; Hager, H.; Yu, P.; Becker, C.M.; Vincent, A. Progressive encephalomyelitis, rigidity, and myoclonus: A novel glycine receptor antibody. Neurology 2008, 71, 1291–1292. [Google Scholar] [CrossRef] [PubMed]
- McKeon, A.; Martinez-Hernandez, E.; Lancaster, E.; Matsumoto, J.Y.; Harvey, R.J.; McEvoy, K.M.; Pittock, S.J.; Lennon, V.A.; Dalmau, J. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013, 70, 44–50. [Google Scholar] [CrossRef]
- Balint, B.; Jarius, S.; Nagel, S.; Haberkorn, U.; Probst, C.; Blocker, I.M.; Bahtz, R.; Komorowski, L.; Stocker, W.; Kastrup, A.; et al. Progressive encephalomyelitis with rigidity and myoclonus: A new variant with DPPX antibodies. Neurology 2014, 82, 1521–1528. [Google Scholar] [CrossRef]
- Martinez-Hernandez, E.; Arino, H.; McKeon, A.; Iizuka, T.; Titulaer, M.J.; Simabukuro, M.M.; Lancaster, E.; Petit-Pedrol, M.; Planaguma, J.; Blanco, Y.; et al. Clinical and Immunologic Investigations in Patients with Stiff-Person Spectrum Disorder. JAMA Neurol. 2016, 73, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Budhram, A.; Sechi, E.; Flanagan, E.P.; Dubey, D.; Zekeridou, A.; Shah, S.S.; Gadoth, A.; Naddaf, E.; McKeon, A.; Pittock, S.J.; et al. Clinical spectrum of high-titre GAD65 antibodies. J. Neurol. Neurosurg. Psychiatry 2021, 92, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Meinck, H.M.; Thompson, P.D. Stiff man syndrome and related conditions. Mov. Disord. 2002, 17, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.P.; Pires, L.A.; Cruzeiro, M.M.; Almeida, A.L.M.; Martins, L.C.; Martins, P.N.; Shigaeff, N.; Vale, T.C. Optical coherence tomography in neurodegenerative disorders. Arq. Neuropsiquiatr. 2022, 80, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Lambe, J.; Rothman, A.; Prince, J.; Saidha, S.; Calabresi, P.A.; Newsome, S.D. Retinal pathology occurs in stiff-person syndrome. Neurology 2020, 94, e2126–e2131. [Google Scholar] [CrossRef]
- Jessen, K.R. GABA and the enteric nervous system. A neurotransmitter function? Mol. Cell Biochem. 1981, 38, 69–76. [Google Scholar] [CrossRef]
- Dumitrascu, O.M.; Tsimerinov, E.I.; Lewis, R.A. Gastrointestinal and Urologic Sphincter Dysfunction in Stiff Person Syndrome. J. Clin. Neuromuscul. Dis. 2016, 18, 92–95. [Google Scholar] [CrossRef]
- Koshorek, J.; Maldonado, D.P.; Reyes-Mantilla, M.; Obando, D.; Wang, Y.; Newsome, S. The evolving spectrum of gastrointestinal dysfunction in stiff person syndrome (4200). Neurology 2021, 96, 4200. [Google Scholar]
- Piepgras, J.; Holtje, M.; Michel, K.; Li, Q.; Otto, C.; Drenckhahn, C.; Probst, C.; Schemann, M.; Jarius, S.; Stocker, W.; et al. Anti-DPPX encephalitis: Pathogenic effects of antibodies on gut and brain neurons. Neurology 2015, 85, 890–897. [Google Scholar] [CrossRef]
- Pimentel Maldonado, D.A.; Balshi, A.; Hu, C.; Fitzgerald, K.C.; Koshorek, J.; Reyes-Mantilla, M.I.; Obando, D.; Wang, Y.; Newsome, S.D. Respiratory symptoms are common in stiff person syndrome spectrum disorders and are associated with number of body regions involved. Eur. J. Neurol. 2023, 30, 2498–2505. [Google Scholar] [CrossRef]
- Allen, A.; Rakocevic, G.; Woodford, M.; Pack, K.; Sexauer, W. Unrecognized Respiratory Manifestations of Stiff Person Syndrome (SPS) (P5.463). Neurology 2018, 90, 15. [Google Scholar]
- Cosentino, G.; Romano, M.; Algeri, M.; Brighina, F.; Alfonsi, E.; Tassorelli, C.; Crescimanno, G. Intranasal midazolam for treating acute respiratory crises in a woman with stiff person syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e715. [Google Scholar] [CrossRef] [PubMed]
