
Hacking the Lipidome: New Ferroptosis Strategies in Cancer Therapy

Abstract
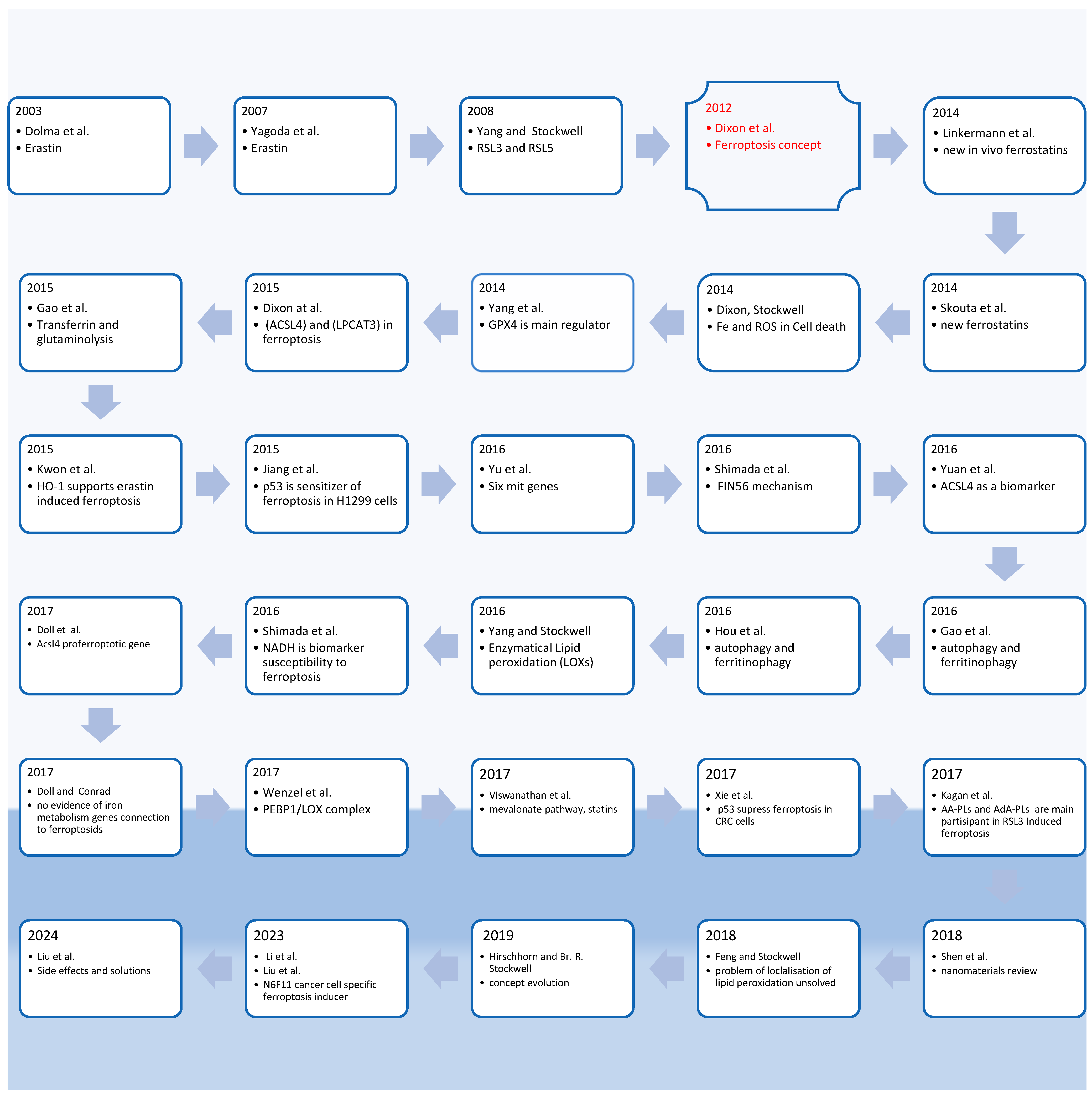
1. Introduction: Ferroptosis History and Concept
Key Points in Ferroptosis Historical Observation and Purpose of This Review
2. Cancer Cells Metabolome and Lipidome Alteration
2.1. Genes Involved in Cancerogenesis That Regulate De Novo Lipogenesis in Cancer
2.2. Ketolysis Is Critical for Acetyl-CoA Production in Cancer
3. Ferroptosis Related Genes and Proteins as Therapeutic Targets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes Regulate Ferroptosis | Influence | Metabolic and Lipidomic Changes | Reference |
|---|---|---|---|
| GPX4 | Suppressor | Recovery of peroxidized lipids | [10,20,95] |
| ACSL4 | Sensitizer | Activation of the long chain PUFAs by CoA before incorporation into the lipid membrane structure | [22] |
| LPCAT3 | Sensitizer | Incorporation of the long chain PUFAs into the membrane phospholipids | [11] |
| ALOX15 | Sensitizer | Peroxidation of the AA and AdA | [84] |
| NOX | Promoter | Producing ROS and increasing of the membrane PUFA-PLs peroxidation | [1] |
| P450 | Sensitizer | Increasing the membrane PUFA-PLs peroxidation | [85] |
| MS4A15 | Suppressor | Elevation MUFA-PL and plasmalogen ether PL, limiting PUFA-PL | [86] |
| HIF-2α | Sensitizer | Enhancement of PUFA-PL | [87] |
| HO-1 | Sensitizer | Elevation of lipid peroxidation | [88] |
| CD71(TfR1) | Sensitizer | Elevation of LIP | [89] |
| FASN | Suppressor | Increasing SFAs and MUFAs in lipids, limiting PUFA-PL | [61] |
| SCD1 | Suppressor | Increasing MUFA/SFA ratio in lipids, limiting PUFA-PL | [70,94] |
| HMGCR | Suppressor | Elevation of the level of CoQ10 (antioxidant) and production of IPP, which participate in the building of GPX4 | [16,20,30] |
4. Lipidomic Changes at Ferroptosis: Keys to Increase Sensitivity to Ferroptosis
- PE-(C18:0/C20:4+2[O])
- PE-(C18:0/C20:4+3[O])
- PE-(C18:0/C22:4+2[O])
- PE-(C18:0/C22:4+3[O]) [27].
5. Chemical Modulators of Ferroptosis
