
Molecular Mechanisms of Ischemic Stroke: A Review Integrating Clinical Imaging and Therapeutic Perspectives
 , , ,
, , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Pathophysiology of Ischemic Stroke
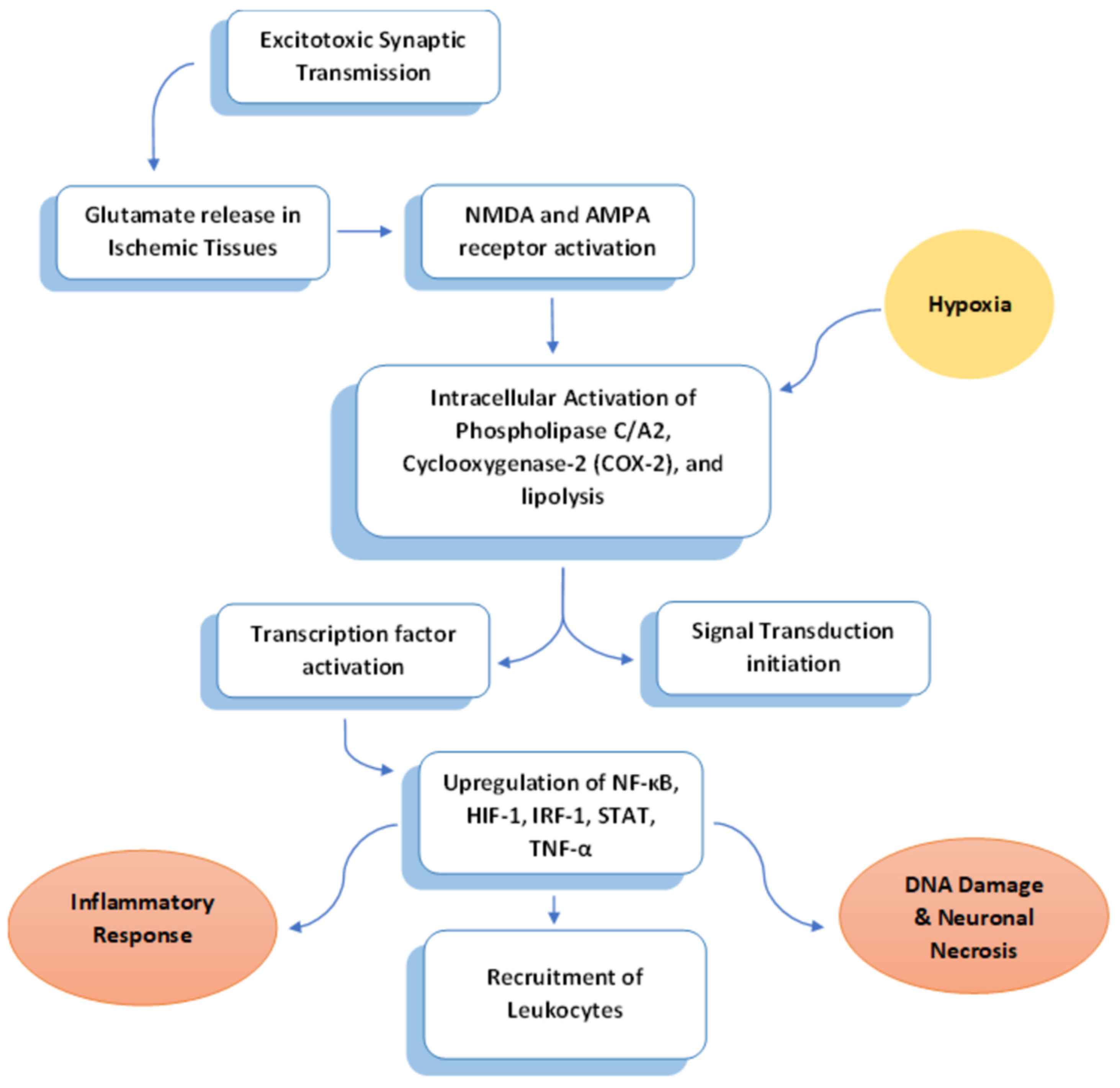
2.1. Excitotoxicity
2.2. Inflammation
2.3. Apoptosis
3. Imaging the Stages of Ischemic Stroke: CT and MRI Correlation
3.1. Stages of Ischemic Stroke on Imaging
3.1.1. Hyperacute Stage
3.1.2. Acute Stage: 12–24 h
3.1.3. Subacute Stage: 2 Days–2 Weeks
3.1.4. Chronic Stage: 2 Weeks–2 Months
4. Advancement in Clinical Imaging Platforms
5. Radiopharmaceuticals in Atherosclerosis Imaging
6. Netrin-1 and Its Regulatory Effect
7. Blood–Brain Barrier Permeability in Stroke
8. Established Pharmaceutical Strategies in Stroke Management
8.1. Antiplatelets in Stroke Management
8.2. Anticoagulants in Stroke Management
9. Interorgan Interaction in Ischemic Events
10. Time-Dependent Excitotoxicity and Inflammation
11. Endothelium Dysfunction and Necroptosis after Ischemia/Reperfusion Injury
11.1. Role of Endothelium in BBB and Neurovascular Unit
11.2. Mechanism of Endothelial Necroptosis after Ischemia/Reperfusion Injury
11.3. Intervention Using RIPK1-Inhibitor and Infliximab as a Potential Therapeutic Drug
12. Imaging Ischemic Penumbra and Future Perspectives
12.1. Characteristics of Ischemic Penumbra
12.2. Imaging Techniques for Evaluating Penumbra Evolution
12.3. Multiparameter Imaging for Accurate Assessment
13. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.B.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [PubMed]
- WHO EMRO|Stroke, Cerebrovascular Accident|Health Topics. Available online: http://www.emro.who.int/health-topics/stroke-cerebrovascular-accident/index.html (accessed on 2 February 2024).