- Nasri, A.; Gharbi, A.; Ouali, U.; Mrabet, S.; Souissi, A.; Jomli, R.; Gargouri, A.; Bendjebara, M.; Kacem, I.; Gouider, R. Psychiatric Symptoms in Stiff-Person Syndrome: A Systematic Review and a Report of Two Cases. J. Acad. Consult. Liaison Psychiatry 2023, 64, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Benavides, D.R.; Newsome, S.D. Serotonin-norepinephrine reuptake inhibitors may exacerbate stiff-person syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e281. [Google Scholar] [CrossRef]
- Meinck, H.M.; Ricker, K.; Conrad, B. The stiff-man syndrome: New pathophysiological aspects from abnormal exteroceptive reflexes and the response to clomipramine, clonidine, and tizanidine. J. Neurol. Neurosurg. Psychiatry 1984, 47, 280–287. [Google Scholar] [CrossRef]
- Chan, C.K.; Pimentel Maldonado, D.A.; Wang, Y.; Obando, D.; Hughes, A.J.; Newsome, S.D. Cognitive and Mood Profiles Among Patients with Stiff Person Syndrome Spectrum Disorders. Front. Neurol. 2022, 13, 865462. [Google Scholar] [CrossRef] [PubMed]
- Barker, M.J.; Greenwood, K.M.; Jackson, M.; Crowe, S.F. Persistence of cognitive effects after withdrawal from long-term benzodiazepine use: A meta-analysis. Arch. Clin. Neuropsychol. 2004, 19, 437–454. [Google Scholar] [CrossRef]
- Brieler, J.A.; Salas, J.; Amick, M.E.; Sheth, P.; Keegan-Garrett, E.A.; Morley, J.E.; Scherrer, J.F. Anxiety disorders, benzodiazepine prescription, and incident dementia. J. Am. Geriatr. Soc. 2023. [Google Scholar] [CrossRef]
- Torres-Bondia, F.; Dakterzada, F.; Galvan, L.; Buti, M.; Besanson, G.; Grill, E.; Buil, R.; de Batlle, J.; Pinol-Ripoll, G. Benzodiazepine and Z-Drug Use and the Risk of Developing Dementia. Int. J. Neuropsychopharmacol. 2022, 25, 261–268. [Google Scholar] [CrossRef]
- Balint, B.; Vincent, A.; Meinck, H.M.; Irani, S.R.; Bhatia, K.P. Movement disorders with neuronal antibodies: Syndromic approach, genetic parallels and pathophysiology. Brain 2018, 141, 13–36. [Google Scholar] [CrossRef]
- Flanagan, E.P.; Geschwind, M.D.; Lopez-Chiriboga, A.S.; Blackburn, K.M.; Turaga, S.; Binks, S.; Zitser, J.; Gelfand, J.M.; Day, G.S.; Dunham, S.R.; et al. Autoimmune Encephalitis Misdiagnosis in Adults. JAMA Neurol. 2023, 80, 30–39. [Google Scholar] [CrossRef]
- Chia, N.H.; McKeon, A.; Dalakas, M.C.; Flanagan, E.P.; Bower, J.H.; Klassen, B.T.; Dubey, D.; Zalewski, N.L.; Duffy, D.; Pittock, S.J. Stiff person spectrum disorder diagnosis, misdiagnosis, and suggested diagnostic criteria. Ann. Clin. Transl. Neurol. 2023, 10, 1083–1094. [Google Scholar] [CrossRef]
- Tsiortou, P.; Alexopoulos, H.; Dalakas, M.C. GAD antibody-spectrum disorders: Progress in clinical phenotypes, immunopathogenesis and therapeutic interventions. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211003486. [Google Scholar] [CrossRef] [PubMed]
- Meinck, H.M.; Faber, L.; Morgenthaler, N.; Seissler, J.; Maile, S.; Butler, M.; Solimena, M.; DeCamilli, P.; Scherbaum, W.A. Antibodies against glutamic acid decarboxylase: Prevalence in neurological diseases. J. Neurol. Neurosurg. Psychiatry 2001, 71, 100–103. [Google Scholar] [CrossRef]
- Ruiz-Garcia, R.; Martinez-Hernandez, E.; Saiz, A.; Dalmau, J.; Graus, F. The Diagnostic Value of Onconeural Antibodies Depends on How They Are Tested. Front. Immunol. 2020, 11, 1482. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Bhatia, K.P.; Dalmau, J. “Antibody of Unknown Significance” (AUS): The Issue of Interpreting Antibody Test Results. Mov. Disord. 2021, 36, 1543–1547. [Google Scholar] [CrossRef] [PubMed]