5.1. Ferroptosis Inducers
5.2. Ferroptosis Inhibitors
6. Therapeutic Strategies Based on the Metabolome and Lipidome Reprogramming
6.1. De Novo Lipogenesis Pathway as Targets for Sensitization to Ferroptosis
6.2. Ketolysis Pathway as a Target for Ferroptosis Sensitisation
6.3. Ferroptosis Sensitizers Which Target Genes Involved to PUFA-PLs Biosynthesis and Metabolism
6.4. Ferroptosis Sensitizers Acting on Different Lipid Metabolism Pathways Reprogramming
6.5. Redox Balance Reprogramming for Ferroptosis Sensitization
6.5.1. Activation of Enzymes Involved in Peroxidation
6.5.2. “Relaxing” Oxidative Stress: Lipidome Reprogramming as New Alternative Paradoxal Key to Ferroptosis Sensitization
6.6. General Strategies to “Hack” the Lipidome for Cancer Treatment by Ferroptosis
- -
- Assess all enzymes involved in lipid metabolism which promote the incorporation of MUFAs and SFAs into cell membrane phospholipids and inhibit these enzymes.
- -
- Assess all enzymes involved in lipid metabolism which promote the incorporation of more PUFAs into cell membrane phospholipids and activate these enzymes.
- -
- Block de novo lipogenesis by inhibiting the responsible enzymes.
- -
- Block the production of Acetyl-CoA, which is the primary substrate for de novo lipogenesis, by inhibiting ketolysis.
- -
- Activate enzymes that generate lipid ROS (such as NOX, LOX, COX, and POR) for synergistic use with ferroptosis inducers.
- -
- Increase PUFA assimilation with nutrients.
- -
- Preserve PUFAs (“relax” ROS) and accumulate them using antioxidants (ferrostatin, sulforaphane, erucin, etc.) or through reversible inhibition of NOX, LOX, COX, POR (for example, using niacin to decrease NOX activity).
- -
- Screen substances that correspond to target genes.
- -
- Assess changes in lipidomics after cell culture treatment (changes in PUFA-PL/MUFA-PL/SFA-PL).
- -
- Determine the order of treatment (pre- or co-treatment).
- -
- Check cell viability after ferroptosis induction.
