
Solid Phase Synthesis and TAR RNA-Binding Activity of Nucleopeptides Containing Nucleobases Linked to the Side Chains via 1,4-Linked-1,2,3-triazole
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
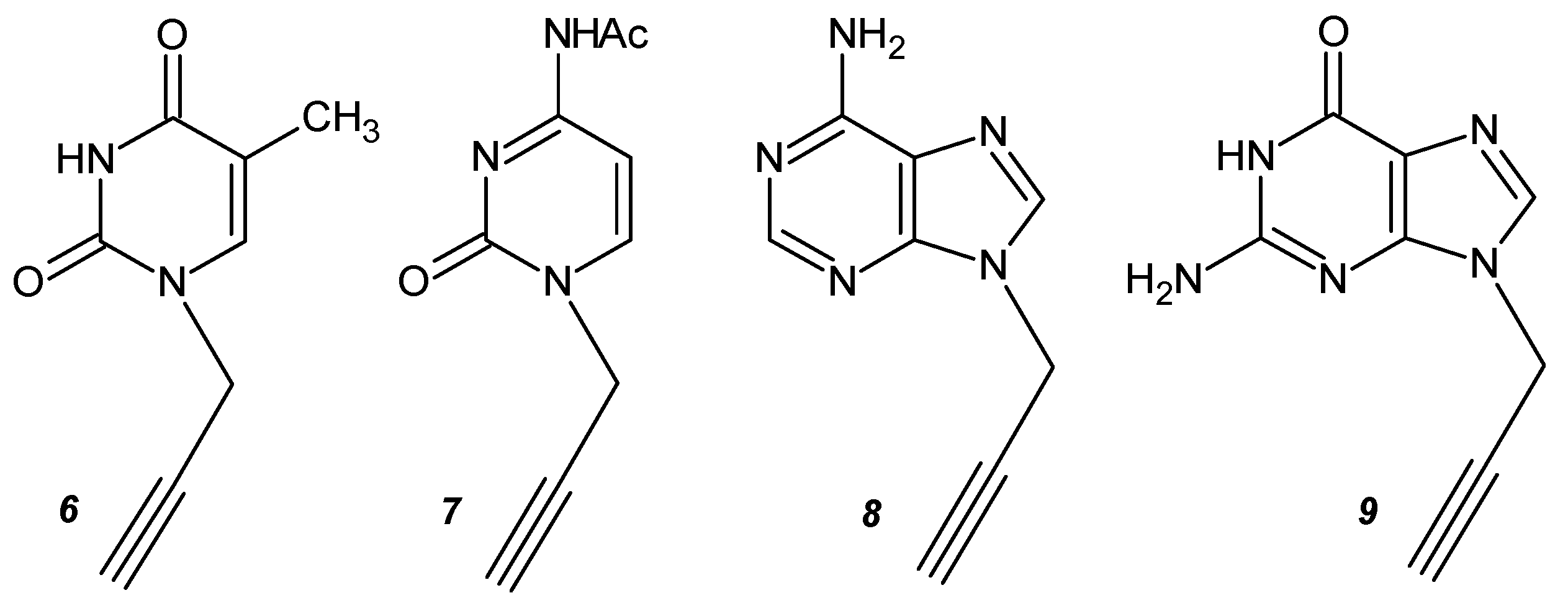
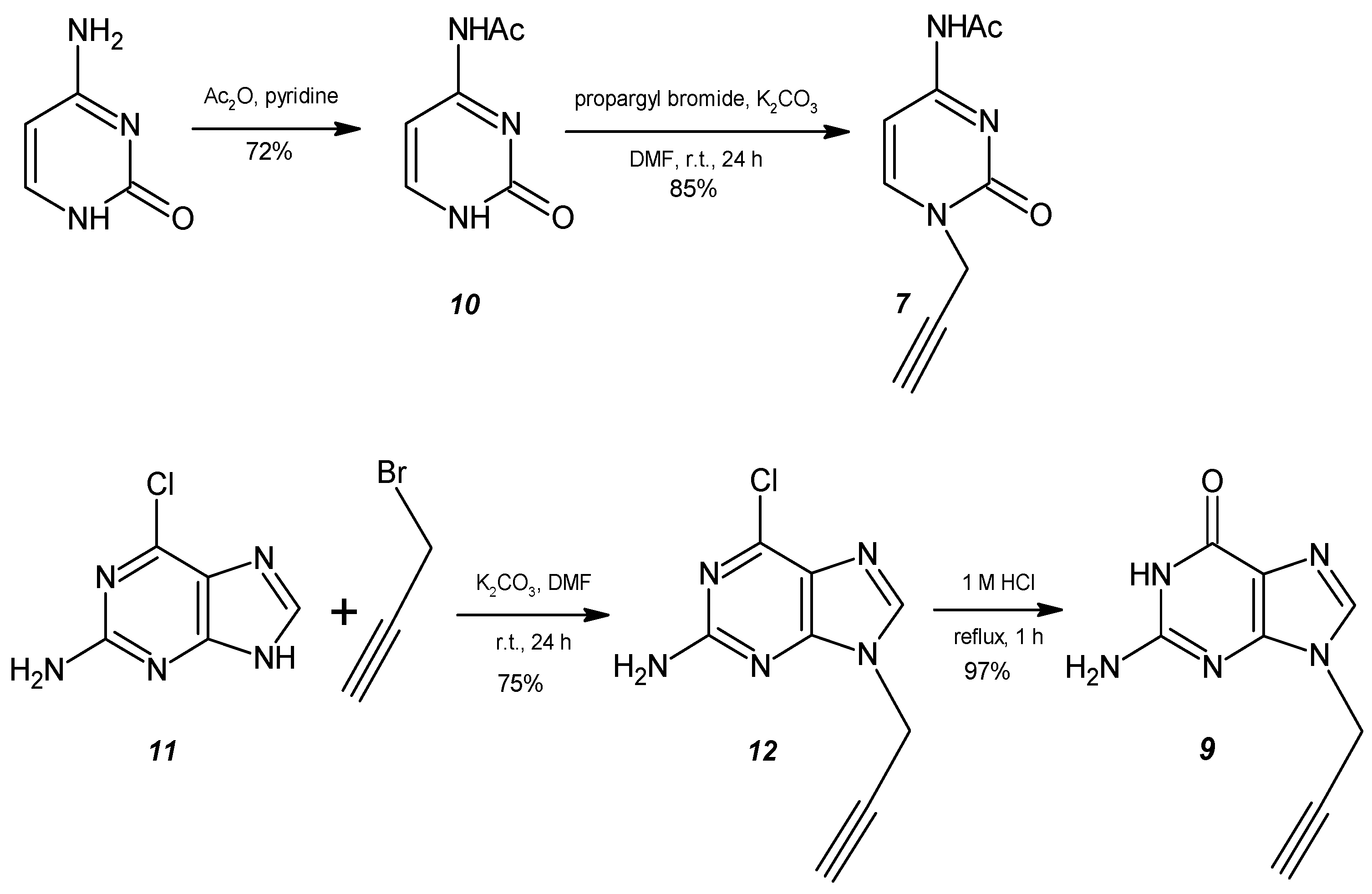
2.1. General Procedure for the Synthesis of the Monopropargyl Nucleobases (5)
2.2. General Procedure for the Synthesis of 1,4-TzlNBAs
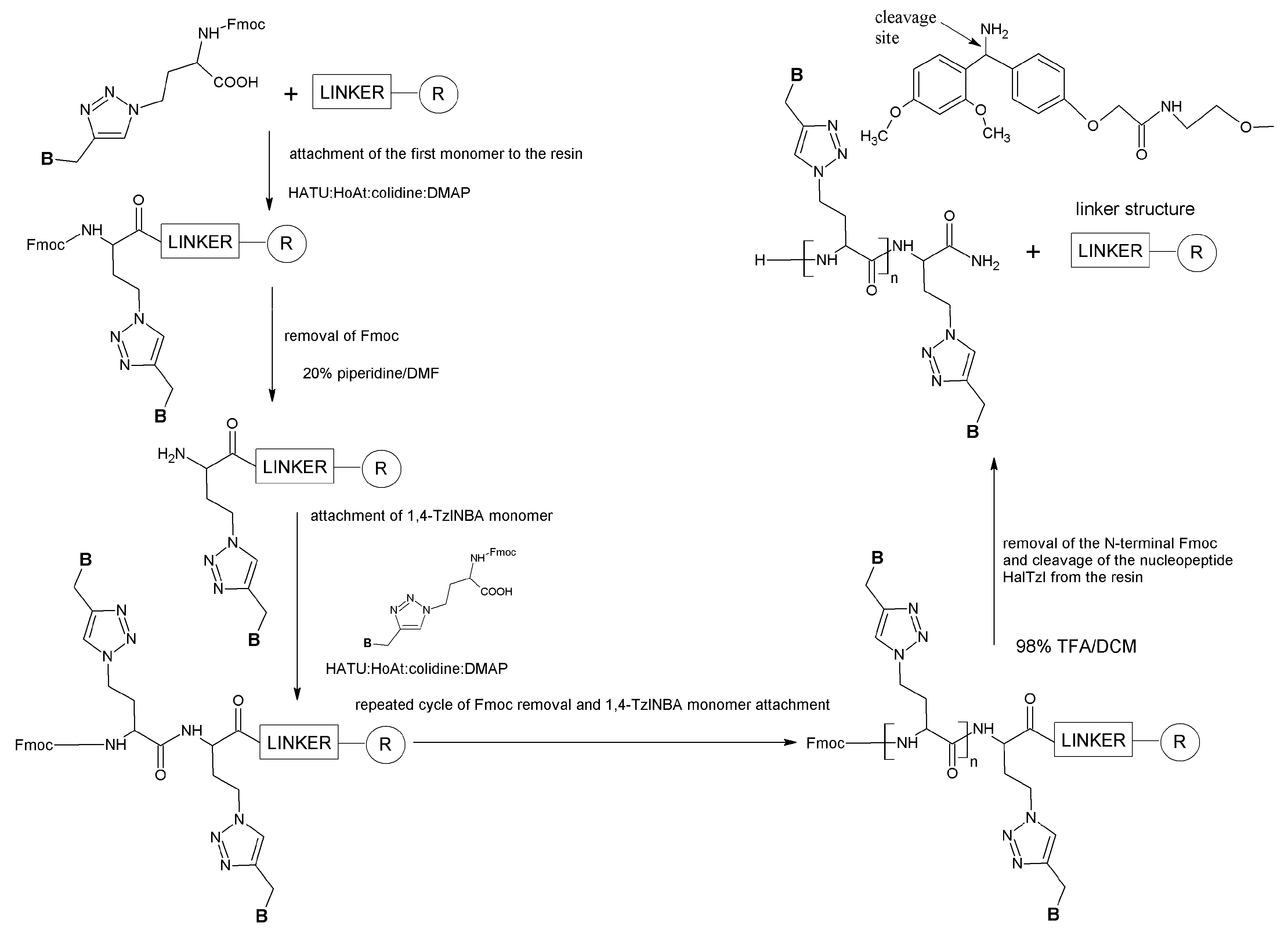
2.3. Solid-Phase Synthesis of HalTzls
2.4. TAR RNA Synthesis
2.5. Circular Dichroism (CD) Spectropolarimetry
2.6. Fluorescence Anisotropy Spectroscopy
3. Results and Discussion
3.1. Synthesis of N-Propargyl Nucleobases and 1,4-TzlNBA Monomers
3.2. HalTzls Synthesis
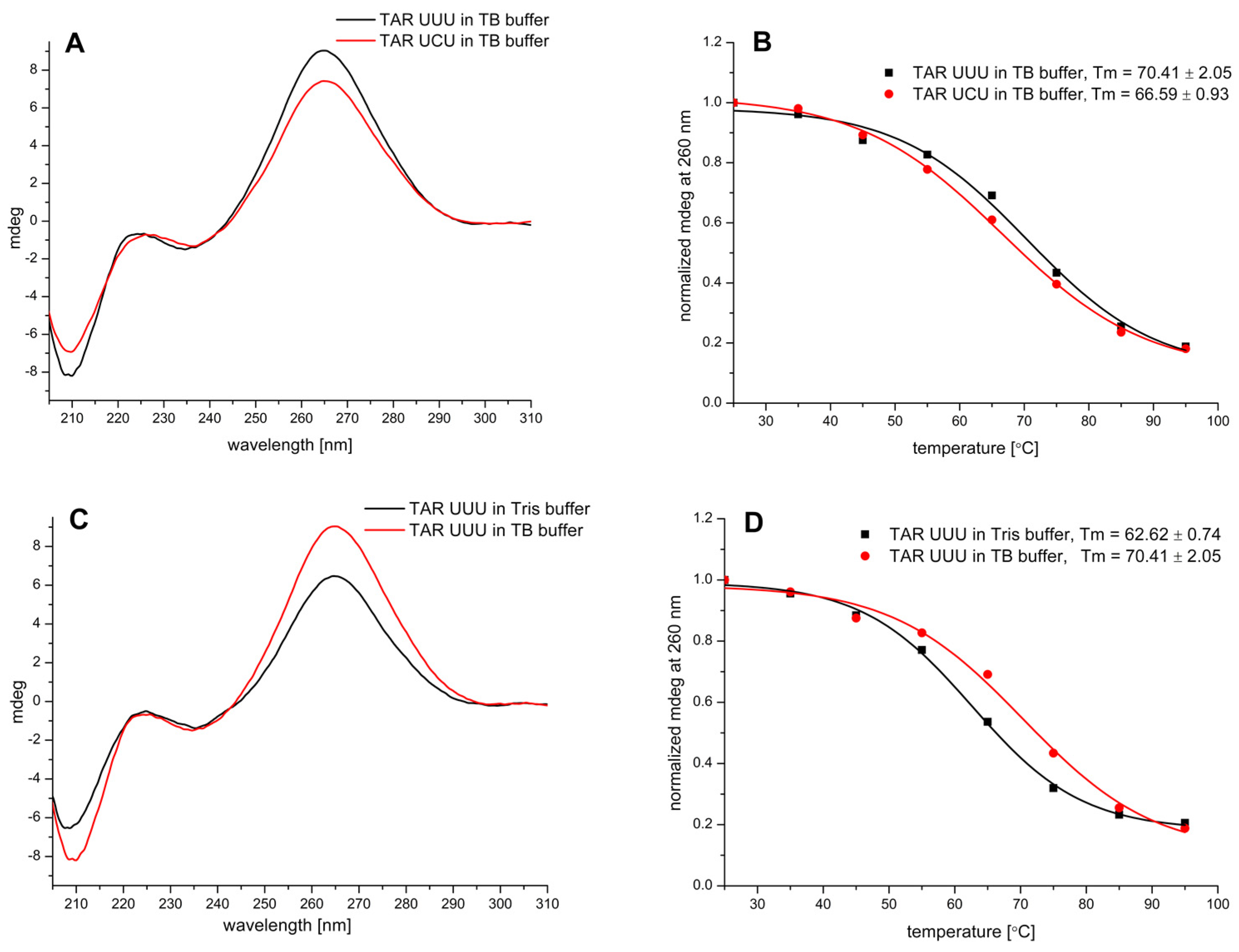
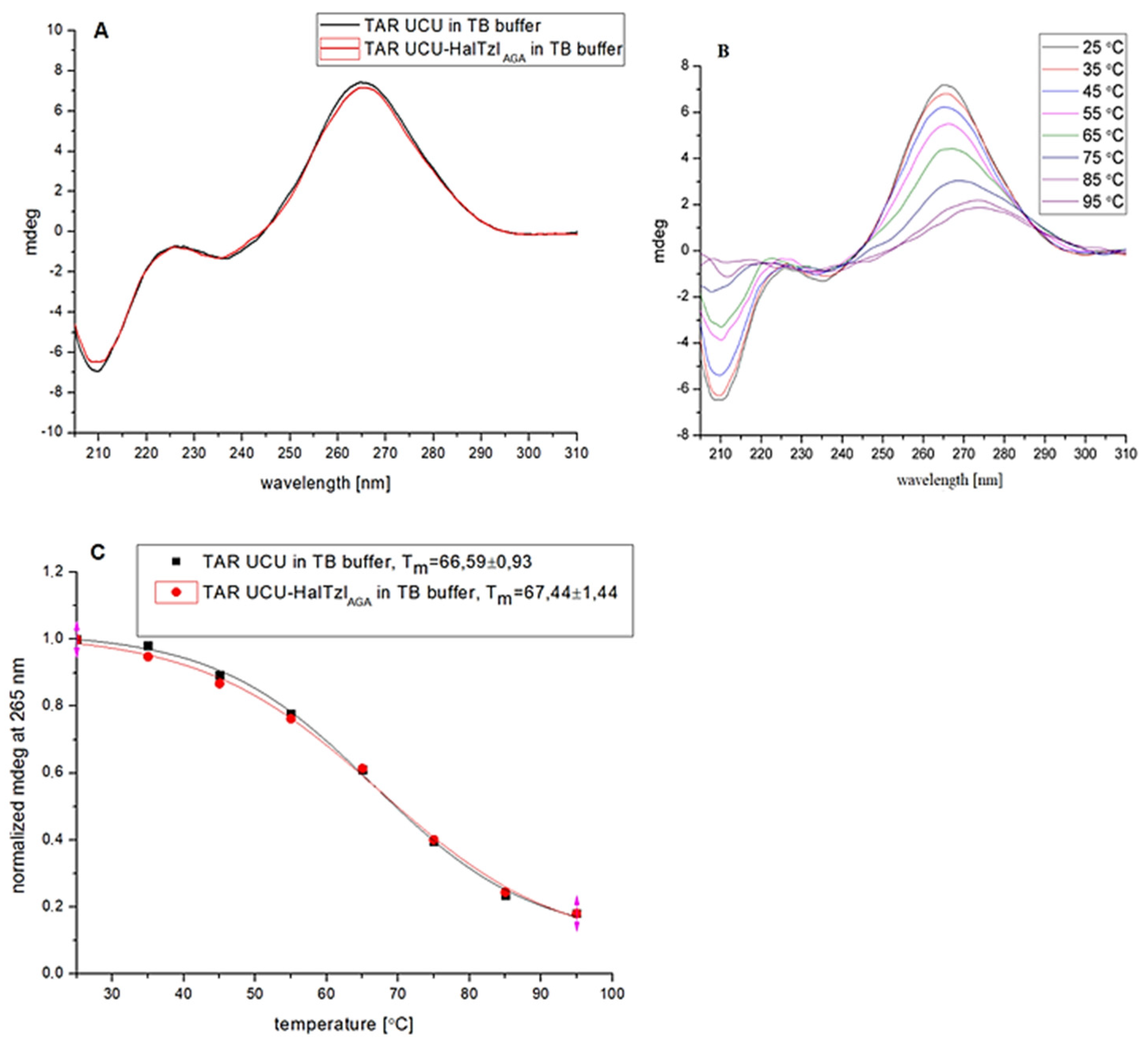
3.3. CD Study of TAR RNA—HalTzls Interactions
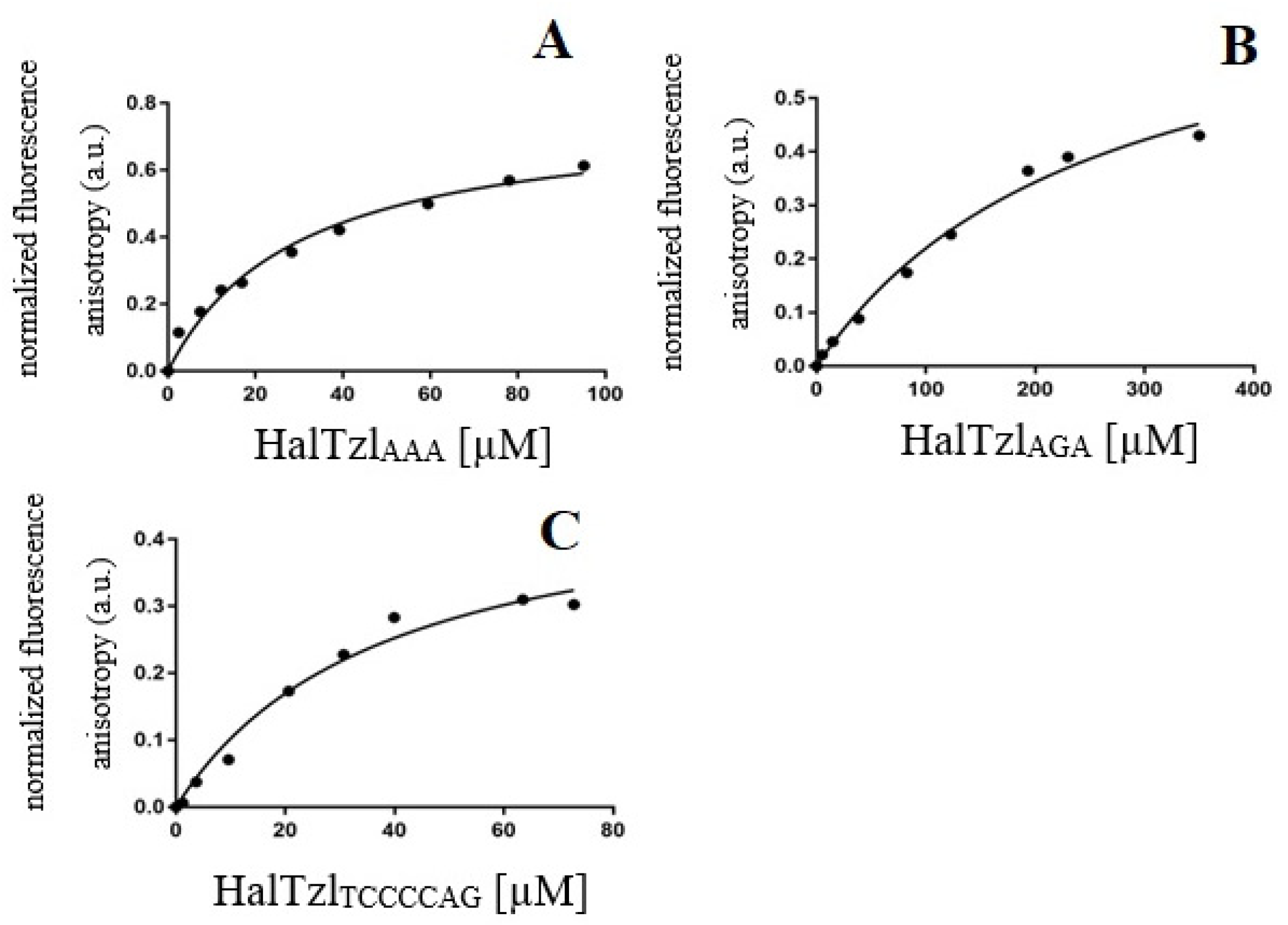
3.4. Fluorescence Anisotropy Study of TAR RNA—HalTzls Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1,4-TzlNBA | nucleobase amino acid bearing of nucleobase attached to the side chain via 1,4-linked 1,2,3-triazole moiety |
| Ac | acetyl |
| ACN | acetonitrile |
| ARR | arginine-rich region |
| B, BH | nucleobase |
| C18 | octadodecane chain |
| CuAAC | Cu(I)-catalyzed azide-alkyne cycloaddition |
| DCM | dichloromethane |
| DMAP | 4-Dimethylaminopyridine |
| DMF | N,N-dimethylformamide |
| FAM | fluorescein |
| Fmoc | fluorenylmethyloxycarbonyl |
| Fmoc-Aha | Fmoc-L-azidohomoalanine |
| Hal | L-homoalanine |
| HalTzl | nucleopeptide designed on the basis of L-azidohomoalanine (Hal), containing nucleobases attached through a 1,4-linked-triazole linker to the Hal’s side chain |
| HATU | [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium;3-hydroxytriazolo[4,5-b]pyridine hexafluorophosphate |
| HIV-1 | human immunodeficiency virus type 1 |
| HOAt | 1-Hydroxy-7-azabenzotriazole |
| MeOH | methanol |
| NBA | nucleobase amino acid |
| NP | nucleopeptide |
| PNA | peptide nucleic acid |
| RNAP II | RNA polymerase II |
| RT | reverse transcriptase |
| SPPS | solid-phase peptide synthesis |
| TAR | trans-activation response element |
| TB | tris/borate buffer |
| t-BuOH | tert-butanol |