- Palace, J.; Fuller, G. Don’t do the blood* test!! Pract. Neurol. 2020, 20, 428–429. [Google Scholar] [CrossRef]
- Blinder, T.; Lewerenz, J. Cerebrospinal Fluid Findings in Patients with Autoimmune Encephalitis-A Systematic Analysis. Front. Neurol. 2019, 10, 804. [Google Scholar] [CrossRef]
- Petit-Pedrol, M.; Armangue, T.; Peng, X.; Bataller, L.; Cellucci, T.; Davis, R.; McCracken, L.; Martinez-Hernandez, E.; Mason, W.P.; Kruer, M.C.; et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: A case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014, 13, 276–286. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Li, M.; Fujii, M.; Jacobowitz, D.M. Stiff person syndrome: Quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology 2001, 57, 780–784. [Google Scholar] [CrossRef]
- Reiber, H.; Peter, J.B. Cerebrospinal fluid analysis: Disease-related data patterns and evaluation programs. J. Neurol. Sci. 2001, 184, 101–122. [Google Scholar] [CrossRef]
- Jarius, S.; Stich, O.; Speck, J.; Rasiah, C.; Wildemann, B.; Meinck, H.M.; Rauer, S. Qualitative and quantitative evidence of anti-glutamic acid decarboxylase-specific intrathecal antibody synthesis in patients with stiff person syndrome. J. Neuroimmunol. 2010, 229, 219–224. [Google Scholar] [CrossRef]
- Nollet, H.; Vanderstraeten, G.; Sustronck, B.; Van Ham, L.; Ziegler, M.; Deprez, P. Suspected case of stiff-horse syndrome. Vet. Rec. 2000, 146, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Cantatore, F.; Marcatili, M.; Withers, J. First Case of Stiff-Horse Syndrome in United Kingdom. J. Equine Vet. Sci. 2022, 116, 104022. [Google Scholar] [CrossRef] [PubMed]
- Pancotto, T.E.; Rossmeisl, J.H., Jr. A case of stiff dog syndrome associated with anti-glutamic acid decarboxylase antibodies. J. Clin. Mov. Disord. 2017, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Purcell, T.B.; Sellers, A.D.; Goehring, L.S. Presumed case of “stiff-horse syndrome” caused by decreased gamma-aminobutyric acid (GABA) production in an American Paint mare. Can. Vet. J. 2012, 53, 75–78. [Google Scholar]
- Hampe, C.S.; Petrosini, L.; De Bartolo, P.; Caporali, P.; Cutuli, D.; Laricchiuta, D.; Foti, F.; Radtke, J.R.; Vidova, V.; Honnorat, J.; et al. Monoclonal antibodies to 65kDa glutamate decarboxylase induce epitope specific effects on motor and cognitive functions in rats. Orphanet J. Rare Dis. 2013, 8, 82. [Google Scholar] [CrossRef]
- Carvajal-Gonzalez, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain 2014, 137 Pt 8, 2178–2192. [Google Scholar] [CrossRef]
- Rakocevic, G.; Raju, R.; Dalakas, M.C. Anti-glutamic acid decarboxylase antibodies in the serum and cerebrospinal fluid of patients with stiff-person syndrome: Correlation with clinical severity. Arch. Neurol. 2004, 61, 902–904. [Google Scholar] [CrossRef]
- Rakocevic, G.; Alexopoulos, H.; Dalakas, M.C. Quantitative clinical and autoimmune assessments in stiff person syndrome: Evidence for a progressive disorder. BMC Neurol. 2019, 19, 1. [Google Scholar] [CrossRef]
- Madlener, M.; Strippel, C.; Thaler, F.S.; Doppler, K.; Wandinger, K.P.; Lewerenz, J.; Ringelstein, M.; Roessling, R.; Menge, T.; Wickel, J.; et al. Glutamic acid decarboxylase antibody-associated neurological syndromes: Clinical and antibody characteristics and therapy response. J. Neurol. Sci. 2023, 445, 120540. [Google Scholar] [CrossRef] [PubMed]