- -
- Conduct toxicology studies and use xenograft models.
- -
- Proceed with clinical development.
7. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Behera, P.K.; Jain, S.; Kumar, A. Visual Exploration of Literature Using Connected Papers: A Practical Approach. Issues Sci. Technol. Librariansh. 2023, 104. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prókai, Á.; Zuchtriegel, G.; Krombach, F.; Welz, P.-S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2016, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.Z.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; Croix, C.M.S.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e626. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv. Mater. 2018, 30, e1704007. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Zhou, Z.; Wu, R.; Chen, X.; Yu, C.; Stockwell, B.; Kroemer, G.; Kang, R.; Tang, D. Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci. Transl. Med. 2023, 15, eadg3049. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Kang, R.; Tang, D. Cell type-specific induction of ferroptosis to boost antitumor immunity. OncoImmunology 2023, 12, 2282252. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. Adverse effects of ferroptotic therapy: Mechanisms and management. Trends Cancer 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Ellis, P.; Moore, L.; Sanders, M.A.; Butler, T.M.; Brunner, S.F.; Lee-Six, H.; Osborne, R.; Farr, B.; Coorens, T.H.H.; Lawson, A.R.J.; et al. Reliable detection of somatic mutations in solid tissues by laser-capture microdissection and low-input DNA sequencing. Nat. Protoc. 2021, 16, 841–871. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, W.; Thompson, P.; Hannun, Y.A. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun. 2018, 9, 3490. [Google Scholar] [CrossRef]
- Balmain, A. The critical roles of somatic mutations and environmental tumor-promoting agents in cancer risk. Nat. Genet. 2020, 52, 1139–1143. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Walter, P.; Raff, M.; Roberts, K. Molecular Biology of the Cell 4th Edition: International Student Edition; Routledge: London, UK, 2002. [Google Scholar]
- Warburg, O.H.; Dickens, F. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology, Berlin-Dahlem, 1st American ed.; Richard R. Smith: New York, NY, USA, 1931. [Google Scholar]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2013, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, A.; Chełstowska, B.; Olejarz, W.; Nowicka, G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers 2020, 12, 1232. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Kokotou, M.G.; Mantzourani, C.; Batsika, C.S.; Mountanea, O.G.; Eleftheriadou, I.; Kosta, O.; Tentolouris, N.; Kokotos, G. Lipidomics Analysis of Free Fatty Acids in Human Plasma of Healthy and Diabetic Subjects by Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS). Biomedicines 2022, 10, 1189. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gu, W.; Wang, Y.; Qin, B.; Ji, W.; Wang, Z.; Liu, S. Untargeted Lipidomics Reveals Characteristic Biomarkers in Patients with Ankylosing Spondylitis Disease. Biomedicines 2023, 11, 47. [Google Scholar] [CrossRef]
- Castelli, F.A.; Rosati, G.; Moguet, C.; Fuentes, C.; Marrugo-Ramírez, J.; Lefebvre, T.; Volland, H.; Merkoçi, A.; Simon, S.; Fenaille, F.; et al. Metabolomics for personalized medicine: The input of analytical chemistry from biomarker discovery to point-of-care tests. Anal. Bioanal. Chem. 2022, 414, 759–789. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Khan, W.; Augustine, D.; Rao, R.S.; Patil, S.; Awan, K.H.; Sowmya, S.V.; Haragannavar, V.C.; Prasad, K. Lipid metabolism in cancer: A systematic review. J. Carcinog. 2021, 20, 4. [Google Scholar] [CrossRef]
