

Potential Causal Association between C-Reactive Protein Levels in Age-Related Macular Degeneration: A Two-Sample Mendelian Randomization Study


Abstract
1. Introduction
2. Materials and Methods
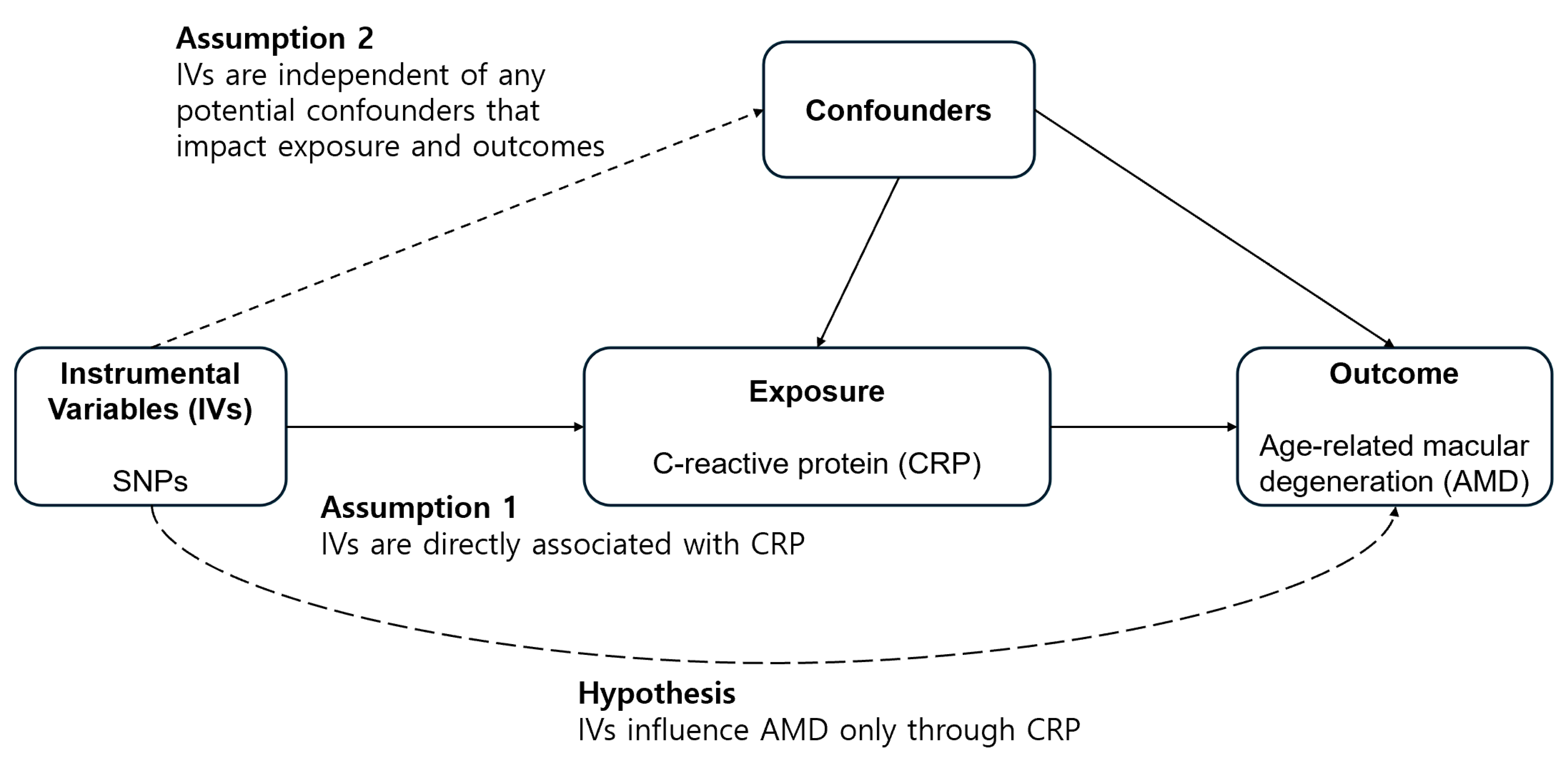
2.1. Study Design
2.2. Data Sources
2.3. Selection of the Genetic Instrumental Variables
2.4. Mendelian Randomization
2.5. Network Analysis of Selected SNP IVs
3. Results
3.1. Genetic Instrumental Variables
3.2. Heterogeneity and Horizontal Pleiotropy of Instrumental Variables
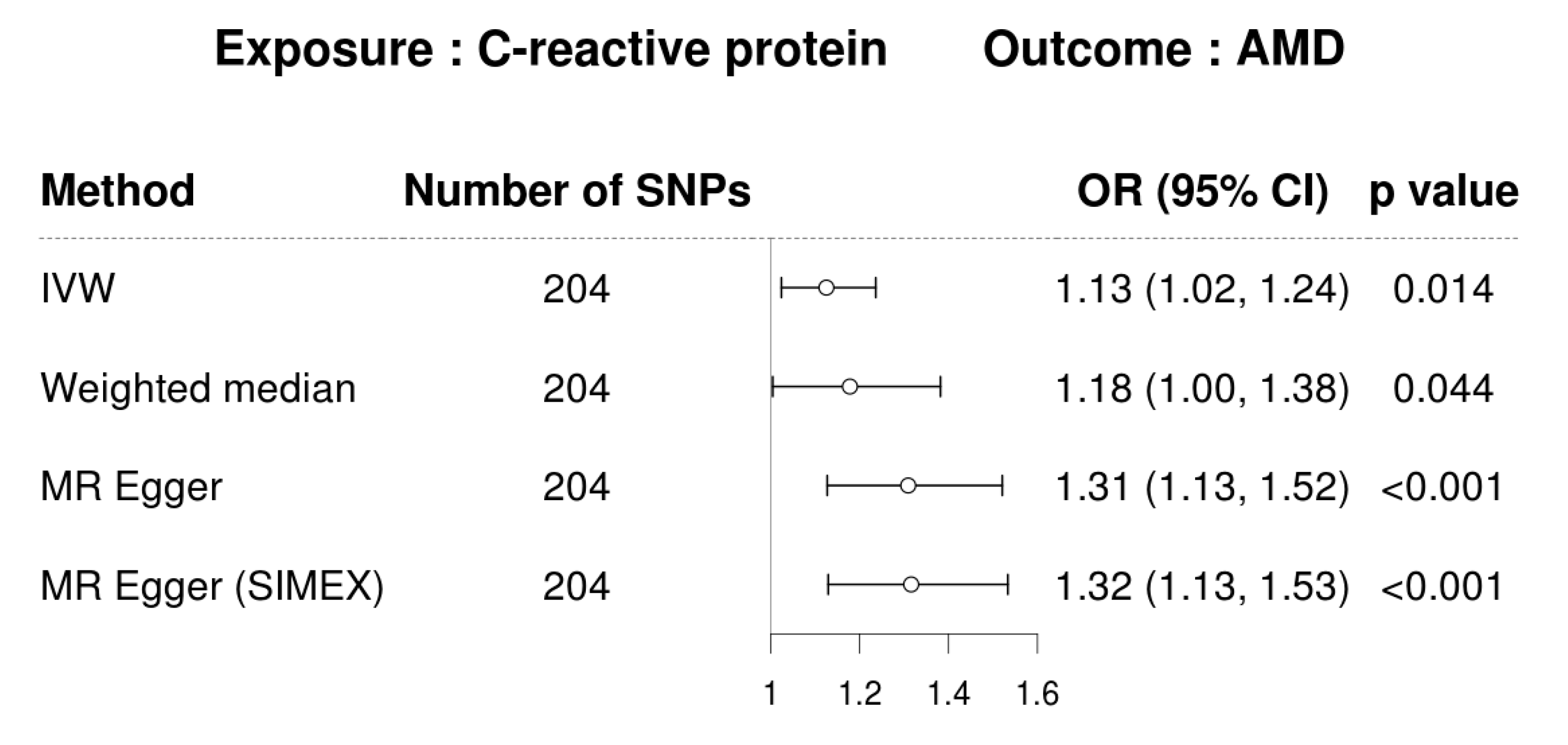
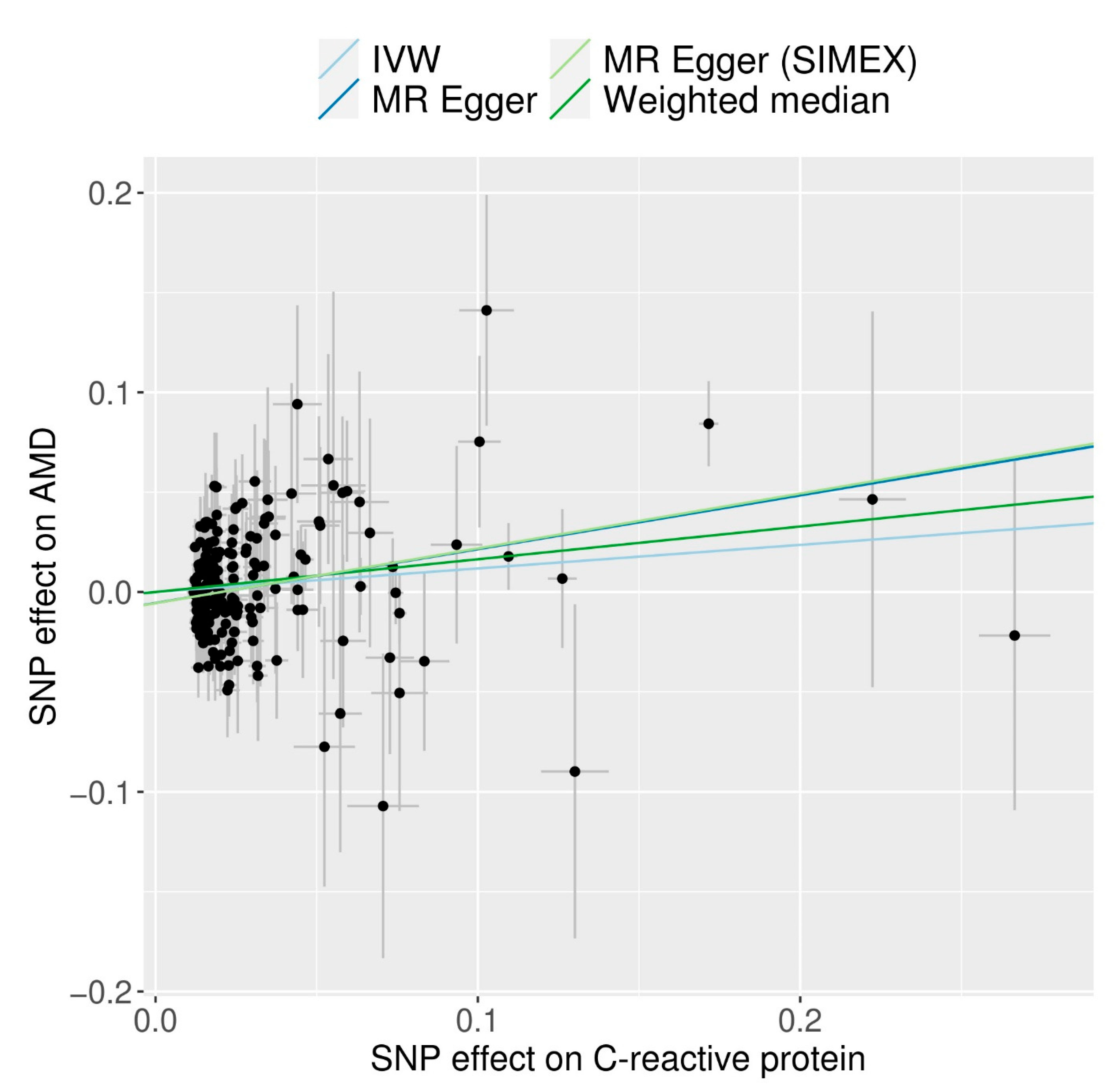
3.3. MR for Analyzing the Causal Association between CRP and AMD
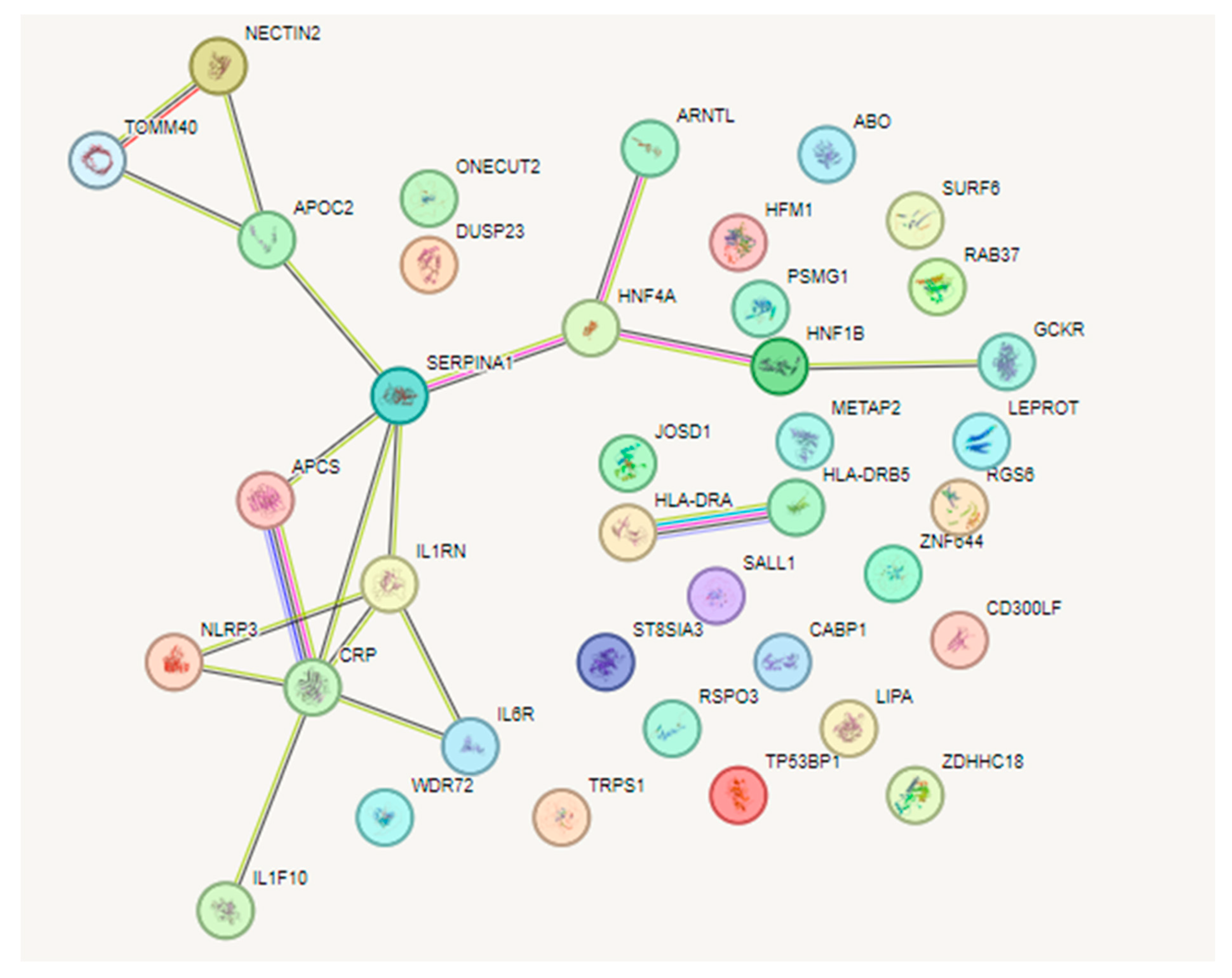
3.4. Network Analysis of SNPs between CRP and AMD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in Europe: A systematic review and meta-analysis. Br. J. Ophthalmol. 2019, 104, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Sobrin, L.; Ripke, S.; Yu, Y.; Fagerness, J.; Bhangale, T.R.; Tan, P.L.; Souied, E.H.; Buitendijk, G.H.; Merriam, J.E.; Richardson, A.J.; et al. Heritability and genome-wide association study to assess genetic differences between advanced age-related macular degeneration subtypes. Ophthalmology 2012, 119, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, M.M.; Owen, L.A.; Morrison, M.A.; Morgan, D.J.; Li, M.; Shakoor, A.; Vitale, A.; Iyengar, S.; Stambolian, D.; Kim, I.K.; et al. Genetics of age-related macular degeneration (AMD). Hum. Mol. Genet. 2017, 26, R45–R50. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Stambolian, D.; Edwards, A.O.; Branham, K.E.; Othman, M.; Jakobsdottir, J.; Tosakulwong, N.; Pericak-Vance, M.A.; Campochiaro, P.A.; Klein, M.L.; et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401–7406. [Google Scholar] [CrossRef] [PubMed]
- Holliday, E.G.; Smith, A.V.; Cornes, B.K.; Buitendijk, G.H.; Jensen, R.A.; Sim, X.; Aspelund, T.; Aung, T.; Baird, P.N.; Boerwinkle, E.; et al. Insights into the genetic architecture of early stage age-related macular degeneration: A genome-wide association study meta-analysis. PLoS ONE 2013, 8, e53830. [Google Scholar] [CrossRef]