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of Ischaemic Stroke: An Integrated View. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Lyden, P.D. Revisiting Cerebral Postischemic Reperfusion Injury: New Insights in Understanding Reperfusion Failure, Hemorrhage, and Edema. Int. J. Stroke 2015, 10, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Wright, P.M.; Markus, R.; Howells, D.W.; Davis, S.M.; Donnan, G.A. Salvaging the Ischaemic Penumbra: More than Just Reperfusion? Clin. Exp. Pharmacol. Physiol. 2002, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Obrenovitch, T.P. The Ischaemic Penumbra: Twenty Years On. Cerebrovasc. Brain Metab. Rev. 1995, 7, 297–323. [Google Scholar]
- Strong, A.J.; Smith, S.E.; Whittington, D.J.; Meldrum, B.S.; Parsons, A.A.; Krupinski, J.; Hunter, A.J.; Patel, S.; Robertson, C. Factors Influencing the Frequency of Fluorescence Transients as Markers of Peri-Infarct Depolarizations in Focal Cerebral Ischemia. Stroke 2000, 31, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Mitsios, N.; Gaffney, J.; Kumar, P.; Krupinski, J.; Kumar, S.; Slevin, M. Pathophysiology of Acute Ischaemic Stroke: An Analysis of Common Signalling Mechanisms and Identification of New Molecular Targets. Pathobiology 2006, 73, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Leker, R.R.; Shohami, E. Cerebral Ischemia and Trauma-Different Etiologies yet Similar Mechanisms: Neuroprotective Opportunities. Brain Res. Brain Res. Rev. 2002, 39, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Alexander, M. Cerebral Ischemia and Inflammation. Curr. Opin. Neurol. 2001, 14, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Kumar, P.; Gaffney, J.; Kumar, S. Gene Activation and Protein Expression Following Ischaemic Stroke: Strategies towards Neuroprotection. J. Cell. Mol. Med. 2005, 9, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Carlson, N.G.; Wieggel, W.A.; Chen, J.; Bacchi, A.; Rogers, S.W.; Gahring, L.C. Inflammatory Cytokines IL-1 Alpha, IL-1 Beta, IL-6, and TNF-Alpha Impart Neuroprotection to an Excitotoxin through Distinct Pathways. J. Immunol. 1999, 163, 3963–3968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chopp, M. Vascular Endothelial Growth Factor and Angiopoietins in Focal Cerebral Ischemia. Trends Cardiovasc. Med. 2002, 12, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Saura, M.; Zaragoza, C.; Bao, C.; McMillan, A.; Lowenstein, C.J. Interaction of Interferon Regulatory Factor-1 and Nuclear Factor kappaB during Activation of Inducible Nitric Oxide Synthase Transcription. J. Mol. Biol. 1999, 289, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.S.; Park, E.J.; Joe, E.; Jou, I. JAK-STAT Signaling Mediates Gangliosides-Induced Inflammatory Responses in Brain Microglial Cells. J. Biol. Chem. 2002, 277, 40594–40601. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.C.; Feuerstein, G.Z. Inflammatory Mediators and Stroke: New Opportunities for Novel Therapeutics. J. Cereb. Blood Flow Metab. 1999, 19, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Pelidou, S.H.; Kostulas, N.; Matusevicius, D.; Kivisäkk, P.; Kostulas, V.; Link, H. High Levels of IL-10 Secreting Cells Are Present in Blood in Cerebrovascular Diseases. Eur. J. Neurol. 1999, 6, 437–442. [Google Scholar] [CrossRef]
- Nathan, C.F.; Prendergast, T.J.; Wiebe, M.E.; Richard Stanley, E.; Platzer, E.; Remold, H.G.; Welte, K.; Rubin, B.Y.; Murray, H.W. Activation of Human Macrophages: Comparison of Other Cytokines with Interferon-γ. J. Exp. Med. 1984, 160, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Baadsgaard, O.; Fisher, G.J.; Voorhees, J.J.; Cooper, K.D. Interactions of Epidermal Cells and T Cells in Inflammatory Skin Diseases. J. Am. Acad. Dermatol. 1990, 23, 1312–1316; discussion 1316–1317. [Google Scholar] [CrossRef] [PubMed]
- Vila, N.; Castillo, J.; Dávalos, A.; Chamorro, A. Proinflammatory Cytokines and Early Neurological Worsening in Ischemic Stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Perini, F.; Morra, M.; Alecci, M.; Galloni, E.; Marchi, M.; Toso, V. Temporal Profile of Serum Anti-Inflammatory and pro-Inflammatory Interleukins in Acute Ischemic Stroke Patients. Neurol. Sci. 2001, 22, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wiessner, C.; Gehrmann, J.; Lindholm, D.; Töpper, R.; Kreutzberg, G.W.; Hossmann, K.A. Expression of Transforming Growth Factor-Beta 1 and Interleukin-1 Beta mRNA in Rat Brain Following Transient Forebrain Ischemia. Acta Neuropathol. 1993, 86, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Clark, R.K.; McDonnell, P.C.; Young, P.R.; White, R.F.; Barone, F.C.; Feuerstein, G.Z. Tumor Necrosis Factor-Alpha Expression in Ischemic Neurons. Stroke 1994, 25, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yue, T.L.; Barone, F.C.; White, R.F.; Gagnon, R.C.; Feuerstein, G.Z. Concomitant Cortical Expression of TNF-Alpha and IL-1 Beta mRNAs Follows Early Response Gene Expression in Transient Focal Ischemia. Mol. Chem. Neuropathol. 1994, 23, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, J.; Losy, J. Early TNF-Alpha Levels Correlate with Ischaemic Stroke Severity. Acta Neurol. Scand. 2001, 104, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Che, X.; Ye, W.; Panga, L.; Wu, D.C.; Yang, G.Y. Monocyte Chemoattractant Protein-1 Expressed in Neurons and Astrocytes during Focal Ischemia in Mice. Brain Res. 2001, 902, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Legos, J.J.; Whitmore, R.G.; Erhardt, J.A.; Parsons, A.A.; Tuma, R.F.; Barone, F.C. Quantitative Changes in Interleukin Proteins Following Focal Stroke in the Rat. Neurosci. Lett. 2000, 282, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Kostulas, N.; Pelidou, S.H.; Kivisäkk, P.; Kostulas, V.; Link, H. Increased IL-1beta, IL-8, and IL-17 mRNA Expression in Blood Mononuclear Cells Observed in a Prospective Ischemic Stroke Study. Stroke 1999, 30, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yue, T.L.; Barone, F.C.; Feuerstein, G.Z. Monocyte Chemoattractant Protein-1 Messenger RNA Expression in Rat Ischemic Cortex. Stroke 1995, 26, 661–665; discussion 665–666. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Ikeda, K.; Mukaida, N.; Harada, A.; Matsumoto, Y.; Yamashita, J.; Matsushima, K. Prevention of Cerebral Edema and Infarct in Cerebral Reperfusion Injury by an Antibody to Interleukin-8. Lab. Investig. 1997, 77, 119–125. [Google Scholar] [PubMed]