- Pakiet, A.; Kobiela, J.; Stepnowski, P.; Sledzinski, T.; Mika, A. Changes in lipids composition and metabolism in colorectal cancer: A review. Lipids Health Dis. 2019, 18, 21. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, 10. [Google Scholar] [CrossRef]
- Bartolacci, C.; Andreani, C.; Vale, G.; Berto, S.; Melegari, M.; Crouch, A.C.; Baluya, D.L.; Kemble, G.; Hodges, K.; Starrett, J.; et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Commun. 2022, 13, 4327. [Google Scholar] [CrossRef] [PubMed]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. CMLS 2005, 62, 1784–1803. [Google Scholar] [CrossRef]
- Romero, P.; Wagg, J.; Green, M.L.; Kaiser, D.; Krummenacker, M.; Karp, P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2005, 6, R2.1–R217. [Google Scholar] [CrossRef]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef] [PubMed]
- Vanauberg, D.; Schulz, C.; Lefebvre, T. Involvement of the pro-oncogenic enzyme fatty acid synthase in the hallmarks of cancer: A promising target in anti-cancer therapies. Oncogenesis 2023, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dales, N.A.; Winther, M.D. Opportunities and Challenges in Developing Stearoyl-Coenzyme A Desaturase-1 Inhibitors as Novel Therapeutics for Human Disease. J. Med. Chem. 2014, 57, 5039–5056. [Google Scholar] [CrossRef] [PubMed]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Coleman, C.; Sen, T. Stearoyl coenzyme A desaturase-1: Multitasker in cancer, metabolism, and ferroptosis. Trends Cancer 2023, 9, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H. Elovl6: A new player in fatty acid metabolism and insulin sensitivity. J. Mol. Med. 2009, 87, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.H.; Chen, W.Y.; Kuo, Y.H.; Tung, C.L.; Tsao, C.J.; Shiau, A.L.; Wu, C.L. Elovl6 is a poor prognostic predictor in breast cancer. Oncol. Lett. 2016, 12, 207–212. [Google Scholar] [CrossRef]
- Su, Y.-C.; Feng, Y.-H.; Wu, H.-T.; Huang, Y.-S.; Tung, C.-L.; Wu, P.; Chang, C.-J.; Shiau, A.-L.; Wu, C.-L. Elovl6 is a negative clinical predictor for liver cancer and knockdown of Elovl6 reduces murine liver cancer progression. Sci. Rep. 2018, 8, 6586. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, X.; Tang, J.; Zhao, H.; Duan, M. Decreased expression levels of ELOVL6 indicate poor prognosis in hepatocellular carcinoma. Oncol. Lett. 2019, 18, 6214–6220. [Google Scholar] [CrossRef] [PubMed]
- Israël, M.; Schwartz, L. Inhibition of the ketolytic acetyl CoA supply to tumors could be their “Achilles heel”. Int. J. Cancer 2020, 147, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- Israël, M.; Schwartz, L. The metabolic rewiring observed in cancer renders tumor cells dependent of ketone bodies and vulnerable to SCOT inhibition. Endocrinol. Diabetes Metab. J. 2020, 4, 1–13. [Google Scholar]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Hou, J.; Jiang, C.; Wen, X.; Li, C.; Xiong, S.; Yue, T.; Long, P.; Shi, J.; Zhang, Z. ACSL4 as a Potential Target and Biomarker for Anticancer: From Molecular Mechanisms to Clinical Therapeutics. Front. Pharmacol. 2022, 13, 949863. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Snodgrass, R.G.; Brüne, B. Regulation and Functions of 15-Lipoxygenases in Human Macrophages. Front. Pharmacol. 2019, 10, 719. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.; Mueller, C.; Pfeiffer, S.; Kraft, V.A.N.; Merl-Pham, J.; Bao, X.; Feederle, R.; Jin, X.; Hauck, S.M.; Schmitt-Kopplin, P.; et al. MS4A15 drives ferroptosis resistance through calcium-restricted lipid remodeling. Cell Death Differ. 2022, 29, 670–686. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.-Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e3417. [Google Scholar] [CrossRef]