| TFA | trifluoroacetic acid |
| Tzl | 1,2,3-triazol |
References
- Harries, L.W. RNA Biology Provides New Therapeutic Targets for Human Disease. Front. Genet. 2019, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. RNA in Biology and Therapeutics. Mol. Cells 2023, 46, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, L.; Wang, X.; Jin, H. RNA-based therapeutics: An overview and prospectus. Cell Death Dis. 2022, 13, 644. [Google Scholar] [CrossRef] [PubMed]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef] [PubMed]
- Mucha, P.; Szyk, A.; Rekowski, P.; Weiss, P.A.; Agris, P.F. Anticodon domain methylated nucleosides of yeast tRNA(Phe) are significant recognition determinants in the binding of a phage display selected peptide. Biochemistry 2001, 40, 14191–14199. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Monga, V. Peptide Nucleic Acids: Recent Developments in the Synthesis and Backbone Modifications. Bioorg. Chem. 2023, 141, 106860. [Google Scholar] [CrossRef]
- Wang, F.; Li, P.; Chu, H.C.; Lo, P.K. Nucleic Acids and Their Analogues for Biomedical Applications. Biosensors 2022, 12, 93. [Google Scholar] [CrossRef]
- Coppock, M.B.; Williams, M.E. Nucleic Acid Mimetics Supramolecular Aspects of Chemical Biology. In Supramolecular Chemistry: From Molecules to Nanomaterials; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Du, X.; Zhou, J.; Li, X.; Xu, B. Self-assembly of nucleopeptides to interact with DNAs. Interface Focus. 2017, 7, 20160116. [Google Scholar] [CrossRef]
- Robles, J.; Pedroso, E.; Grandas, A. Solid-phase synthesis of a nucleopeptide from the linking site of adenovirus-2 nucleoprotein, -Ser(p5′CATCAT)-Gly-Asp-. Convergent versus stepwise strategy. Nucleic Acids Res. 1995, 23, 4151–4161. [Google Scholar] [CrossRef]
- Tomassi, S.; Ieranò, C.; Del Bene, A.; D’Aniello, A.; Napolitano, M.; Rea, G.; Auletta, F.; Portella, L.; Capiluongo, A.; Mazzarella, V.; et al. Tailoring the Structure of Cell Penetrating DNA and RNA Binding Nucleopeptides. Int. J. Mol. Sci. 2022, 23, 8504. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Ruczyński, J.; Rekowski, P.; Alenowicz, M.; Mucha, P.; Pieszko, M.; Miszka, A.; Dobkowski, M.; Bluijssen, H. Synthesis and hybridization studies of a new CPP-PNA conjugate as a potential therapeutic agent in atherosclerosis treatment. Protein Pept. Lett. 2014, 21, 672–678. [Google Scholar] [CrossRef]