- Shin, H.T.; Yoon, B.W.; Seo, J.H. Comparison of risk allele frequencies of single nucleotide polymorphisms associated with age-related macular degeneration in different ethnic groups. BMC Ophthalmol. 2021, 21, 97. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, J.L. Age-related macular degeneration. JAMA 2002, 288, 2233–2236. [Google Scholar] [CrossRef] [PubMed]
- De Jong, P.T. Age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Kanda, A.; Abecasis, G.; Swaroop, A. Inflammation in the pathogenesis of age-related macular degeneration. Br. J. Ophthalmol. 2008, 92, 448–450. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nowak, J.Z. Age-related macular degeneration (AMD): Pathogenesis and therapy. Pharmacol. Rep. 2006, 58, 353–363. [Google Scholar] [PubMed]
- Tillett, W.S.; Francis, T. Serological Reactions in Pneumonia with a Non-Protein Somatic Fraction of Pneumococcus. J. Exp. Med. 1930, 52, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B.; Bader, A.; Poli, V.; Ruther, U. Interleukin-6 is necessary, but not sufficient, for induction of the humanC-reactive protein gene in vivo. Biochem. J. 1997, 325, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Szalai, A.J.; van Ginkel, F.W.; Dalrymple, S.A.; Murray, R.; McGhee, J.R.; Volanakis, J.E. Testosterone and IL-6 requirements for human C-reactive protein gene expression in transgenic mice. J. Immunol. 1998, 160, 5294–5299. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [PubMed]
- Healy, B.; Freedman, A. Infections. BMJ 2006, 332, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Osman, R.; L’Allier, P.L.; Elgharib, N.; Tardif, J.C. Critical appraisal of C-reactive protein throughout the spectrum of cardiovascular disease. Vasc. Health Risk Manag. 2006, 2, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Soinio, M.; Marniemi, J.; Laakso, M.; Lehto, S.; Ronnemaa, T. High-sensitivity C-reactive protein and coronary heart disease mortality in patients with type 2 diabetes: A 7-year follow-up study. Diabetes Care 2006, 29, 329–333. [Google Scholar] [CrossRef]