- Candelaria, P.V.; Leoh, L.S.; Penichet, M.L.; Daniels-Wells, T.R. Antibodies Targeting the Transferrin Receptor 1 (TfR1) as Direct Anti-cancer Agents. Front. Immunol. 2021, 12, 607692. [Google Scholar] [CrossRef]
- Shan, G.; Zhang, H.; Bi, G.; Bian, Y.; Liang, J.; Valeria, B.; Zeng, D.; Yao, G.; Zhan, C.; Fan, H. Multi-Omics Analysis of Cancer Cell Lines with High/Low Ferroptosis Scores and Development of a Ferroptosis-Related Model for Multiple Cancer Types. Front. Cell Dev. Biol. 2021, 9, 794475. [Google Scholar] [CrossRef]
- Wohlhieter, C.A.; Richards, A.L.; Uddin, F.; Hulton, C.H.; Quintanal-Villalonga, À.; Martin, A.; de Stanchina, E.; Bhanot, U.; Asher, M.; Shah, N.S.; et al. Concurrent Mutations in STK11 and KEAP1 Promote Ferroptosis Protection and SCD1 Dependence in Lung Cancer. Cell Rep. 2020, 33, 108444. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Carbone, M.; Melino, G. Stearoyl CoA Desaturase Regulates Ferroptosis in Ovarian Cancer Offering New Therapeutic Perspectives. Cancer Res. 2019, 79, 5149–5150. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Wiernicki, B.; Dubois, H.; Tyurina, Y.Y.; Hassannia, B.; Bayir, H.; Kagan, V.E.; Vandenabeele, P.; Wullaert, A.; Vanden Berghe, T. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020, 11, 922. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, J.; Kang, R.; Yang, M.; Tang, D. Lipid Metabolism in Ferroptosis. Adv. Biol. 2021, 5, e2100396. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.Y.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e429. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, W.K.; Bae, K.H.; Lee, S.C.; Lee, E.W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef]
- Lippmann, J.; Petri, K.; Fulda, S.; Liese, J. Redox Modulation and Induction of Ferroptosis as a New Therapeutic Strategy in Hepatocellular Carcinoma. Transl. Oncol. 2020, 13, 100785. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Wu, Z.; Zhong, M.; Liu, Y.; Xiong, Y.; Gao, Z.; Ma, J.; Zhuang, G.; Hong, X. Application of natural products for inducing ferroptosis in tumor cells. Biotechnol. Appl. Biochem. 2022, 69, 190–197. [Google Scholar] [CrossRef]
- Stepanić, V.; Kučerová-Chlupáčová, M. Review and Chemoinformatic Analysis of Ferroptosis Modulators with a Focus on Natural Plant Products. Molecules 2023, 28, 475. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.-Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021, 38, 101744. [Google Scholar] [CrossRef] [PubMed]
- Delesderrier, E.; Monteiro, J.D.C.; Freitas, S.; Pinheiro, I.C.; Batista, M.S.; Citelli, M. Can Iron and Polyunsaturated Fatty Acid Supplementation Induce Ferroptosis? Cell Physiol. Biochem. 2023, 57, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Benavente, C.A.; Jacobson, E.L. Niacin restriction upregulates NADPH oxidase and reactive oxygen species (ROS) in human keratinocytes. Free Radic. Biol. Med. 2008, 44, 527–537. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Wu, X.; Xu, F.; Ma, H.; Wu, M.; Xia, Y. Targeting Ferroptosis Pathway to Combat Therapy Resistance and Metastasis of Cancer. Front. Pharmacol. 2022, 13, 909821. [Google Scholar] [CrossRef]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniëls, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. Febs J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Chugh, V.; Gupta, A.K. Essential fatty acids as functional components of foods- a review. J. Food Sci. Technol. 2014, 51, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Worthmann, A.; Ridder, J.; Piel, S.Y.L.; Evangelakos, I.; Musfeldt, M.; Voß, H.; O’Farrell, M.; Fischer, A.W.; Adak, S.; Sundd, M.; et al. Fatty acid synthesis suppresses dietary polyunsaturated fatty acid use. Nat. Commun. 2024, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Li, E.-Q.; Zhao, W.; Zhang, C.; Qin, L.-Z.; Liu, S.-J.; Feng, Z.-Q.; Wen, X.; Chen, C.-P. Synthesis and anti-cancer activity of ND-646 and its derivatives as acetyl-CoA carboxylase 1 inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase—Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Lupu, R.; Menendez, J.A. Pharmacological inhibitors of Fatty Acid Synthase (FASN)--catalyzed endogenous fatty acid biogenesis: A new family of anti-cancer agents? Curr. Pharm. Biotechnol. 2006, 7, 483–493. [Google Scholar] [CrossRef]