- Nandhini, K.P.; Shaer, D.A.; Albericio, F.; de la Torre, B.G. The challenge of peptide nucleic acid synthesis. Chem. Soc. Rev. 2023, 52, 2764–2789. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef]
- Warner, K.; Hajdin, C.; Weeks, K. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Falese, J.P.; Donlic, A.; Hargrove, A.E. Targeting RNA with small molecules: From fundamental principles towards the clinic. Chem. Soc. Rev. 2021, 50, 2224–2243. [Google Scholar] [CrossRef]
- Pant, S.; Jena, N.R. Inhibition of the RNA-dependent RNA Polymerase of the SARS-CoV-2 by Short Peptide Inhibitors. Eur. J. Pharm. Sci. 2021, 167, 106012. [Google Scholar] [CrossRef]
- Mucha, P.; Szyk, A.; Rekowski, P.; Barciszewski, J. Structural requirements for conserved Arg52 residue for interaction of the human immunodeficiency virus type 1 trans-activation responsive element with trans-activator of transcription protein (49-57). Capillary electrophoresis mobility shift assay. J. Chrom. A. 2002, 968, 211–220. [Google Scholar] [CrossRef]
- Musumeci, D.; Roviello, V.; Roviello, G.N. DNA- and RNA-binding ability of oligoDapT, a nucleobase-decorated peptide, for biomedical applications. Int. J. Nanomed. 2018, 13, 2613–2629. [Google Scholar] [CrossRef]
- Mercurio, M.E.; Tomassi, S.; Gaglione, M.; Russo, R.; Chambery, A.; Lama, S.; Stiuso, P.; Cosconati, S.; Novellino, E.; Di Maro, S.; et al. Switchable Protecting Strategy for Solid Phase Synthesis of DNA and RNA Interacting Nucleopeptides. J. Org. Chem. 2016, 81, 11612–11625. [Google Scholar] [CrossRef]
- van der Heden van Noort, G.J.; van Delft, P.; Meeuwenoord, N.J.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V. Fully automated sequential solid phase approach towards viral RNA-nucleopeptides. Chem. Commun. 2012, 48, 8093–8095. [Google Scholar] [CrossRef]
- Verona, M.; Verdolino, V.; Palazzesi, F.; Corradini, R. Focus on PNA Flexibility and RNA Binding using Molecular Dynamics and Metadynamics. Sci. Rep. 2017, 7, 42799. [Google Scholar] [CrossRef]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef]