- Gresa-Arribas, N.; Arino, H.; Martinez-Hernandez, E.; Petit-Pedrol, M.; Sabater, L.; Saiz, A.; Dalmau, J.; Graus, F. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS ONE 2015, 10, e0121364. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.R.; Baquet, Z.; Eisenbarth, G.S.; Tisch, R.; Smeyne, R.; Workman, C.J.; Vignali, D.A. Central nervous system destruction mediated by glutamic acid decarboxylase-specific CD4+ T cells. J. Immunol. 2010, 184, 4863–4870. [Google Scholar] [CrossRef]
- Chang, T.; Alexopoulos, H.; McMenamin, M.; Carvajal-Gonzalez, A.; Alexander, S.K.; Deacon, R.; Erdelyi, F.; Szabo, G.; Lang, B.; Blaes, F.; et al. Neuronal surface and glutamic acid decarboxylase autoantibodies in Nonparaneoplastic stiff person syndrome. JAMA Neurol. 2013, 70, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.; Honnorat, J.; Hampe, C.S.; Guerra-Narbona, R.; Lopez-Ramos, J.C.; Delgado-Garcia, J.M.; Saitow, F.; Suzuki, H.; Yanagawa, Y.; Mizusawa, H.; et al. Disease-specific monoclonal antibodies targeting glutamate decarboxylase impair GABAergic neurotransmission and affect motor learning and behavioral functions. Front. Behav. Neurosci. 2015, 9, 78. [Google Scholar] [CrossRef]
- Fouka, P.; Alexopoulos, H.; Akrivou, S.; Trohatou, O.; Politis, P.K.; Dalakas, M.C. GAD65 epitope mapping and search for novel autoantibodies in GAD-associated neurological disorders. J. Neuroimmunol. 2015, 281, 73–77. [Google Scholar] [CrossRef]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blumcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J.; et al. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 2012, 135 Pt 5, 1622–1638. [Google Scholar] [CrossRef]
- Meinck, H.M.; Ricker, K.; Hulser, P.J.; Schmid, E.; Peiffer, J.; Solimena, M. Stiff man syndrome: Clinical and laboratory findings in eight patients. J. Neurol. 1994, 241, 157–166. [Google Scholar] [CrossRef]
- Korn, T.; Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 2017, 17, 179–194. [Google Scholar]
- Thaler, F.S.; Thaller, A.L.; Biljecki, M.; Schuh, E.; Winklmeier, S.; Mahler, C.F.; Gerhards, R.; Volk, S.; Schnorfeil, F.; Subklewe, M.; et al. Abundant glutamic acid decarboxylase (GAD)-reactive B cells in gad-antibody-associated neurological disorders. Ann. Neurol. 2019, 85, 448–454. [Google Scholar] [CrossRef]
- Biljecki, M.; Eisenhut, K.; Beltran, E.; Winklmeier, S.; Mader, S.; Thaller, A.; Eichhorn, P.; Steininger, P.; Flierl-Hecht, A.; Lewerenz, J.; et al. Antibodies Against Glutamic Acid Decarboxylase 65 Are Locally Produced in the CSF and Arise During Affinity Maturation. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200090. [Google Scholar] [CrossRef]
- Graus, F.; Saiz, A.; Dalmau, J. GAD antibodies in neurological disorders—Insights and challenges. Nat. Rev. Neurol. 2020, 16, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Mariottini, A.; Bulgarini, G.; Cornacchini, S.; Damato, V.; Saccardi, R.; Massacesi, L. Hematopoietic Stem Cell Transplantation for the Treatment of Autoimmune Neurological Diseases: An Update. Bioengineering 2023, 10, 176. [Google Scholar] [CrossRef]
- Pugliese, A.; Solimena, M.; Awdeh, Z.L.; Alper, C.A.; Bugawan, T.; Erlich, H.A.; De Camilli, P.; Eisenbarth, G.S. Association of HLA-DQB1*0201 with stiff-man syndrome. J. Clin. Endocrinol. Metab. 1993, 77, 1550–1553. [Google Scholar] [PubMed]