- Jiang, R.; Wu, J.; Rosenblatt, M.; Dai, W.; Rodriguez, R.X.; Sui, J.; Qi, S.; Liang, Q.; Xu, B.; Meng, Q.; et al. Elevated C-reactive protein mediates the liver-brain axis: A preliminary study. EBioMedicine 2023, 93, 104679. [Google Scholar] [CrossRef]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain—From eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Chirco, K.R.; Whitmore, S.S.; Wang, K.; Potempa, L.A.; Halder, J.A.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Monomeric C-reactive protein and inflammation in age-related macular degeneration. J. Pathol. 2016, 240, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O.; Ritter, R., 3rd; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef]
- Seddon, J.M.; Gensler, G.; Milton, R.C.; Klein, M.L.; Rifai, N. Association between C-reactive protein and age-related macular degeneration. JAMA 2004, 291, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; George, S.; Rosner, B.; Rifai, N. Progression of age-related macular degeneration: Prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch. Ophthalmol. 2005, 123, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Knudtson, M.D.; Klein, B.E.; Wong, T.Y.; Cotch, M.F.; Liu, K.; Cheng, C.Y.; Burke, G.L.; Saad, M.F.; Jacobs, D.R., Jr.; et al. Inflammation, complement factor h, and age-related macular degeneration: The Multi-ethnic Study of Atherosclerosis. Ophthalmology 2008, 115, 1742–1749. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Tan, A.G.; Mitchell, P.; Wang, J.J. A review and meta-analysis of the association between C-reactive protein and age-related macular degeneration. Surv. Ophthalmol. 2011, 56, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Mitta, V.P.; Christen, W.G.; Glynn, R.J.; Semba, R.D.; Ridker, P.M.; Rimm, E.B.; Hankinson, S.E.; Schaumberg, D.A. C-reactive protein and the incidence of macular degeneration: Pooled analysis of 5 cohorts. JAMA Ophthalmol. 2013, 131, 507–513. [Google Scholar] [CrossRef] [PubMed]
- McGwin, G.; Hall, T.A.; Xie, A.; Owsley, C. The relation between C reactive protein and age related macular degeneration in the Cardiovascular Health Study. Br. J. Ophthalmol. 2005, 89, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.H.; Tan, A.G.; Rochtchina, E.; Favaloro, E.J.; Williams, A.; Mitchell, P.; Wang, J.J. Circulating inflammatory markers and hemostatic factors in age-related maculopathy: A population-based case-control study. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Despriet, D.D.; Klaver, C.C.; Witteman, J.C.; Bergen, A.A.; Kardys, I.; de Maat, M.P.; Boekhoorn, S.S.; Vingerling, J.R.; Hofman, A.; Oostra, B.A.; et al. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA 2006, 296, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Schaumberg, D.A.; Christen, W.G.; Kozlowski, P.; Miller, D.T.; Ridker, P.M.; Zee, R.Y. A prospective assessment of the Y402H variant in complement factor H, genetic variants in C-reactive protein, and risk of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2336–2340. [Google Scholar] [CrossRef]
- Kim, I.K.; Ji, F.; Morrison, M.A.; Adams, S.; Zhang, Q.; Lane, A.M.; Capone, A.; Dryja, T.P.; Ott, J.; Miller, J.W.; et al. Comprehensive analysis of CRP, CFH Y402H and environmental risk factors on risk of neovascular age-related macular degeneration. Mol. Vis. 2008, 14, 1487–1495. [Google Scholar] [PubMed]
- Cipriani, V.; Hogg, R.E.; Sofat, R.; Moore, A.T.; Webster, A.R.; Yates, J.R.W.; Fletcher, A.E.; European Eye Study, G. Association of C-Reactive Protein Genetic Polymorphisms With Late Age-Related Macular Degeneration. JAMA Ophthalmol. 2017, 135, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ong, J.S.; An, J.; Hewitt, A.W.; Gharahkhani, P.; MacGregor, S. Using Mendelian randomization to evaluate the causal relationship between serum C-reactive protein levels and age-related macular degeneration. Eur. J. Epidemiol. 2020, 35, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. Multivariable Mendelian randomization: The use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 2015, 181, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Minelli, C.; Del Greco, M.F.; van der Plaat, D.A.; Bowden, J.; Sheehan, N.A.; Thompson, J. The use of two-sample methods for Mendelian randomization analyses on single large datasets. Int. J. Epidemiol. 2021, 50, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Winkler, T.W.; Grassmann, F.; Brandl, C.; Kiel, C.; Gunther, F.; Strunz, T.; Weidner, L.; Zimmermann, M.E.; Korb, C.A.; Poplawski, A.; et al. Genome-wide association meta-analysis for early age-related macular degeneration highlights novel loci and insights for advanced disease. BMC Med. Genom. 