- Kiliszek, A.; Banaszak, K.; Dauter, Z.; Rypniewski, W. The first crystal structures of RNA-PNA duplexes and a PNA-PNA duplex containing mismatches--toward anti-sense therapy against TREDs. Nucleic Acids Res. 2016, 44, 1937–1943. [Google Scholar] [CrossRef]
- Quijano, E.; Bahal, R.; Ricciardi, A.; Saltzman, W.M.; Glazer, P.M. Therapeutic Peptide Nucleic Acids: Principles, Limitations, and Opportunities. Yale J. Biol. Med. 2017, 90, 583–598. [Google Scholar]
- MacLelland, V.; Kravitz, M.; Gupta, A. Therapeutic and diagnostic applications of antisense peptide nucleic acids. Mol. Ther. Nucleic Acids 2023, 35, 102086. [Google Scholar] [CrossRef]
- Musumeci, D.; Mokhir, A.; Roviello, G.N. Synthesis and nucleic acid binding evaluation of a thyminyl l-diaminobutanoic acid-based nucleopeptide. Bioorg. Chem. 2020, 100, 103862. [Google Scholar] [CrossRef]
- Roviello, G.N.; Musumeci, D. Synthetic approaches to nucleopeptides containing all four nucleobases, and nucleic acid-binding studies on a mixed-sequence nucleo-oligolysine. RSC Adv. 2016, 6, 63578–63585. [Google Scholar] [CrossRef]
- Roviello, G.N.; Vicidomini, C.; Di Gaetano, S.; Capasso, D.; Musumeci, D.; Roviello, V. Solid phase synthesis and RNA-binding activity of an arginine-containing nucleopeptide. RSC Adv. 2016, 6, 14140–14148. [Google Scholar] [CrossRef]
- Roviello, G.N.; Musumeci, D.; D’Alessandro, C.; Pedone, C. Binding ability of a thymine-functionalized oligolysine towards nucleic acids. Bioorg. Med. Chem. 2014, 22, 997–1002. [Google Scholar] [CrossRef]
- Tomassi, S.; Montalban, F.F.; Russo, R.; Novellino, E.; Messere, A.; Di Maro, S. Investigation of the Stereochemical-Dependent DNA and RNA Binding of Arginine-Based Nucleopeptides. Symmetry 2019, 11, 567. [Google Scholar] [CrossRef]
- Kriek, N.M.; Meeuwenoord, N.J.; van den Elst, H.; Heus, H.A.; van der Marel, G.A.; Filippov, D.V. Chemical synthesis of picornaviral protein primers of RNA replication. Org. Biomol. Chem. 2006, 4, 3576–3586. [Google Scholar] [CrossRef]
- Gotora, P.T.; van der Sluis, R.; Williams, M.E. HIV-1 Tat amino acid residues that influence Tat-TAR binding affinity: A scoping review. BMC Infect. Dis. 2023, 23, 164. [Google Scholar] [CrossRef]