- Liimatainen, S.; Honnorat, J.; Pittock, S.J.; McKeon, A.; Manto, M.; Radtke, J.R.; Biobank, T.D.E.; Hampe, C.S. GAD65 autoantibody characteristics in patients with co-occurring type 1 diabetes and epilepsy may help identify underlying epilepsy etiologies. Orphanet J. Rare Dis. 2018, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Belbezier, A.; Joubert, B.; Montero-Martin, G.; Fernandez-Vina, M.; Fabien, N.; Rogemond, V.; Mignot, E.; Honnorat, J. Multiplex family with GAD65-Abs neurologic syndromes. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e416. [Google Scholar] [CrossRef]
- Thaler, F.S.; Bangol, B.; Biljecki, M.; Havla, J.; Schumacher, A.M.; Kumpfel, T. Possible link of genetic variants to autoimmunity in GAD-antibody-associated neurological disorders. J. Neurol. Sci. 2020, 413, 116860. [Google Scholar] [CrossRef]
- Muniz-Castrillo, S.; Ambati, A.; Dubois, V.; Vogrig, A.; Joubert, B.; Rogemond, V.; Picard, G.; Lin, L.; Fabien, N.; Mignot, E.; et al. Primary DQ effect in the association between HLA and neurological syndromes with anti-GAD65 antibodies. J. Neurol. 2020, 267, 1906–1911. [Google Scholar] [CrossRef]
- Marinovic, I.; Pivalica, D.; Aljinovic, J.; Vlak, T.; Skoric, E.; Martinovic Kaliterna, D. Extremely rare coincidence of non-radiographic axial spondyloarthropathy HLA-B27 positive and Stiff Person Syndrome—Rheumatologist point of view. Mod. Rheumatol. 2016, 26, 278–280. [Google Scholar] [CrossRef]
- Gutmann, B.; Crivellaro, C.; Mitterer, M.; Zingerle, H.; Egarter-Vigl, E.; Wiedermann, C.J. Paraneoplastic stiff-person syndrome, heterotopic soft tissue ossification and gonarthritis in a HLA B27-positive woman preceding the diagnosis of Hodgkin’s lymphoma. Haematologica 2006, 91 (Suppl. 12), ECR59. [Google Scholar]
- Strippel, C.; Herrera-Rivero, M.; Wendorff, M.; Tietz, A.K.; Degenhardt, F.; Witten, A.; Schroeter, C.; Nelke, C.; Golombeck, K.S.; Madlener, M.; et al. A genome-wide association study in autoimmune neurological syndromes with anti-GAD65 autoantibodies. Brain 2023, 146, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Thaler, F.S.; Zimmermann, L.; Kammermeier, S.; Strippel, C.; Ringelstein, M.; Kraft, A.; Suhs, K.W.; Wickel, J.; Geis, C.; Markewitz, R.; et al. Rituximab Treatment and Long-term Outcome of Patients with Autoimmune Encephalitis: Real-world Evidence from the GENERATE Registry. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1088. [Google Scholar] [CrossRef]
- Lancaster, E.; Martinez-Hernandez, E.; Dalmau, J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011, 77, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Meinck, H.M. Pragmatic Treatment of Stiff Person Spectrum Disorders. Mov. Disord. Clin. Pr. 2018, 5, 394–401. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Fujii, M.; Li, M.; Lutfi, B.; Kyhos, J.; McElroy, B. High-dose intravenous immune globulin for stiff-person syndrome. N. Engl. J. Med. 2001, 345, 1870–1876. [Google Scholar] [CrossRef]
- Yi, J.; Dalakas, M.C. Long-term Effectiveness of IVIg Maintenance Therapy in 36 Patients with GAD Antibody-Positive Stiff-Person Syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e200011. [Google Scholar] [CrossRef]
- Aljarallah, S.; Newsome, S.D. Use of subcutaneous immunoglobulin in stiff person syndrome: Case series. Medicine 2021, 100, e25260. [Google Scholar] [CrossRef] [PubMed]
- Nelke, C.; Spatola, M.; Schroeter, C.B.; Wiendl, H.; Lunemann, J.D. Neonatal Fc Receptor-Targeted Therapies in Neurology. Neurotherapeutics 2022, 19, 729–740. [Google Scholar] [CrossRef]
- Remlinger, J.; Madarasz, A.; Guse, K.; Hoepner, R.; Bagnoud, M.; Meli, I.; Feil, M.; Abegg, M.; Linington, C.; Shock, A.; et al. Antineonatal Fc Receptor Antibody Treatment Ameliorates MOG-IgG-Associated Experimental Autoimmune Encephalomyelitis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1134. [Google Scholar] [CrossRef]