- Bitsch, A.; Klene, W.; Murtada, L.; Prange, H.; Rieckmann, P. A Longitudinal Prospective Study of Soluble Adhesion Molecules in Acute Stroke. Stroke 1998, 29, 2129–2135. [Google Scholar] [CrossRef]
- Clark, W.M.; Coull, B.M.; Briley, D.P.; Mainolfi, E.; Rothlein, R. Circulating Intercellular Adhesion Molecule-1 Levels and Neutrophil Adhesion in Stroke. J. Neuroimmunol. 1993, 44, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Onodera, H.; Shiga, Y.; Shozuhara, H.; Ninomiya, M.; Kihara, T.; Tamatani, T.; Miyasaka, M.; Kogure, K. Role of Cell Adhesion Molecules in Brain Injury after Transient Middle Cerebral Artery Occlusion in the Rat. Brain Res. 1994, 656, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Mössner, R.; Motsch, L.; Kischka, U.; Grau, A.; Hennerici, M. Circulating Selectin- and Immunoglobulin-Type Adhesion Molecules in Acute Ischemic Stroke. Stroke 1995, 26, 1361–1364. [Google Scholar] [CrossRef]
- Huang, J.; Choudhri, T.F.; Winfree, C.J.; McTaggart, R.A.; Kiss, S.; Mocco, J.; Kim, L.J.; Protopsaltis, T.S.; Zhang, Y.; Pinsky, D.J.; et al. Postischemic Cerebrovascular E-Selectin Expression Mediates Tissue Injury in Murine Stroke. Stroke 2000, 31, 3047–3053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.L.; Chopp, M.; Jiang, N.; Tang, W.X.; Prostak, J.; Manning, A.M.; Anderson, D.C. Anti-Intercellular Adhesion Molecule-1 Antibody Reduces Ischemic Cell Damage after Transient but Not Permanent Middle Cerebral Artery Occlusion in the Wistar Rat. Stroke 1995, 26, 1438–1442; discussion 1443. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Hayashi, T.; Tojo, S.J.; Kitagawa, H.; Kimura, K.; Mizugaki, M.; Itoyama, Y.; Abe, K. Anti-P-Selectin Antibody Attenuates Rat Brain Ischemic Injury. Neurosci. Lett. 1999, 265, 163–166. [Google Scholar] [CrossRef]
- Toyoda, T.; Suzuki, S.; Kassell, N.F.; Lee, K.S. Intraischemic Hypothermia Attenuates Neutrophil Infiltration in the Rat Neocortex after Focal Ischemia-Reperfusion Injury. Neurosurgery 1996, 39, 1200–1205. [Google Scholar] [CrossRef]
- Dawson, J.; Hurtenbach, U.; MacKenzie, A. Cyclosporin A Inhibits the in Vivo Production of Interleukin-1beta and Tumour Necrosis Factor Alpha, but Not Interleukin-6, by a T-Cell-Independent Mechanism. Cytokine 1996, 8, 882–888. [Google Scholar] [CrossRef]
- Vila, N.; Castillo, J.; Dávalos, A.; Esteve, A.; Planas, A.M.; Chamorro, A. Levels of Anti-Inflammatory Cytokines and Neurological Worsening in Acute Ischemic Stroke. Stroke 2003, 34, 671–675. [Google Scholar] [CrossRef]
- Spera, P.A.; Ellison, J.A.; Feuerstein, G.Z.; Barone, F.C. IL-10 Reduces Rat Brain Injury Following Focal Stroke. Neurosci. Lett. 1998, 251, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, W.D.; Busto, R.; Bethea, J.R. Postischemic Hypothermia and IL-10 Treatment Provide Long-Lasting Neuroprotection of CA1 Hippocampus Following Transient Global Ischemia in Rats. Exp. Neurol. 1999, 158, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Strle, K.; Zhou, J.H.; Shen, W.H.; Broussard, S.R.; Johnson, R.W.; Freund, G.G.; Dantzer, R.; Kelley, K.W. Interleukin-10 in the Brain. Crit. Rev. Immunol. 2001, 21, 427–449. [Google Scholar] [CrossRef] [PubMed]
- Le, D.A.; Wu, Y.; Huang, Z.; Matsushita, K.; Plesnila, N.; Augustinack, J.C.; Hyman, B.T.; Yuan, J.; Kuida, K.; Flavell, R.A.; et al. Caspase Activation and Neuroprotection in Caspase-3-Deficient Mice after In Vivo Cerebral Ischemia and In Vitro Oxygen Glucose Deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15188–15193. [Google Scholar] [CrossRef]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.; Sasaki, T.; Elia, A.J.; Cheng, H.Y.; Ravagnan, L.; et al. Essential Role of the Mitochondrial Apoptosis-Inducing Factor in Programmed Cell Death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Eldadah, B.A.; Faden, A.I. Caspase Pathways, Neuronal Apoptosis, and CNS Injury. J. Neurotrauma 2000, 17, 811–829. [Google Scholar] [CrossRef] [PubMed]
- Ekshyyan, O.; Aw, T.Y. Apoptosis: A Key in Neurodegenerative Disorders. Curr. Neurovasc. Res. 2004, 1, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical Pathways of Caspase Activation during Apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef]
- Velier, J.J.; Ellison, J.A.; Kikly, K.K.; Spera, P.A.; Barone, F.C.; Feuerstein, G.Z. Caspase-8 and Caspase-3 Are Expressed by Different Populations of Cortical Neurons Undergoing Delayed Cell Death after Focal Stroke in the Rat. J. Neurosci. 1999, 19, 5932–5941. [Google Scholar] [CrossRef]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor Suppressor P53 Is a Regulator of Bcl-2 and Bax Gene Expression In Vitro and In Vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar] [PubMed]
- Kanekar, S.G.; Zacharia, T.; Roller, R. Imaging of Stroke: Part 2, Pathophysiology at the Molecular and Cellular Levels and Corresponding Imaging Changes. AJR Am. J. Roentgenol. 2012, 198, 63–74. [Google Scholar] [CrossRef]
- Tomandl, B.F.; Klotz, E.; Handschu, R.; Stemper, B.; Reinhardt, F.; Huk, W.J.; Eberhardt, K.E.; Fateh-Moghadam, S. Comprehensive Imaging of Ischemic Stroke with Multisection CT. Radiographics 2003, 23, 565–592. [Google Scholar] [CrossRef]
- Magnetic Resonance Imaging of the Brain and Spine, 4th Ed., Vol. 1 and 2. AJNR Am. J. Neuroradiol. 2009, 30, e76–e77. [CrossRef]
- Provenzale, J.M.; Jahan, R.; Naidich, T.P.; Fox, A.J. Assessment of the Patient with Hyperacute Stroke: Imaging and Therapy. Radiology 2003, 229, 347–359. [Google Scholar] [CrossRef]
- Schaefer, P.W.; Grant, P.E.; Gonzalez, R.G. Diffusion-Weighted MR Imaging of the Brain. Radiology 2000, 217, 331–345. [Google Scholar] [CrossRef]
- Wu, L.; Liu, J.; Li, P.; Tang, B.; James, T.D. Two-Photon Small-Molecule Fluorescence-Based Agents for Sensing, Imaging, and Therapy within Biological Systems. Chem. Soc. Rev. 2021, 50, 702–734. [Google Scholar] [CrossRef]
- He, X.; Zhang, L.; Qi, H.; Yu, P.; Fei, J.; Mao, L. Improving the Fluorescence Detection Limit with Positively Charged Carbon Nanostructures as a Low Background Signal Platform. Analyst 2014, 139, 2114–2117. [Google Scholar] [CrossRef]