- Singh, R.R.; Mondal, I.; Janjua, T.; Popat, A.; Kulshreshtha, R. Engineered smart materials for RNA based molecular therapy to treat Glioblastoma. Bioact. Mater. 2023, 33, 396–423. [Google Scholar] [CrossRef]
- Yu, A.M.; Choi, Y.H.; Tu, M.J. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef]
- Garner, A.L. Contemporary Progress and Opportunities in RNA-Targeted Drug Discovery. ACS Med. Chem. Lett. 2023, 14, 251–259. [Google Scholar] [CrossRef]
- Abulwerdi, F.A.; Le Grice, S.F.J. Recent Advances in Targeting the HIV-1 Tat/TAR Complex. Curr. Pharm. Des. 2017, 23, 4112–4121. [Google Scholar] [CrossRef]
- Ne, E.; Palstra, R.J.; Mahmoudi, T. Transcription: Insights From the HIV-1 Promoter. Int. Rev. Cell Mol. Biol. 2018, 335, 191–243. [Google Scholar]
- Rice, A.P. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr. Pharm. Des. 2017, 23, 4098–4102. [Google Scholar] [CrossRef]
- Schulze-Gahmen, U.; Hurley, J.H. Structural mechanism for HIV-1 TAR loop recognition by Tat and the super elongation complex. Proc. Natl. Acad. Sci. USA 2018, 115, 12973–12978. [Google Scholar] [CrossRef]
- Ronsard, L.; Rai, T.; Rai, D.; Ramachandran, V.G.; Banerjea, A.C. In silico Analyses of Subtype Specific HIV-1 Tat-TAR RNA Interaction Reveals the Structural Determinants for Viral Activity. Front. Microbiol. 2017, 8, 1467. [Google Scholar] [CrossRef]
- Obayashi, C.M.; Shinohara, Y.; Masuda, T.; Kawai, G. Influence of the 5′-terminal sequences on the 5′-UTR structure of HIV-1 genomic RNA. Sci. Rep. 2021, 11, 10920. [Google Scholar] [CrossRef]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef]
- Dutilleul, A.; Rodari, A.; Van Lint, C. Depicting HIV-1 Transcriptional Mechanisms: A Summary of What We Know. Viruses 2020, 12, 1385. [Google Scholar] [CrossRef]
- Chavali, S.S.; Bonn-Breach, R.; Wedekind, J.E. Face-time with TAR: Portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery. J. Biol. Chem. 2019, 294, 9326–9341. [Google Scholar] [CrossRef]