- Sun, W.; Khare, P.; Wang, X.; Challa, D.K.; Greenberg, B.M.; Ober, R.J.; Ward, E.S. Selective Depletion of Antigen-Specific Antibodies for the Treatment of Demyelinating Disease. Mol. Ther. 2021, 29, 1312–1323. [Google Scholar] [CrossRef]
- Albahra, S.; Yates, S.G.; Joseph, D.; De Simone, N.; Burner, J.D.; Sarode, R. Role of plasma exchange in stiff person syndrome. Transfus. Apher. Sci. 2019, 58, 310–312. [Google Scholar] [CrossRef]
- Mercure-Corriveau, N.; Roy, S.; Hu, C.; Crowe, E.P.; Zhu, X.; Obando, D.; Patel, E.U.; Tobian, A.A.R.; Wang, Y.; Bloch, E.M.; et al. Therapeutic plasma exchange in the management of stiff person syndrome spectrum disorders: A case series and review of the literature. Ther. Adv. Neurol. Disord. 2023, 16, 17562864231180736. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Rakocevic, G.; Dambrosia, J.M.; Alexopoulos, H.; McElroy, B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann. Neurol. 2017, 82, 271–277. [Google Scholar] [CrossRef]
- Aljarallah, S.; Wang, Y.; Shoemaker, T.; Newsome, S. Long-term Rituximab Use Benefits Patients with Stiff Person Syndrome (P2.2-033). Neurology 2019, 92, 15. [Google Scholar]
- Husari, K.; Tardo, L.; Greenberg, B. Bortezomib for the Treatment of Refractory Stiff Person Spectrum Disorder (P2.2-037). Neurology 2019, 92 (Suppl. 15), 15. [Google Scholar]
- Scheibe, F.; Pruss, H.; Mengel, A.M.; Kohler, S.; Numann, A.; Kohnlein, M.; Ruprecht, K.; Alexander, T.; Hiepe, F.; Meisel, A. Bortezomib for treatment of therapy-refractory anti-NMDA receptor encephalitis. Neurology 2017, 88, 366–370. [Google Scholar] [CrossRef]
- Scheibe, F.; Ostendorf, L.; Reincke, S.M.; Pruss, H.; von Brunneck, A.C.; Kohnlein, M.; Alexander, T.; Meisel, C.; Meisel, A. Daratumumab treatment for therapy-refractory anti-CASPR2 encephalitis. J. Neurol. 2020, 267, 317–323. [Google Scholar] [CrossRef]
- Ratuszny, D.; Skripuletz, T.; Wegner, F.; Gross, M.; Falk, C.; Jacobs, R.; Ruschulte, H.; Stangel, M.; Suhs, K.W. Case Report: Daratumumab in a Patient with Severe Refractory Anti-NMDA Receptor Encephalitis. Front. Neurol. 2020, 11, 602102. [Google Scholar] [CrossRef]
- Ringelstein, M.; Ayzenberg, I.; Lindenblatt, G.; Fischer, K.; Gahlen, A.; Novi, G.; Hayward-Konnecke, H.; Schippling, S.; Rommer, P.S.; Kornek, B.; et al. Interleukin-6 Receptor Blockade in Treatment-Refractory MOG-IgG-Associated Disease and Neuromyelitis Optica Spectrum Disorders. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1100. [Google Scholar] [CrossRef]
- Sormani, M.P.; Muraro, P.A.; Schiavetti, I.; Signori, A.; Laroni, A.; Saccardi, R.; Mancardi, G.L. Autologous hematopoietic stem cell transplantation in multiple sclerosis: A meta-analysis. Neurology 2017, 88, 2115–2122. [Google Scholar] [CrossRef]
- Sanders, S.; Bredeson, C.; Pringle, C.E.; Martin, L.; Allan, D.; Bence-Bruckler, I.; Hamelin, L.; Hopkins, H.S.; Sabloff, M.; Sheppard, D.; et al. Autologous stem cell transplantation for stiff person syndrome: Two cases from the Ottawa blood and marrow transplant program. JAMA Neurol. 2014, 71, 1296–1299. [Google Scholar] [CrossRef]
- Georges, G.; McSweeney, P.; Bowen, J.; Pearlman, M.; Wundes, A.; von Geldern, G.; Kraft, G.; Weiss, M.; Sytsma, J.; McLaughlin, B. Autologous hematopoietic stem cell transplantation may be highly effective treatment for severe, treatment refractory stiff person syndrome (S31. 007). Neurology 2018, 90, S120. [Google Scholar]