- Wang, P.; Yu, L.; Gong, J.; Xiong, J.; Zi, S.; Xie, H.; Zhang, F.; Mao, Z.; Liu, Z.; Kim, J.S. An Activity-Based Fluorescent Probe for Imaging Fluctuations of Peroxynitrite (ONOO−) in the Alzheimer’s Disease Brain. Angew. Chem. Int. Ed. 2022, 61, e202206894. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, Q.; Xu, F.; Li, Y.; Kim, D.; Chung, J.; Baek, G.; Wu, X.; Hillman, P.F.; Lee, E.Y.; et al. An Activatable AIEgen Probe for High-Fidelity Monitoring of Overexpressed Tumor Enzyme Activity and Its Application to Surgical Tumor Excision. Angew. Chem. Int. Ed. Engl. 2020, 59, 10186–10195. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.-B.; Wang, Z.-Y.; Xiang, Z.; Lu, P.; Lai, H.-H.; Yuan, L.; Zhang, X.-B.; Tan, W. A General Strategy for Development of Activatable NIR-II Fluorescent Probes for In Vivo High-Contrast Bioimaging. Angew. Chem. Int. Ed. Engl. 2021, 60, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Zhu, A.; Tian, Y. Functional Surface Engineering of C-Dots for Fluorescent Biosensing and In Vivo Bioimaging. Acc. Chem. Res. 2014, 47, 20–30. [Google Scholar] [CrossRef]
- Tarkin, J.M.; Joshi, F.R.; Rajani, N.K.; Rudd, J.H.F. PET Imaging of Atherosclerosis. Future Cardiol. 2015, 11, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis--an Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Wentzel, J.J.; Chatzizisis, Y.S.; Gijsen, F.J.H.; Giannoglou, G.D.; Feldman, C.L.; Stone, P.H. Endothelial Shear Stress in the Evolution of Coronary Atherosclerotic Plaque and Vascular Remodelling: Current Understanding and Remaining Questions. Cardiovasc. Res. 2012, 96, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial Infarction Accelerates Atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef]
- Munjal, A.; Gupta, N. Radiopharmaceuticals. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Amsallem, M.; Saito, T.; Tada, Y.; Dash, R.; McConnell, M.V. Magnetic Resonance Imaging and Positron Emission Tomography Approaches to Imaging Vascular and Cardiac Inflammation. Circ. J. 2016, 80, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- LaForest, R.; Woodard, P.K.; Gropler, R.J. Cardiovascular PET/MRI: Challenges and Opportunities. Cardiol. Clin. 2016, 34, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Bengel, F.M.; Gambhir, S.S. Cardiovascular Molecular Imaging. Radiology 2007, 244, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Osborn, E.A.; Kessinger, C.W.; Tawakol, A.; Jaffer, F.A. Metabolic and Molecular Imaging of Atherosclerosis and Venous Thromboembolism. J. Nucl. Med. 2017, 58, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Marnane, M.; Merwick, A.; Sheehan, O.C.; Hannon, N.; Foran, P.; Grant, T.; Dolan, E.; Moroney, J.; Murphy, S.; O’Rourke, K.; et al. Carotid Plaque Inflammation on 18F-Fluorodeoxyglucose Positron Emission Tomography Predicts Early Stroke Recurrence. Ann. Neurol. 2012, 71, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Chimowitz, M.; Brown, R.D.; Lal, B.K.; Meschia, J.F. The Urgent Need for Contemporary Clinical Trials in Patients with Asymptomatic Carotid Stenosis. Neurology 2016, 87, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Tawakol, A.; Singh, P.; Rudd, J.H.F.; Soffer, J.; Cai, G.; Vucic, E.; Brannan, S.P.; Tarka, E.A.; Shaddinger, B.C.; Sarov-Blat, L.; et al. Effect of Treatment for 12 Weeks with Rilapladib, a Lipoprotein-Associated Phospholipase A2 Inhibitor, on Arterial Inflammation as Assessed with 18F-Fluorodeoxyglucose-Positron Emission Tomography Imaging. J. Am. Coll. Cardiol. 2014, 63, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Abdelbaky, A.; Corsini, E.; Figueroa, A.L.; Fontanez, S.; Subramanian, S.; Ferencik, M.; Brady, T.J.; Hoffmann, U.; Tawakol, A. Focal Arterial Inflammation Precedes Subsequent Calcification in the Same Location: A Longitudinal FDG-PET/CT Study. Circ. Cardiovasc. Imaging 2013, 6, 747–754. [Google Scholar] [CrossRef]
- Figueroa, A.L.; Abdelbaky, A.; Truong, Q.A.; Corsini, E.; MacNabb, M.H.; Lavender, Z.R.; Lawler, M.A.; Grinspoon, S.K.; Brady, T.J.; Nasir, K.; et al. Measurement of Arterial Activity on Routine FDG PET/CT Images Improves Prediction of Risk of Future CV Events. JACC Cardiovasc. Imaging 2013, 6, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, A.L.; Takx, R.A.P.; MacNabb, M.H.; Abdelbaky, A.; Lavender, Z.R.; Kaplan, R.S.; Truong, Q.A.; Lo, J.; Ghoshhajra, B.B.; Grinspoon, S.K.; et al. Relationship Between Measures of Adiposity, Arterial Inflammation, and Subsequent Cardiovascular Events. Circ. Cardiovasc. Imaging 2016, 9, e004043. [Google Scholar] [CrossRef] [PubMed]
- Tarkin, J.M.; Joshi, F.R.; Evans, N.R.; Chowdhury, M.M.; Figg, N.L.; Shah, A.V.; Starks, L.T.; Martin-Garrido, A.; Manavaki, R.; Yu, E.; et al. Detection of Atherosclerotic Inflammation by 68Ga-DOTATATE PET Compared to [18F]FDG PET Imaging. J. Am. Coll. Cardiol. 2017, 69, 1774–1791. [Google Scholar] [CrossRef] [PubMed]
- Bozhko, D.; Osborn, E.A.; Rosenthal, A.; Verjans, J.W.; Hara, T.; Kellnberger, S.; Wissmeyer, G.; Ovsepian, S.V.; McCarthy, J.R.; Mauskapf, A.; et al. Quantitative Intravascular Biological Fluorescence-Ultrasound Imaging of Coronary and Peripheral Arteries In Vivo. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Kim, J.W.; Shishkov, M.; Namati, E.; Morse, T.; Shubochkin, R.; McCarthy, J.R.; Ntziachristos, V.; Bouma, B.E.; Jaffer, F.A.; et al. Intra-Arterial Catheter for Simultaneous Microstructural and Molecular Imaging In Vivo. Nat. Med. 2011, 17, 1680–1684. [Google Scholar] [CrossRef]
- Boyer, N.P.; Gupton, S.L. Revisiting Netrin-1: One Who Guides (Axons). Front. Cell. Neurosci. 2018, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Lee, W.-H.; Bae, Y.C.; Suk, K. Axon Guidance Molecules Guiding Neuroinflammation. Exp. Neurobiol. 2019, 28, 311–319. [Google Scholar] [CrossRef]
- Shabani, M.; Haghani, M.; Tazangi, P.E.; Bayat, M.; Shid Moosavi, S.M.; Ranjbar, H. Netrin-1 Improves the Amyloid-β-Mediated Suppression of Memory and Synaptic Plasticity. Brain Res. Bull. 2017, 131, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, J.; Gong, J.; Shen, H.; Wang, X. Expression of Netrin-1 and Its Receptors, Deleted in Colorectal Cancer and Uncoordinated Locomotion-5 Homolog B, in Rat Brain Following Focal Cerebral Ischemia Reperfusion Injury. Neural Regen. Res. 2013, 8, 64–69. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, Y.; Lu, H.; Zhang, Z.; Wang, Y.; Yang, G.-Y. Netrin-1 Overexpression Promotes White Matter Repairing and Remodeling after Focal Cerebral Ischemia in Mice. J. Cereb. Blood Flow Metab. 2013, 33, 1921–1927. [Google Scholar] [CrossRef]