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G.; CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 2011, 40, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Davey Smith, G.; Sheehan, N.; Thompson, J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med. 2017, 36, 1783–1802. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, Y.A.; Seo, J.H. Causal Association of Obesity and Dyslipidemia with Type 2 Diabetes: A Two-Sample Mendelian Randomization Study. Genes 2022, 13, 2407. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Davey Smith, G.; Sheehan, N.A.; Thompson, J.R. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: The role of the I2 statistic. Int. J. Epidemiol. 2016, 45, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Publisher Correction: Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 1196. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Davey Smith, G.; Davies, N.M.; Dudbridge, F.; Gill, D.; Glymour, M.M.; Hartwig, F.P.; Holmes, M.V.; Minelli, C.; Relton, C.L.; et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2019, 4, 186. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.F.; Minelli, C.; Sheehan, N.A.; Thompson, J.R. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 2015, 34, 2926–2940. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Lee, S.; Won, S. Causal Evaluation of Laboratory Markers in Type 2 Diabetes on Cancer and Vascular Diseases Using Various Mendelian Randomization Tools. Front. Genet. 2020, 11, 597420. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F. Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.H.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 2002, 134, 411–431. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, D.; Douglas, I.; Smeeth, L.; Fletcher, A. Age-related macular degeneration and complement activation-related diseases: A population-based case-control study. Ophthalmology 2008, 115, 1904–1910. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [PubMed]
- Kawa, M.P.; Machalinska, A.; Roginska, D.; Machalinski, B. Complement system in pathogenesis of AMD: Dual player in degeneration and protection of retinal tissue. J. Immunol. Res. 2014, 2014, 483960. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Gensler, G.; Rosner, B. C-reactive protein and CFH, ARMS2/HTRA1 gene variants are independently associated with risk of macular degeneration. Ophthalmology 2010, 117, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Gensler, G.; Klein, M.L.; Milton, R.C. C-reactive protein and homocysteine are associated with dietary and behavioral risk factors for age-related macular degeneration. Nutrition 2006, 22, 441–443. [Google Scholar] [CrossRef]