- Kass-Iliyya, L.; Snowden, J.A.; Thorpe, A.; Jessop, H.; Chantry, A.D.; Sarrigiannis, P.G.; Hadjivassiliou, M.; Sharrack, B. Autologous haematopoietic stem cell transplantation for refractory stiff-person syndrome: The UK experience. J. Neurol. 2021, 268, 265–275. [Google Scholar] [CrossRef]
- Burt, R.K.; Balabanov, R.; Han, X.; Quigley, K.; Arnautovic, I.; Helenowski, I.; Rose, J.; Siddique, T. Autologous hematopoietic stem cell transplantation for stiff-person spectrum disorder: A clinical trial. Neurology 2021, 96, e817–e830. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Limited Benefits Halt Enrollment in Hematopoietic Stem Cell Transplantation Trial for Stiff-Person Syndrome: Should There Be More to Come? Neurology 2021, 96, 239–240. [Google Scholar] [CrossRef] [PubMed]
- Clow, E.C.; Couban, S.; Grant, I.A. Stiff-person syndrome associated with multiple myeloma following autologous bone marrow transplantation. Muscle Nerve 2008, 38, 1649–1652. [Google Scholar] [CrossRef]
- He, A.; Merkel, B.; Brown, J.W.L.; Zhovits Ryerson, L.; Kister, I.; Malpas, C.B.; Sharmin, S.; Horakova, D.; Kubala Havrdova, E.; Spelman, T.; et al. Timing of high-efficacy therapy for multiple sclerosis: A retrospective observational cohort study. Lancet Neurol. 2020, 19, 307–316. [Google Scholar] [CrossRef]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kümpfel, T. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef]
- Wegmann, T.G.; Lin, H.; Guilbert, L.; Mosmann, T.R. Bidirectional cytokine interactions in the maternal-fetal relationship: Is successful pregnancy a TH2 phenomenon? Immunol. Today 1993, 14, 353–356. [Google Scholar] [CrossRef]
- Confavreux, C.; Hutchinson, M.; Hours, M.M.; Cortinovis-Tourniaire, P.; Moreau, T.; Group, P.i.M.S. Rate of pregnancy-related relapse in multiple sclerosis. N. Engl. J. Med. 1998, 339, 285–291. [Google Scholar] [CrossRef]
- Bourre, B.; Marignier, R.; Zephir, H.; Papeix, C.; Brassat, D.; Castelnovo, G.; Collongues, N.; Vukusic, S.; Labauge, P.; Outteryck, O.; et al. Neuromyelitis optica and pregnancy. Neurology 2012, 78, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.E.; Newsome, S.D. Improvement of stiff-person syndrome symptoms in pregnancy: Case series and literature review. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Galati, A.; McElrath, T.; Bove, R. Use of B-cell–depleting therapy in women of childbearing potential with multiple sclerosis and neuromyelitis optica spectrum disorder. Neurol. Clin. Pract. 2022, 12, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Yeshokumar, A.K.; Sun, L.R.; Newsome, S.D. Defining the expanding clinical spectrum of pediatric-onset stiff person syndrome. Pediatr. Neurol. 2021, 114, 11–15. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlad, B.; Wang, Y.; Newsome, S.D.; Balint, B. Stiff Person Spectrum Disorders—An Update and Outlook on Clinical, Pathophysiological and Treatment Perspectives. Biomedicines 2023, 11, 2500. https://doi.org/10.3390/biomedicines11092500
Vlad B, Wang Y, Newsome SD, Balint B. Stiff Person Spectrum Disorders—An Update and Outlook on Clinical, Pathophysiological and Treatment Perspectives. Biomedicines. 2023; 11(9):2500. https://doi.org/10.3390/biomedicines11092500
Chicago/Turabian StyleVlad, Benjamin, Yujie Wang, Scott D. Newsome, and Bettina Balint. 2023. "Stiff Person Spectrum Disorders—An Update and Outlook on Clinical, Pathophysiological and Treatment Perspectives" Biomedicines 11, no. 9: 2500. https://doi.org/10.3390/biomedicines11092500