- Lu, H.; Song, X.; Wang, F.; Wang, G.; Wu, Y.; Wang, Q.; Wang, Y.; Yang, G.-Y.; Zhang, Z. Hyperexpressed Netrin-1 Promoted Neural Stem Cells Migration in Mice after Focal Cerebral Ischemia. Front. Cell. Neurosci. 2016, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.D.; Ii, M.; Park, K.W.; Suli, A.; Sorensen, L.K.; Larrieu-Lahargue, F.; Urness, L.D.; Suh, W.; Asai, J.; Kock, G.A.H.; et al. Netrins Promote Developmental and Therapeutic Angiogenesis. Science 2006, 313, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Crouse, D.; Lee, M.; Karnik, S.K.; Sorensen, L.K.; Murphy, K.J.; Kuo, C.J.; Li, D.Y. The Axonal Attractant Netrin-1 Is an Angiogenic Factor. Proc. Natl. Acad. Sci. USA 2004, 101, 16210–16215. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, F.; Chen, Y.; Hao, Q.; Liu, W.; Su, H.; Young, W.L.; Yang, G.-Y. Overexpression of Netrin-1 Induces Neovascularization in the Adult Mouse Brain. J. Cereb. Blood Flow Metab. 2008, 28, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Cai, H. Netrin-1 Induces Angiogenesis via a DCC-Dependent ERK1/2-eNOS Feed-Forward Mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 6530–6535. [Google Scholar] [CrossRef] [PubMed]
- Navankasattusas, S.; Whitehead, K.J.; Suli, A.; Sorensen, L.K.; Lim, A.H.; Zhao, J.; Park, K.W.; Wythe, J.D.; Thomas, K.R.; Chien, C.-B.; et al. The Netrin Receptor UNC5B Promotes Angiogenesis in Specific Vascular Beds. Development 2008, 135, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Castets, M.; Coissieux, M.-M.; Delloye-Bourgeois, C.; Bernard, L.; Delcros, J.-G.; Bernet, A.; Laudet, V.; Mehlen, P. Inhibition of Endothelial Cell Apoptosis by Netrin-1 during Angiogenesis. Dev. Cell 2009, 16, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Larrivée, B.; Freitas, C.; Trombe, M.; Lv, X.; Delafarge, B.; Yuan, L.; Bouvrée, K.; Bréant, C.; Del Toro, R.; Bréchot, N.; et al. Activation of the UNC5B Receptor by Netrin-1 Inhibits Sprouting Angiogenesis. Genes Dev. 2007, 21, 2433–2447. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Le Noble, F.; Yuan, L.; Jiang, Q.; De Lafarge, B.; Sugiyama, D.; Bréant, C.; Claes, F.; De Smet, F.; Thomas, J.-L.; et al. The Netrin Receptor UNC5B Mediates Guidance Events Controlling Morphogenesis of the Vascular System. Nature 2004, 432, 179–186. [Google Scholar] [CrossRef]
- Ding, Q.; Liao, S.-J.; Yu, J. Axon Guidance Factor Netrin-1 and Its Receptors Regulate Angiogenesis after Cerebral Ischemia. Neurosci. Bull. 2014, 30, 683–691. [Google Scholar] [CrossRef]
- Yang, Y.; Zou, L.; Wang, Y.; Xu, K.-S.; Zhang, J.-X.; Zhang, J.-H. Axon Guidance Cue Netrin-1 Has Dual Function in Angiogenesis. Cancer Biol. Ther. 2007, 6, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, Y.; He, X.; Yuan, F.; Lin, X.; Xie, B.; Tang, G.; Huang, J.; Tang, Y.; Jin, K.; et al. Netrin-1 Hyperexpression in Mouse Brain Promotes Angiogenesis and Long-Term Neurological Recovery after Transient Focal Ischemia. Stroke 2012, 43, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Le, T.; Chang, T.T.J.; Habib, A.; Wu, S.; Shen, F.; Young, W.L.; Su, H.; Liu, J. AAV-Mediated Netrin-1 Overexpression Increases Peri-Infarct Blood Vessel Density and Improves Motor Function Recovery after Experimental Stroke. Neurobiol. Dis. 2011, 44, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.A.; Cao, M.; Kang, B.Y.; Liu, Y.; Mehta, J.L.; Hermonat, P.L. Systemic Human Netrin-1 Gene Delivery by Adeno-Associated Virus Type 8 Alters Leukocyte Accumulation and Atherogenesis In Vivo. Gene Ther. 2011, 18, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-J.; Gong, Q.; Chen, X.-R.; Ye, L.-X.; Ding, Q.; Zeng, J.-S.; Yu, J. Netrin-1 Rescues Neuron Loss by Attenuating Secondary Apoptosis in Ipsilateral Thalamic Nucleus Following Focal Cerebral Infarction in Hypertensive Rats. Neuroscience 2013, 231, 225–232. [Google Scholar] [CrossRef]
- Chen, J.; Du, H.; Zhang, Y.; Chen, H.; Zheng, M.; Lin, P.; Lan, Q.; Yuan, Q.; Lai, Y.; Pan, X.; et al. Netrin-1 Prevents Rat Primary Cortical Neurons from Apoptosis via the DCC/ERK Pathway. Front. Cell. Neurosci. 2017, 11, 387. [Google Scholar] [CrossRef]
- Yin, J.-W.; Li, J.; Ren, Y.-M.; Li, Y.; Wang, R.-X.; Wang, S.; Zuo, Y.-X. Dexmedetomidine and Netrin-1 Combination Therapy Inhibits Endoplasmic Reticulum Stress by Regulating the ERK5/MEF2A Pathway to Attenuate Cerebral Ischemia Injury. Front. Neurosci. 2021, 15, 641345. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Chen, R.; Chen, H.; Zhang, Y.; Chen, J.; Lin, P.; Lan, Q.; Yuan, Q.; Lai, Y.; Jiang, X.; et al. Netrin-1 Promotes Synaptic Formation and Axonal Regeneration via JNK1/c-Jun Pathway after the Middle Cerebral Artery Occlusion. Front. Cell. Neurosci. 2018, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, S.; Li, B.; Wang, X.; Sun, C.; Qin, H.; Sun, H. Netrin-1 Overexpression Improves Neurobehavioral Outcomes and Reduces Infarct Size via Inhibition of the Notch1 Pathway Following Experimental Stroke. J. Neurosci. Res. 2017, 95, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Li, G.; Yu, J.; Xu, K.; Wu, W. Receptors, Channel Proteins, and Enzymes Involved in Microglia-Mediated Neuroinflammation and Treatments by Targeting Microglia in Ischemic Stroke. Neuroscience 2021, 460, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, H.; Xu, Y. Crosstalk between Microglia and T Cells Contributes to Brain Damage and Recovery after Ischemic Stroke. Neurol. Res. 2016, 38, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.-Q.; Ma, X.-T.; Hu, Z.-W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.-J.; Tian, D.-S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Wicks, E.E.; Ran, K.R.; Kim, J.E.; Xu, R.; Lee, R.P.; Jackson, C.M. The Translational Potential of Microglia and Monocyte-Derived Macrophages in Ischemic Stroke. Front. Immunol. 2022, 13, 897022. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Zhong, W.; Li, Y.; Zhang, W. Netrin-1 Controls Inflammation in Response to Ischemic Stroke through Altering Microglia Phenotype. Front. Immunol. 2023, 14, 1178638. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Hu, G.; Yao, Q.; Wu, J.; He, Z.; Law, B.Y.-K.; Hu, G.; Zhou, X.; Du, J.; Wu, A.; et al. Microglia Autophagy in Ischemic Stroke: A Double-Edged Sword. Front. Immunol. 2022, 13, 1013311. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Zhong, W.; Li, Y.; Zhang, W. Netrin-1 Attenuates Cerebral Ischemia/Reperfusion Injury by Limiting Mitochondrial ROS and Ca2+ Levels via Activation of AKT Phosphorylation and Mitochondrial m-AAA Protease AFG3L2. FASEB J. 2023, 37, e22805. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, Y.; Yuan, F.; Liu, J.; Zeng, L.; Yang, G.-Y. Overexpression of Netrin-1 Improves Neurological Outcomes in Mice Following Transient Middle Cerebral Artery Occlusion. Front. Med. 2011, 5, 86–93. [Google Scholar] [CrossRef]