- Bhutto, I.A.; Baba, T.; Merges, C.; Juriasinghani, V.; McLeod, D.S.; Lutty, G.A. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br. J. Ophthalmol. 2011, 95, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Molins, B.; Romero-Vazquez, S.; Fuentes-Prior, P.; Adan, A.; Dick, A.D. C-Reactive Protein as a Therapeutic Target in Age-Related Macular Degeneration. Front. Immunol. 2018, 9, 808. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Krogh Nielsen, M.; Sorensen, T.L.; Subhi, Y. Systemic levels of C-reactive protein in patients with age-related macular degeneration: A systematic review with meta-analyses. Mech. Ageing Dev. 2020, 191, 111353. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, S.; Niu, L.; Huang, S. High-Throughput Sequencing Data Reveal an Antiangiogenic Role of HNF4A-Mediated CACNA1A/VEGFA Axis in Proliferative Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2023, 64, 32. [Google Scholar] [CrossRef] [PubMed]
- Baba, K.; Piano, I.; Lyuboslavsky, P.; Chrenek, M.A.; Sellers, J.T.; Zhang, S.; Gargini, C.; He, L.; Tosini, G.; Iuvone, P.M. Removal of clock gene Bmal1 from the retina affects retinal development and accelerates cone photoreceptor degeneration during aging. Proc. Natl. Acad. Sci. USA 2018, 115, 13099–13104. [Google Scholar] [CrossRef] [PubMed]
- Gambella, A.; Kalantari, S.; Cadamuro, M.; Quaglia, M.; Delvecchio, M.; Fabris, L.; Pinon, M. The Landscape of HNF1B Deficiency: A Syndrome Not Yet Fully Explored. Cells 2023, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Gomer, R.H. The Development of Serum Amyloid P. as a Possible Therapeutic. Front. Immunol. 2018, 9, 2328. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.G.; Reeder, E.L.; Collins, S.M.; Gopalan, P.; Robson, M.J. Contributions of Interleukin-1 Receptor Signaling in Traumatic Brain Injury. Front. Behav. Neurosci. 2019, 13, 287. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W.; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.W.; Casanova, R.; Saldana, S.; Kuchibhatla, M.; Plassman, B.L.; Hayden, K.M. Analysis of pleiotropic genetic effects on cognitive impairment, systemic inflammation, and plasma lipids in the Health and Retirement Study. Neurobiol. Aging 2019, 80, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chang, S.C.; Lee, Y.S.; Ho, W.M.; Huang, Y.H.; Wu, Y.Y.; Chu, Y.C.; Wu, K.H.; Wei, L.S.; Wang, H.L.; et al. TOMM40 Genetic Variants Cause Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 4085. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Shaw, M.; Todd, K.; Khrestian, M.; D’Aleo, G.; Barnard, P.J.; Zahratka, J.; Pillai, J.; Yu, C.E.; Keene, C.D.; et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 2018, 63, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, X.; Escames, G.; Lei, W.; Zhang, X.; Li, M.; Jing, T.; Yao, Y.; Qiu, Z.; Wang, Z.; et al. The NLRP3 inflammasome: Contributions to inflammation-related diseases. Cell. Mol. Biol. Lett. 2023, 28, 51. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, D.N.; Pathak, A.; Agrawal, A. IL-6 regulates induction of C-reactive protein gene expression by activating STAT3 isoforms. Mol. Immunol. 2022, 146, 50–56. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Data Source | No. of Participants | Population | No. of Variants | Reference |
|---|---|---|---|---|---|
| CRP | BBJ Project + UKB | 436,491 | East Asian and European | 20,529,698 | [46] |
| AMD | Eleven sources of data including the IAMDGC and UKB | 105,248 (14,034 cases + 91,214 controls) | European | 11,703,383 | [47] |
| Exposure | Heterogeneity | Horizontal Pleiotropy | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cochran’s Q Test from IVW | Rücker’s Q Test from MR-Egger | MR-PRESSO Global Test | MR-Egger | MR-Egger (SIMEX) | ||||||
| N | F | I2 (%) | p-Value | p-Value | p-Value | Intercept, β (SE) | p-Value | Intercept, β (SE) | p-Value | |
| C-reactive protein | 204 | 113.47 | 97.45 | 0.042 | 0.074 | 0.032 | −0.006 (0.002) | 0.012 | −0.006 (0.002) | 0.012 |
| SNP | Chr | Position | Nearby Gene | Effect Allele | Other Allele | Effect Allele Frequency | β | SE | p-Value | F |
|---|---|---|---|---|---|---|---|---|---|---|