- Chavali, S.S.; Mali, S.M.; Jenkins, J.L.; Fasan, R.; Wedekind, J.E. Co-crystal structures of HIV TAR RNA bound to lab-evolved proteins show key roles for arginine relevant to the design of cyclic peptide TAR inhibitors. J. Biol. Chem. 2020, 295, 16470–16486. [Google Scholar] [CrossRef]
- Chavali, S.S.; Mali, S.M.; Bonn, R.; Saseendran Anitha, A.; Bennett, R.P.; Smith, H.C.; Fasan, R.; Wedekind, J.E. Cyclic peptides with a distinct arginine-fork motif recognize the HIV trans-activation response RNA in vitro and in cells. J. Biol. Chem. 2021, 297, 101390. [Google Scholar] [CrossRef]
- Kaushik, N.; Basu, A.; Palumbo, P.; Myers, R.L.; Pandey, V.N. Anti-TAR polyamide nucleotide analog conjugated with a membrane-permeating peptide inhibits human immunodeficiency virus type 1 production. J. Virol. 2002, 76, 3881–3891. [Google Scholar] [CrossRef]
- Chaubey, B.; Tripathi, S.; Ganguly, S.; Harris, D.; Casale, R.A.; Pandey, V.N. A PNA-transportan conjugate targeted to the TAR region of the HIV-1 genome exhibits both antiviral and virucidal properties. Virology 2005, 331, 418–428. [Google Scholar] [CrossRef]
- Tan, X.; Bruchez, M.P.; Armitage, B.A. Closing the Loop: Constraining TAT Peptide by γPNA Hairpin for Enhanced Cellular Delivery of Biomolecules. Bioconjug Chem. 2018, 29, 2892–2898. [Google Scholar] [CrossRef]
- Lazrek, H.B.; Taourirte, M.; Oulih, T.; Barascut, J.L.; Imbach, J.L.; Pannecouque, C.; Witrouw, M.; De Clercq, E. Synthesis and anti-HIV activity of new modified 1,2,3-triazole acyclonucleosides. Nucleosides Nucleotides Nucleic Acids 2001, 20, 1949–1960. [Google Scholar] [CrossRef]
- Dobkowski, M.; Szychowska, A.; Pieszko, M.; Barciszewski, J.; Mucha, P. ‘Click’ chemistry synthesis and capillary electrophoresis study of 1,4-linked 1,2,3-triazole AZT-systemin conjugate. J. Pep. Sci. 2014, 20, 696–703. [Google Scholar] [CrossRef]
- Qi, L.; Zhang, J.; Liu, Q.; Gao, X. Ligand “switching on” fluorescence of HIV-1 RNA-templated copper nanoclusters for ligand-RNA interaction assays. Int. J. Biol. Macromol. 2023, 256, 127779. [Google Scholar] [CrossRef]
- Kumar, S.; Maiti, S. The effect of N-acetylation and N-methylation of lysine residue of Tat peptide on its interaction with HIV-1 TAR RNA. PLoS ONE 2013, 8, e77595. [Google Scholar] [CrossRef]