- Nehra, G.; Bauer, B.; Hartz, A.M.S. Blood-Brain Barrier Leakage in Alzheimer’s Disease: From Discovery to Clinical Relevance. Pharmacol. Ther. 2022, 234, 108119. [Google Scholar] [CrossRef]
- Villringer, K.; Sanz Cuesta, B.E.; Ostwaldt, A.-C.; Grittner, U.; Brunecker, P.; Khalil, A.A.; Schindler, K.; Eisenblätter, O.; Audebert, H.; Fiebach, J.B. DCE-MRI Blood-Brain Barrier Assessment in Acute Ischemic Stroke. Neurology 2017, 88, 433–440. [Google Scholar] [CrossRef]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood-Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672. [Google Scholar] [CrossRef] [PubMed]
- Kassner, A.; Merali, Z. Assessment of Blood-Brain Barrier Disruption in Stroke. Stroke 2015, 46, 3310–3315. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Stanwell, P.; Cordato, D.; Attia, J.; Levi, C. Reperfusion Therapy in Acute Ischemic Stroke: Dawn of a New Era? BMC Neurol. 2018, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Laufs, U.; Huang, Z.; Nakamura, T.; Huang, P.; Moskowitz, M.A.; Liao, J.K. Stroke Protection by 3-Hydroxy-3-Methylglutaryl (HMG)-CoA Reductase Inhibitors Mediated by Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 1998, 95, 8880–8885. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Demarco, K.M.; Sanchez-Covarrubias, L.; Solinsky, C.M.; Davis, T.P. Transforming Growth Factor-Beta Signaling Alters Substrate Permeability and Tight Junction Protein Expression at the Blood-Brain Barrier during Inflammatory Pain. J. Cereb. Blood Flow Metab. 2009, 29, 1084–1098. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Salayandia, V.M.; Thompson, J.F.; Yang, L.Y.; Estrada, E.Y.; Yang, Y. Attenuation of Acute Stroke Injury in Rat Brain by Minocycline Promotes Blood–Brain Barrier Remodeling and Alternative Microglia/Macrophage Activation during Recovery. J. Neuroinflammation 2015, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: Direct CNS Effects of Sphingosine 1-Phosphate (S1P) Receptor Modulation and Implications in Multiple Sclerosis Therapy. J. Neurol. Sci. 2013, 328, 9–18. [Google Scholar] [CrossRef]
- Tian, D.; Shi, K.; Zhu, Z.; Yao, J.; Yang, X.; Su, L.; Zhang, S.; Zhang, M.; Gonzales, R.J.; Liu, Q.; et al. Fingolimod Enhances the Efficacy of Delayed Alteplase Administration in Acute Ischemic Stroke by Promoting Anterograde Reperfusion and Retrograde Collateral Flow. Ann. Neurol. 2018, 84, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Davis, T.P. Targeting Transporters: Promoting Blood-Brain Barrier Repair in Response to Oxidative Stress Injury. Brain Res. 2015, 1623, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix Metalloproteinases in the Brain and Blood-Brain Barrier: Versatile Breakers and Makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, H.; Tsuchimine, S.; O’Hashi, K.; Sakai, K.; Hattori, K.; Hidese, S.; Nakajima, S.; Chiba, S.; Yoshimura, A.; Fukuzato, N.; et al. Association between Vascular Endothelial Growth Factor-Mediated Blood-Brain Barrier Dysfunction and Stress-Induced Depression. Mol. Psychiatry 2022, 27, 3822–3832. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-Brain Barrier Dysfunction in Ischemic Stroke: Targeting Tight Junctions and Transporters for Vascular Protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Inflammatory Mediators and Modulation of Blood-Brain Barrier Permeability. Cell. Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef]
- Díaz-Castro, B.; Robel, S.; Mishra, A. Astrocyte Endfeet in Brain Function and Pathology: Open Questions. Annu. Rev. Neurosci. 2023, 46, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Bálint, A.; Tornyos, D.; El Alaoui El Abdallaoui, O.; Kupó, P.; Komócsi, A. Network Meta-Analysis of Ticagrelor for Stroke Prevention in Patients at High Risk for Cardiovascular or Cerebrovascular Events. Stroke 2021, 52, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; et al. Aspirin in the Primary and Secondary Prevention of Vascular Disease: Collaborative Meta-Analysis of Individual Participant Data from Randomised Trials. Lancet 2009, 373, 1849–1860. [Google Scholar] [CrossRef]
- Kornfeld, M.; Munsayac, J.R. Antiplatelet Medications. In Inpatient Anticoagulation; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; pp. 47–65. ISBN 978-1-118-06717-8. [Google Scholar]
- Antiplatelet Trialists’ Collaboration. Collaborative Overview of Randomised Trials of Antiplatelet Therapy—I: Prevention of Death, Myocardial Infarction, and Stroke by Prolonged Antiplatelet Therapy in Various Categories of Patients. BMJ 1994, 308, 81–106. [Google Scholar] [CrossRef]
- Canadian Cooperative Study Group A Randomized Trial of Aspirin and Sulfinpyrazone in Threatened Stroke. N. Engl. J. Med. 1978, 299, 53–59. [CrossRef] [PubMed]
- Diener, H.C.; Cunha, L.; Forbes, C.; Sivenius, J.; Smets, P.; Lowenthal, A. European Stroke Prevention Study. 2. Dipyridamole and Acetylsalicylic Acid in the Secondary Prevention of Stroke. J. Neurol. Sci. 1996, 143, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Antithrombotic Trialists’ Collaboration. Collaborative Meta-Analysis of Randomised Trials of Antiplatelet Therapy for Prevention of Death, Myocardial Infarction, and Stroke in High Risk Patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef] [PubMed]
- The Dutch TIA Study Group. The Dutch TIA Trial: Protective Effects of Low-Dose Aspirin and Atenolol in Patients with Transient Ischemic Attacks or Nondisabling Stroke. Stroke 1988, 19, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Farrell, B.; Godwin, J.; Richards, S.; Warlow, C. The United Kingdom Transient Ischaemic Attack (UK-TIA) Aspirin Trial: Final Results. J. Neurol. Neurosurg. Psychiatry 1991, 54, 1044–1054. [Google Scholar] [CrossRef]
- Hansson, L.; Zanchetti, A.; Carruthers, S.G.; Dahlöf, B.; Elmfeldt, D.; Julius, S.; Ménard, J.; Rahn, K.H.; Wedel, H.; Westerling, S. Effects of Intensive Blood-Pressure Lowering and Low-Dose Aspirin in Patients with Hypertension: Principal Results of the Hypertension Optimal Treatment (HOT) Randomised Trial. HOT Study Group. Lancet 1998, 351, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- The European Stroke Prevention Study (ESPS). Principal End-Points. The ESPS Group. Lancet 1987, 2, 1351–1354. [Google Scholar]