- von Roemeling, C.A.; Caulfield, T.R.; Marlow, L.; Bok, I.; Wen, J.; Miller, J.L.; Hughes, R.; Hazlehurst, L.; Pinkerton, A.B.; Radisky, D.C.; et al. Accelerated bottom-up drug design platform enables the discovery of novel stearoyl-CoA desaturase 1 inhibitors for cancer therapy. Oncotarget 2018, 9, 3–20. [Google Scholar] [CrossRef]
- Guo, S.; Wang, Y.; Zhou, D.; Li, Z. Significantly increased monounsaturated lipids relative to polyunsaturated lipids in six types of cancer microenvironment are observed by mass spectrometry imaging. Sci. Rep. 2014, 4, 5959. [Google Scholar] [CrossRef]
- Hess, D.; Chisholm, J.W.; Igal, R.A. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS ONE 2010, 5, e11394. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Batchuluun, B.; Pinkosky, S.L.; Steinberg, G.R. Lipogenesis inhibitors: Therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 2022, 21, 283–305. [Google Scholar] [CrossRef] [PubMed]
- Israël, M.; Berg, E.; Tenenbaum, G. Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing. J. Clin. Med. 2023, 12, 1589. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, R.; Berg, E.; Tenenbaum, G.; Israël, M. Inhibition of SCOT and Ketolysis Decreases Tumor Growth and Inflammation in the Lewis Cancer Model. J. Oncol. Clin. Res. 2022, 3, 1–12. [Google Scholar]
- Kan, C.F.; Singh, A.B.; Dong, B.; Shende, V.R.; Liu, J. PPARδ activation induces hepatic long-chain acyl-CoA synthetase 4 expression in vivo and in vitro. Biochim. Biophys. Acta 2015, 1851, 577–587. [Google Scholar] [CrossRef]
- Shao, G.; Qian, Y.; Lu, L.; Liu, Y.; Wu, T.; Ji, G.; Xu, H. Research progress in the role and mechanism of LPCAT3 in metabolic related diseases and cancer. J. Cancer 2022, 13, 2430–2439. [Google Scholar] [CrossRef]
- Gao, H.; Song, Y.; Li, D.; Feng, W.; Liu, J. Saikosaponin A inhibits IL-1β-induced inflammatory mediators in human osteoarthritis chondrocytes by activating LXRα. Oncotarget 2017, 8, 88941–88950. [Google Scholar] [CrossRef]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef]
- Oh, M.; Jang, S.Y.; Lee, J.-Y.; Kim, J.W.; Jung, Y.; Kim, J.; Seo, J.; Han, T.-S.; Jang, E.; Son, H.Y.; et al. The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism. Nat. Commun. 2023, 14, 5728. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Guo, L.; Ghassemi, S.; Snyder, N.W.; Worth, A.J.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef]
- Thupari, J.N.; Pinn, M.L.; Kuhajda, F.P. Fatty acid synthase inhibition in human breast cancer cells leads to malonyl-CoA-induced inhibition of fatty acid oxidation and cytotoxicity. Biochem. Biophys. Res. Commun. 2001, 285, 217–223. [Google Scholar] [CrossRef]
- Folmes, C.D.; Lopaschuk, G.D. Role of malonyl-CoA in heart disease and the hypothalamic control of obesity. Cardiovasc. Res. 2007, 73, 278–287. [Google Scholar] [CrossRef] [PubMed]
- NaveenKumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by Melatonin. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Almahi, W.A.; Yu, K.N.; Mohammed, F.; Kong, P.; Han, W. Hemin enhances radiosensitivity of lung cancer cells through ferroptosis. Exp. Cell Res. 2022, 410, 112946. [Google Scholar] [CrossRef] [PubMed]