- Garg, A.; Heinemann, U. A novel form of RNA double helix based on G·U and C·A+ wobble base pairing. RNA 2018, 24, 209–218. [Google Scholar] [CrossRef]
- Carter-O’Connell, I.; Booth, D.; Eason, B.; Grover, N. Thermodynamic examination of trinucleotide bulged RNA in the context of HIV-1 TAR RNA. RNA 2008, 14, 2550–2556. [Google Scholar] [CrossRef]
- Zhao, R.; Zhu, J.; Jiang, X.; Bai, R. Click chemistry-aided drug discovery: A retrospective and prospective outlook. Eur. J. Med. Chem. 2024, 264, 116037. [Google Scholar] [CrossRef]
- Manoharan, A.; Jayan, J.; Rangarajan, T.M.; Bose, K.; Benny, F.; Ipe, R.S.; Kumar, S.; Kukreti, N.; Abdelgawad, M.A.; Ghoneim, M.M.; et al. “Click Chemistry”: An Emerging Tool for Developing a New Class of Structural Motifs against Various Neurodegenerative Disorders. ACS Omega 2023, 8, 44437–44457. [Google Scholar] [CrossRef]
- Kaur, J.; Saxena, M.; Rishi, N. An Overview of Recent Advances in Biomedical Applications of Click Chemistry. Bioconjugate Chem. 2021, 32, 1455–1471. [Google Scholar] [CrossRef]
- Guarrochena, X.; Kaudela, B.; Mindt, T.L. Automated solid-phase synthesis of metabolically stabilized triazolo-peptidomimetics. J. Pept. Sci. 2023, 29, e3488. [Google Scholar] [CrossRef]
- Carter, E.P.; Ang, C.G.; Chaiken, I.M. Peptide Triazole Inhibitors of HIV-1: Hijackers of Env Metastability. Curr. Protein Pept. Sci. 2023, 24, 59–77. [Google Scholar] [CrossRef]
- Baraniak, D.; Boryski, J. Triazole-Modified Nucleic Acids for the Application in Bioorganic and Medicinal Chemistry. Biomedicines 2021, 9, 628. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, T.; Gong, W.; Desaulniers, J.P. Chemical architecture and applications of nucleic acid derivatives containing 1,2,3-triazole functionalities synthesized via click chemistry. Molecules 2012, 17, 12665–12703. [Google Scholar] [CrossRef] [PubMed]
- Hornum, M.; Kumar, P.; Podsiadly, P.; Nielsen, P. Increasing the Stability of DNA/RNA Duplexes by Introducing Stacking Phenyl-Substituted Pyrazole, Furan, and Triazole Moieties in the Major Groove. J. Org. Chem. 2015, 80, 9592–9602. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Hornum, M.; Nielsen, L.J.; Enderlin, G.; Andersen, N.K.; Len, C.; Herve, G.; Sartori, G.; Nielsen, P. High-affinity RNA targeting by oligonucleotides displaying aromatic stacking and amino groups in the major groove. Comparison of triazoles and phenyl substituents. J. Org. Chem. 2014, 79, 2854–2863. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleopeptide Label | [M+H]+ | RP-HPLC | Quantity [mg] (Yield %) | ||