- Bousser, M.G.; Eschwege, E.; Haguenau, M.; Lefaucconnier, J.M.; Thibult, N.; Touboul, D.; Touboul, P.J. “AICLA” Controlled Trial of Aspirin and Dipyridamole in the Secondary Prevention of Athero-Thrombotic Cerebral Ischemia. Stroke 1983, 14, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.C.; Easton, J.D.; Farrant, M.; Barsan, W.; Conwit, R.A.; Elm, J.J.; Kim, A.S.; Lindblad, A.S.; Palesch, Y.Y.; Clinical Research Collaboration, Neurological Emergencies Treatment Trials Network, and the POINT Investigators. Clopidogrel and Aspirin in Acute Ischemic Stroke and High-Risk TIA. N. Engl. J. Med. 2018, 379, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Gent, M.; Blakely, J.A.; Easton, J.D.; Ellis, D.J.; Hachinski, V.C.; Harbison, J.W.; Panak, E.; Roberts, R.S.; Sicurella, J.; Turpie, A.G. The Canadian American Ticlopidine Study (CATS) in Thromboembolic Stroke. Lancet 1989, 1, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.L.; Adams, R.; Albers, G.; Alberts, M.J.; Benavente, O.; Furie, K.; Goldstein, L.B.; Gorelick, P.; Halperin, J.; Harbaugh, R.; et al. Guidelines for Prevention of Stroke in Patients with Ischemic Stroke or Transient Ischemic Attack: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association Council on Stroke: Co-Sponsored by the Council on Cardiovascular Radiology and Intervention: The American Academy of Neurology Affirms the Value of This Guideline. Stroke 2006, 37, 577–617. [Google Scholar] [CrossRef] [PubMed]
- CAPRIE Steering Committee A Randomised, Blinded, Trial of Clopidogrel versus Aspirin in Patients at Risk of Ischaemic Events (CAPRIE). CAPRIE Steering Committee. Lancet 1996, 348, 1329–1339. [Google Scholar] [CrossRef]
- Tornyos, D.; Komócsi, A.; Bálint, A.; Kupó, P.; El Abdallaoui, O.E.A.; Szapáry, L.; Szapáry, L.B. Antithrombotic Therapy for Secondary Prevention in Patients with Stroke or Transient Ischemic Attack: A Multiple Treatment Network Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2022, 17, e0273103. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishida, Y.; Nakayama, T.; Asai, S. Comparative Effect of Clopidogrel and Aspirin versus Aspirin Alone on Laboratory Parameters: A Retrospective, Observational, Cohort Study. Cardiovasc. Diabetol. 2013, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Goto, S. Cilostazol: Potential Mechanism of Action for Antithrombotic Effects Accompanied by a Low Rate of Bleeding. Atheroscler. Suppl. 2005, 6, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Oh, G.T.; Park, S.Y.; Choi, J.-H.; Park, J.-G.; Kim, C.D.; Lee, W.S.; Rhim, B.Y.; Shin, Y.W.; Hong, K.W. Cilostazol Reduces Atherosclerosis by Inhibition of Superoxide and Tumor Necrosis Factor-Alpha Formation in Low-Density Lipoprotein Receptor-Null Mice Fed High Cholesterol. J. Pharmacol. Exp. Ther. 2005, 313, 502–509. [Google Scholar] [CrossRef]
- Rybalkin, S.D.; Rybalkina, I.; Beavo, J.A.; Bornfeldt, K.E. Cyclic Nucleotide Phosphodiesterase 1C Promotes Human Arterial Smooth Muscle Cell Proliferation. Circ. Res. 2002, 90, 151–157. [Google Scholar] [CrossRef]
- Otsuki, M.; Saito, H.; Xu, X.; Sumitani, S.; Kouhara, H.; Kurabayashi, M.; Kasayama, S. Cilostazol Represses Vascular Cell Adhesion Molecule-1 Gene Transcription via Inhibiting NF-kappaB Binding to Its Recognition Sequence. Atherosclerosis 2001, 158, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.K.; Kim, Y.K.; Kim, K.Y.; Lee, J.H.; Hong, K.W. Remnant Lipoprotein Particles Induce Apoptosis in Endothelial Cells by NAD(P)H Oxidase-Mediated Production of Superoxide and Cytokines via Lectin-like Oxidized Low-Density Lipoprotein Receptor-1 Activation: Prevention by Cilostazol. Circulation 2004, 109, 1022–1028. [Google Scholar] [CrossRef]
- Choi, J.M.; Shin, H.K.; Kim, K.Y.; Lee, J.H.; Hong, K.W. Neuroprotective Effect of Cilostazol against Focal Cerebral Ischemia via Antiapoptotic Action in Rats. J. Pharmacol. Exp. Ther. 2002, 300, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ouyang, M.; Yang, J.; Song, L.; Yang, M.; Anderson, C.S. Anticoagulants for Acute Ischaemic Stroke. Cochrane Database Syst. Rev. 2021, 10, CD000024. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 128, e240–e327. [Google Scholar] [CrossRef] [PubMed]
- Furie, K.L.; Kasner, S.E.; Adams, R.J.; Albers, G.W.; Bush, R.L.; Fagan, S.C.; Halperin, J.L.; Johnston, S.C.; Katzan, I.; Kernan, W.N.; et al. Guidelines for the Prevention of Stroke in Patients with Stroke or Transient Ischemic Attack: A Guideline for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2011, 42, 227–276. [Google Scholar] [CrossRef] [PubMed]
- Chimowitz, M.I.; Lynn, M.J.; Howlett-Smith, H.; Stern, B.J.; Hertzberg, V.S.; Frankel, M.R.; Levine, S.R.; Chaturvedi, S.; Kasner, S.E.; Benesch, C.G.; et al. Comparison of Warfarin and Aspirin for Symptomatic Intracranial Arterial Stenosis. N. Engl. J. Med. 2005, 352, 1305–1316. [Google Scholar] [CrossRef]
- Kay, R.; Wong, K.S.; Yu, Y.L.; Chan, Y.W.; Tsoi, T.H.; Ahuja, A.T.; Chan, F.L.; Fong, K.Y.; Law, C.B.; Wong, A. Low-Molecular-Weight Heparin for the Treatment of Acute Ischemic Stroke. N. Engl. J. Med. 1995, 333, 1588–1593. [Google Scholar] [CrossRef] [PubMed]
- CLOTS (Clots in Legs Or sTockings after Stroke) Trials Collaboration; Dennis, M.; Sandercock, P.; Reid, J.; Graham, C.; Forbes, J.; Murray, G. Effectiveness of Intermittent Pneumatic Compression in Reduction of Risk of Deep Vein Thrombosis in Patients Who Have Had a Stroke (CLOTS 3): A Multicentre Randomised Controlled Trial. Lancet 2013, 382, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhou, W.; Cheng, W.; Liu, Y.; Chang, R. Short-Term and Long-Term Safety and Efficacy of Treatment of Acute Ischemic Stroke with Low-Molecular-Weight Heparin: Meta-Analysis of 19 Randomized Controlled Trials. World Neurosurg. 2020, 141, e26–e41. [Google Scholar] [CrossRef]
- Chen, Z.; Venkat, P.; Seyfried, D.; Chopp, M.; Yan, T.; Chen, J. Brain-Heart Interaction: Cardiac Complications After Stroke. Circ. Res. 2017, 121, 451–468. [Google Scholar] [CrossRef] [PubMed]