| rs12972970 | 19 | 45387596 | NECTIN2 | A | G | 0.152 | −0.172 | 0.0030 | 1.00 × 10−200 | 3271.84 |
| rs117310449 | 19 | 45393516 | TOMM40 | T | C | 0.012 | −0.267 | 0.0111 | 2.74 × 10−128 | 576.43 |
| rs10420434 | 19 | 45451190 | APOC4-APOC2 | A | G | 0.047 | 0.059 | 0.0057 | 1.65 × 10−25 | 108.60 |
| rs28929474 | 14 | 94844947 | SERPINA1 | T | C | 0.020 | −0.103 | 0.0085 | 2.27 × 10−33 | 145.98 |
| rs1800961 | 20 | 43042364 | HNF4A | T | C | 0.027 | −0.101 | 0.0066 | 3.55 × 10−52 | 231.87 |
| rs1037169 | 11 | 13361005 | ARNTL | C | T | 0.688 | 0.029 | 0.0026 | 2.68 × 10−29 | 127.00 |
| rs17138478 | 17 | 36073320 | HNF1B | A | C | 0.144 | 0.032 | 0.0030 | 2.14 × 10−25 | 112.36 |
| rs1260326 | 2 | 27730940 | GCKR | C | T | 0.575 | −0.064 | 0.0022 | 3.85 × 10−187 | 835.74 |
| rs1811471 | 1 | 159642599 | APCS | A | G | 0.258 | 0.110 | 0.0028 | 1.00 × 10−200 | 1529.37 |
| rs55709272 | 2 | 113867288 | IL1F10; IL1RN | C | T | 0.364 | 0.043 | 0.0024 | 2.27 × 10−73 | 319.52 |
| rs4133213 | 1 | 154395212 | IL6R | A | C | 0.442 | −0.076 | 0.0022 | 1.00 × 10−200 | 1183.99 |
| rs56015600 | 1 | 247601886 | NLRP3 | G | A | 0.622 | 0.034 | 0.0022 | 4.09 × 10−52 | 233.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, B.W.; Lee, Y.; Seo, J.H. Potential Causal Association between C-Reactive Protein Levels in Age-Related Macular Degeneration: A Two-Sample Mendelian Randomization Study. Biomedicines 2024, 12, 807. https://doi.org/10.3390/biomedicines12040807
Yoon BW, Lee Y, Seo JH. Potential Causal Association between C-Reactive Protein Levels in Age-Related Macular Degeneration: A Two-Sample Mendelian Randomization Study. Biomedicines. 2024; 12(4):807. https://doi.org/10.3390/biomedicines12040807
Chicago/Turabian StyleYoon, Byung Woo, Young Lee, and Je Hyun Seo. 2024. "Potential Causal Association between C-Reactive Protein Levels in Age-Related Macular Degeneration: A Two-Sample Mendelian Randomization Study" Biomedicines 12, no. 4: 807. https://doi.org/10.3390/biomedicines12040807
APA StyleYoon, B. W., Lee, Y., & Seo, J. H. (2024). Potential Causal Association between C-Reactive Protein Levels in Age-Related Macular Degeneration: A Two-Sample Mendelian Randomization Study. Biomedicines, 12(4), 807. https://doi.org/10.3390/biomedicines12040807