|---|---|---|---|---|---|
| Calculated | Determined | Analytical RP HPLC Gradient | tR [min] | ||
| HalTzlAGA | 931.40 | 931.40 | 0–100%B in 30 min | 7.32 | 3.90 (33.5) |
| HalTzlAAA | 915.42 | 915.42 | 0–100%B in 30 min | 7.06 | 4.20 (36.7) |
| HalTzlTCCCAG | 1747.74 | 874.43 * | 0–100%B in 30 min | 8.59 | 3.20 (16.6) |
| Free/Complexed TAR | Buffer | Tm [°C] | ΔTm [°C] |
|---|---|---|---|
| TAR UUU | Tris | 62.62 ± 0.74 | - |
| TAR UUU—HalTzlAAA | 63.31 ± 0.85 | 0.69 | |
| TAR UUU | TB | 70.41 ± 2.05 | - |
| TAR UUU—HalTzlAAA | 70.27 ± 2.07 | −0.14 | |
| TAR UCU | TB | 66.59 ± 0.93 | - |
| TAR UCU—HalTzlAGA | 67.44 ± 1.44 | 0.85 | |
| TARUCU—HalTzlTCCCAG | 68.34 ± 0.99 | 1.75 |
| Complex | Kd [µM] |
|---|---|
| TAR UUU—HalTzlAAA | 30.07 ± 4.76 |
| TAR UCU—HalTzlAGA | 255.9 ± 53.19 |
| TAR UCU—HalTzlTCCCAG | 38.27 ± 9.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mucha, P.; Pieszko, M.; Bylińska, I.; Wiczk, W.; Ruczyński, J.; Prochera, K.; Rekowski, P. Solid Phase Synthesis and TAR RNA-Binding Activity of Nucleopeptides Containing Nucleobases Linked to the Side Chains via 1,4-Linked-1,2,3-triazole. Biomedicines 2024, 12, 570. https://doi.org/10.3390/biomedicines12030570
Mucha P, Pieszko M, Bylińska I, Wiczk W, Ruczyński J, Prochera K, Rekowski P. Solid Phase Synthesis and TAR RNA-Binding Activity of Nucleopeptides Containing Nucleobases Linked to the Side Chains via 1,4-Linked-1,2,3-triazole. Biomedicines. 2024; 12(3):570. https://doi.org/10.3390/biomedicines12030570
Chicago/Turabian StyleMucha, Piotr, Małgorzata Pieszko, Irena Bylińska, Wiesław Wiczk, Jarosław Ruczyński, Katarzyna Prochera, and Piotr Rekowski. 2024. "Solid Phase Synthesis and TAR RNA-Binding Activity of Nucleopeptides Containing Nucleobases Linked to the Side Chains via 1,4-Linked-1,2,3-triazole" Biomedicines 12, no. 3: 570. https://doi.org/10.3390/biomedicines12030570