- Samuels, M.A. The Brain-Heart Connection. Circulation 2007, 116, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.; Baur, L.H.B.; Pisters, R.; Kamp, O.; Verheugt, F.W.A.; Smeets, J.L.R.M.; Cheriex, E.C.; Tieleman, R.G.; Prins, M.H.; Crijns, H.J.G.M.; et al. Feasibility of TEE-Guided Stroke Risk Assessment in Atrial Fibrillation-Background, Aims, Design and Baseline Data of the TIARA Pilot Study. Neth. Heart J. 2011, 19, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Bascuñana, P.; Hess, A.; Borchert, T.; Wang, Y.; Wollert, K.C.; Bengel, F.M.; Thackeray, J.T. 11C-Methionine PET Identifies Astroglia Involvement in Heart–Brain Inflammation Networking After Acute Myocardial Infarction. J. Nucl. Med. 2020, 61, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Grotta, J.C.; Alexandrov, A.V. tPA-Associated Reperfusion after Acute Stroke Demonstrated by SPECT. Stroke 1998, 29, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Hervella, P.; Rodríguez-Castro, E.; Rodríguez-Yáñez, M.; Arias, S.; Santamaría-Cadavid, M.; López-Dequidt, I.; Estany-Gestal, A.; Maqueda, E.; López-Loureiro, I.; Sobrino, T.; et al. Intra- and Extra-Hospital Improvement in Ischemic Stroke Patients: Influence of Reperfusion Therapy and Molecular Mechanisms. Sci. Rep. 2020, 10, 3513. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, A.V.; Hall, C.E.; Labiche, L.A.; Wojner, A.W.; Grotta, J.C. Ischemic Stunning of the Brain: Early Recanalization Without Immediate Clinical Improvement in Acute Ischemic Stroke. Stroke 2004, 35, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Jia, B.; Zhang, X.; Huo, X.; Chen, J.; Gui, L.; Cai, Y.; Guo, Z.; Han, Y.; Peng, Z.; et al. Efficacy and Safety of Butylphthalide in Patients with Acute Ischemic Stroke: A Randomized Clinical Trial. JAMA Neurol. 2023, 80, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Lang, W.; Stadler, C.H.; Poljakovic, Z.; Fleet, D.; Lyse Study Group. A Prospective, Randomized, Placebo-Controlled, Double-Blind Trial about Safety and Efficacy of Combined Treatment with Alteplase (Rt-PA) and Cerebrolysin in Acute Ischaemic Hemispheric Stroke. Int. J. Stroke 2013, 8, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-Interacting Protein Kinase 1 (RIPK1) as a Therapeutic Target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Li, Y.; Zou, C.; Chen, C.; Li, S.; Zhu, Z.; Fan, Q.; Pang, R.; Li, F.; Chen, Z.; Wang, Z.; et al. Myeloid-Derived MIF Drives RIPK1-Mediated Cerebromicrovascular Endothelial Cell Death to Exacerbate Ischemic Brain Injury. Proc. Natl. Acad. Sci. USA 2023, 120, e2219091120. [Google Scholar] [CrossRef] [PubMed]
- Heit, J.J.; Wintermark, M. Imaging Selection for Reperfusion Therapy in Acute Ischemic Stroke. Curr. Treat. Options Neurol. 2015, 17, 332. [Google Scholar] [CrossRef]
- Astrup, J.; Siesjö, B.K.; Symon, L. Thresholds in Cerebral Ischemia—The Ischemic Penumbra. Stroke 1981, 12, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Meerwaldt, R.; Slart, R.H.J.A.; van Dam, G.M.; Luijckx, G.-J.; Tio, R.A.; Zeebregts, C.J. PET/SPECT Imaging: From Carotid Vulnerability to Brain Viability. Eur. J. Radiol. 2010, 74, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Shimosegawa, E.; Hatazawa, J.; Ibaraki, M.; Toyoshima, H.; Suzuki, A. Metabolic Penumbra of Acute Brain Infarction: A Correlation with Infarct Growth. Ann. Neurol. 2005, 57, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Heiss, W.-D. The Ischemic Penumbra: Correlates in Imaging and Implications for Treatment of Ischemic Stroke. The Johann Jacob Wepfer Award 2011. Cerebrovasc. Dis. 2011, 32, 307–320. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Stage | Timeframe | Imaging Features |
|---|---|---|
| Hyperacute Stage | Less Than 12 h |
|
| Acute Stage | 12–24 h |
|
| Subacute Stage | 2 Days–2 Weeks |
|
| Chronic Stage | 2 Weeks–2 Months |
|
| Method | Type of Imaging | Invasiveness | Imaging Contrast Agents | Resolution |
|---|---|---|---|---|
| MRI | Structural/ functional | Non-invasive | MNPs, Gd- nanoparticles | 1–2 mm |
| PET | Structural/ functional | Non-invasive | 18F, 89Zr, nanoparticles | 4–5 mm |
| SPECT | Structural/ functional | Non-invasive | 18F, 64Cu, 11C Tracers/99mTc, 123/124/125/131I, 111In | 4–15 mm |
| CT | Structural/ functional | Non-invasive | AuNPs, iodine-based nanoparticles | 1 mm |
| Angiography | Structural | Invasive | iodine-based | 0.16 mm |
| OCT | Structural | Invasive | ICAM-1-targeting gold nanoshells | 0.005–0.02 mm |
| IVUS | Structural | Invasive | - | 0.1 mm |
| OFDI | Structural | Invasive | Imaging agents with emission wavelengths between 650 nm and 1000 nm | 0.01–0.02 mm |
| NIRF | Functional | Invasive | Indocyanine green (ICG) and methylene blue | 0.012 mm |
| Molecular Imaging Agent | Imaging Modality | Target Molecule/Process | Clinical Applications |
|---|---|---|---|
| Fluorodeoxyglucose (FDG) | PET | Glucose uptake | Assess inflammation in atherosclerotic plaques |
| 18F-Sodium Fluoride (NaF) | PET | Microcalcifications | Assess plaque stability and vulnerability to rupture |
| 99mTc-Annexin V | SPECT/PET | Phosphatidylserine | Detect plaque vulnerability and assess risk of rupture |
| 99mTc-DTPA-mannosyl-dextran | SPECT | Mannose receptor | Target macrophages within atherosclerotic plaques |
| 18F-Fluoromethylcholine (FCH) | PET | Choline uptake | Assess macrophage infiltration in atherosclerotic plaques |
| 68Ga-dotatate | PET | Somatostatin receptor subtype 2 | Detect plaque inflammation |
| Gadolinium-based contrast agents (GBCAs) | MRI | N/A | Visualize plaque morphology and composition |
| Drug Discovery | Description |
|---|---|
| Matrix Metalloproteinases (MMPs) [124] |
|
| |
| Vascular Endothelial Growth Factor (VEGF) [125] |
|
| |
| Endothelial Cell Tight Junction Proteins [126] |
|
| |
| Inflammatory Mediators [127] |
|
| |
| Astrocytic Endfeet [128] |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, S.; Nadeem, A.; Akram, U.; Sarwar, A.; Quraishi, A.; Siddiqui, H.; Malik, M.A.J.; Nabi, M.; Ul Haq, I.; Cho, A.; et al. Molecular Mechanisms of Ischemic Stroke: A Review Integrating Clinical Imaging and Therapeutic Perspectives. Biomedicines 2024, 12, 812. https://doi.org/10.3390/biomedicines12040812
Rehman S, Nadeem A, Akram U, Sarwar A, Quraishi A, Siddiqui H, Malik MAJ, Nabi M, Ul Haq I, Cho A, et al. Molecular Mechanisms of Ischemic Stroke: A Review Integrating Clinical Imaging and Therapeutic Perspectives. Biomedicines. 2024; 12(4):812. https://doi.org/10.3390/biomedicines12040812
Chicago/Turabian StyleRehman, Sana, Arsalan Nadeem, Umar Akram, Abeer Sarwar, Ammara Quraishi, Hina Siddiqui, Muhammad Abdullah Javed Malik, Mehreen Nabi, Ihtisham Ul Haq, Andrew Cho, and et al. 2024. "Molecular Mechanisms of Ischemic Stroke: A Review Integrating Clinical Imaging and Therapeutic Perspectives" Biomedicines 12, no. 4: 812. https://doi.org/10.3390/biomedicines12040812
APA StyleRehman, S., Nadeem, A., Akram, U., Sarwar, A., Quraishi, A., Siddiqui, H., Malik, M. A. J., Nabi, M., Ul Haq, I., Cho, A., Mazumdar, I., Kim, M., Chen, K., Sepehri, S., Wang, R., Balar, A. B., Lakhani, D. A., & Yedavalli, V. S. (2024). Molecular Mechanisms of Ischemic Stroke: A Review Integrating Clinical Imaging and Therapeutic Perspectives. Biomedicines, 12(4), 812. https://doi.org/10.3390/biomedicines12040812