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The Role of Reactive Oxygen Species and Ferroptosis in Heme-Mediated Activation of Human Platelets. ACS Chem. Biol. 2018, 13, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.; Hermann, E.; Lin, D.; Chowanadisai, W.; Hull, E.; Montgomery, M. HDAC inhibition induces EMT and alterations in cellular iron homeostasis to augment ferroptosis sensitivity in SW13 cells. Redox Biol. 2021, 47, 102149. [Google Scholar] [CrossRef] [PubMed]
- Shiose, A.; Sumimoto, H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J. Biol. Chem. 2000, 275, 13793–13801. [Google Scholar] [CrossRef] [PubMed]
- Regier, D.S.; Waite, K.A.; Wallin, R.; McPhail, L.C. A phosphatidic acid-activated protein kinase and conventional protein kinase C isoforms phosphorylate p22(phox), an NADPH oxidase component. J. Biol. Chem. 1999, 274, 36601–36608. [Google Scholar] [CrossRef]
- Reyes, L.A.; Boslett, J.; Varadharaj, S.; De Pascali, F.; Hemann, C.; Druhan, L.J.; Ambrosio, G.; El-Mahdy, M.; Zweier, J.L. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc. Natl. Acad. Sci. USA 2015, 112, 11648–11653. [Google Scholar] [CrossRef]
- Stoeckler, J.D.; Stoeckler, H.A.; Kouttab, N.; Maizel, A.L. 1alpha,25-Dihydroxyvitamin D3 modulates CD38 expression on human lymphocytes. J. Immunol. 1996, 157, 4908–4917. [Google Scholar] [CrossRef]
- Zhu, Y.; Murthy, D.; Kollala, S.S.; Kosmacek, E.A.; Chatterjee, A.; McDowell, J.A.; Singh, P.K.; Oberley-Deegan, R.E. The central role of NADPH depletion in MnTE-2-PyP-induced prostate cancer cell growth inhibition. Adv. Redox Res. 2021, 3, 100025. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Liu, Y.W.; Huang, P.; Li, Z.; Xu, C.H.; Wang, H.Z.; Jia, B.Q.; Gong, A.H.; Xu, M. Vitamin C Sensitizes Pancreatic Cancer Cells to Erastin-Induced Ferroptosis by Activating the AMPK/Nrf2/HMOX1 Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 15. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, K.J.; Poire, A.; Zhang, D.; Tsang, Y.H.; Blucher, A.S.; Mills, G.B. Irreversible HER2 inhibitors overcome resistance to the RSL3 ferroptosis inducer in non-HER2 amplified luminal breast cancer. Cell Death Dis. 2023, 14, 532. [Google Scholar] [CrossRef]
- Popazova, O.; Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Oksenych, V.; Kamyshnyi, A. Cardioprotective Activity of Pharmacological Agents Affecting NO Production and Bioavailability in the Early Postnatal Period after Intrauterine Hypoxia in Rats. Biomedicines 2023, 11, 2854. [Google Scholar] [CrossRef] [PubMed]
- SamelİUk, Y.; Kaplaushenko, A.; Nedorezaniuk, N.; Ostretsova, L.; DİAkova, F.; Gutyj, B. Prospects for the search for new biologically active compounds among the derivatives of the heterocyclic system of 1,2,4-triazole. Hacet. Univ. J. Fac. Pharm. 2022, 42, 175–186. [Google Scholar] [CrossRef]
- Ihnatova, T.; Kaplaushenko, A.; Frolova, Y.; Pryhlo, E. Synthesis and antioxidant properties of some new 5-phenethyl-3-thio-1,2,4-triazoles. Pharmacia 2021, 68, 129–133. [Google Scholar] [CrossRef]
- Dovbnya, D.V.; Kaplaushenko, A.H.; Frolova, Y.S.; Pruglo, E.S. Synthesis and antioxidant properties of new (2,4- and 3,4-dimethoxyphenyl)-1,2,4-triazoles. Pharmacia 2022, 69, 135–142. [Google Scholar] [CrossRef]
- Gotsulya, A. Synthesis and Antiradical Activity of Alkyl Derivatives of 5-(5-Methyl-1H-Pyrazol-3-Yl)-4-Phenyl-4H-1,2,4-Triazole-3-Thiol. J. Fac. Pharm. Ank. Univ. 2020, 44, 211–219. [Google Scholar] [CrossRef]
- Shcherbyna, R.; Vashchyk, Y. The Research of 1,2,4-Triazole Derivatives Hepatoprotective Activity under Tetracycline and Infectious Hepatitis. J. Fac. Pharm. Ank. Univ. 2019, 43, 135–146. [Google Scholar] [CrossRef]
- Safonov, A.A.; Nosulenko, I.S. Antiradical activity of novel 4-amino-5-(thiophen-2-ylmethyl)-4H-1,2,4-triazole-3-thiol derivatives. Curr. Issues Pharm. Med. Sci. Pract. 2021, 14, 162–166. [Google Scholar] [CrossRef]
- Yousefian, M.; Shakour, N.; Hosseinzadeh, H.; Hayes, A.W.; Hadizadeh, F.; Karimi, G. The natural phenolic compounds as modulators of NADPH oxidases in hypertension. Phytomedicine 2019, 55, 200–213. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]






| Oncogenes Regulate Lipid Metabolism | Metabolic and Lipidomic Changes | Reference |
|---|---|---|
| ACC1 | synthesis of malonyl-CoA | [63] |
| FASN | synthesis of fatty acids | [67] |
| SCD1 | desaturation of FA | [69] |
| ELOVL6 | FA elongation | [71] |
| Small Molecules | Origin | Mechanism | Reference |
|---|---|---|---|
| erastin | synthetic | blocks cystine uptake which leads to depletion of glutathione | [1,4,5] |
| RSL3 | synthetic | binds GPX4 and inhibits it | [6] |
| BSO | synthetic | depletes glutathione | [10] |
| sulfasalazine | synthetic | inhibits system xc- | [103] |
| sorafenib | synthetic | inhibits system xc- | [103] |
| artesunate | natural | elevates labile iron pool | [104] |
| dehydroartemisisnin | natural | decreases GSH level | [104] |
| withaferin A | natural | medium dose NRF2 activator; high dose GPX4 inhibitor | [106] |
| N6F11 | synthetic | TRIM25 agonist, leads to degradation of GPX4 | [30] |
| Target Protein or Pathway | Small Molecules | Origin | Perspective | Reference |
|---|---|---|---|---|
| ACC1 (i) * | soraphen A | natural | potential | [113] |
| ACC1 (i) | ND-646 | synthetic | potential | [118] |
| FASN (i) | C75 | synthetic | potential | [120] |
| FASN (i) | orlistat | natural | potential | [120] |
| FASN (i) | epigallocatechin-3-gallate | natural | potential | [120] |
| SCD1 (i) | CVT11127 | synthetic | tested | [92] |
| SCD1 (i) | SSI-4 | synthetic | potential | [121] |
| SCD1 (i) | aramchol (icomidocholic acid) | natural | potential | [70] |
| SCOT (i) | lithostat | synthetic | potential | [126,127] |
| SCOT-ACAT1 (i) | 3- alpha R lipoic acid | natural | potential | [126,127] |
| SCOT-ACAT1 (i) | hydroxycitrate (from Garcinia Cambogia); | natural | potential | [126,127] |
| SCOT-ACAT1 (i) ACS (i) | allicin | natural | potential | [126,127] |
| Target Protein | Small Molecules | Origin | Perspective | Reference |
|---|---|---|---|---|
| PPARδ (a) */ACSL4 | L-165041 | synthetic | potential | [128] |
| LXR (a)/LPCAT3 | saikosaponin A | natural | potential | [130] |
| LXR (a)/LPCAT3 | 24-hydroxycholesterol | natural | potential | [131] |
| CPT1a (i) ** | etomoxir | synthetic | tested | [27] |
| CPT1 (i) | malonyl-CoA | synthetic | potential | [134,135] |
| HO-1 (a) | heme | synthetic | tested | [138] |
| HMGCR (i) | statins | synthetic | tested | [25] |
| HDAC (i)/Ferroportin | romidepsin | natural | tested | [140] |
| Target Protein | Small Molecules | Origin | Perspective | Reference |
|---|---|---|---|---|
| NOX (a) * | Arachidonic acid (AA) | natural | potential | [141,146] |
| NOX (a) | Phosphatidic acids (PA) | natural | potential | [142] |
| NOX (a) | 8,11,14-eicosatrienoic acid | natural | potential | [141] |
| ALOX15 (a) | PKUMDL_MH_1001 | synthetic | tested | [80] |
| PEBP1 (a)/15LOX | locostatin | synthetic | tested | [24] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varynskyi, B.; Schick, J.A. Hacking the Lipidome: New Ferroptosis Strategies in Cancer Therapy. Biomedicines 2024, 12, 541. https://doi.org/10.3390/biomedicines12030541
Varynskyi B, Schick JA. Hacking the Lipidome: New Ferroptosis Strategies in Cancer Therapy. Biomedicines. 2024; 12(3):541. https://doi.org/10.3390/biomedicines12030541
Chicago/Turabian StyleVarynskyi, Borys, and Joel A. Schick. 2024. "Hacking the Lipidome: New Ferroptosis Strategies in Cancer Therapy" Biomedicines 12, no. 3: 541. https://doi.org/10.3390/biomedicines12030541
APA StyleVarynskyi, B., & Schick, J. A. (2024). Hacking the Lipidome: New Ferroptosis Strategies in Cancer Therapy. Biomedicines, 12(3), 541. https://doi.org/10.3390/biomedicines12030541